

Abstract
We report herein the synthesis and evaluation of a series of new pramipexole derivatives as highly potent and selective dopamine-3 (D3) receptor agonists. A number of these new compounds bind to the D3 receptor with subnanomolar affinities and show excellent selectivity (>10,000) for the D3 receptor over the D1 and D2 receptors. Compound 23 for example, binds to the D3 receptor with a Ki value of 0.53 nM and shows a selectivity of >20,000 over the D2 receptor and the D1 receptor in the binding assays using a rat brain preparation. It has excellent stability in human liver microsomes and in vitro functional assays showed it to be a full agonist for the human D3 receptor.
Keywords: pramipexole derivatives, dopamine 3, receptor, agonists, microsomal stability
Introduction
The dopamine 3 (D3) receptor subtype has been identified as a major target for several agents currently used clinically for the treatment of schizophrenia, Parkinson’s disease, depression, and other neurological diseases.[1–3] Because all of the clinically approved drugs target not only the D3 receptor, but also have high affinities for the D2 receptor and have other off-target effects[4, 5] there is a need to design potent and selective D3 ligands. Such new compounds can be used to further investigate the role of the D3 receptor in different biological and pharmacological processes and to validate unambiguously the D3 receptor as an important therapeutic target in preclinical and clinical studies.
Due to the high degree of sequence homology between the D2 and D3 receptors and nearly identical amino acid sequences that form their binding sites, design of potent and highly selective D3 ligands has been a challenge for many years.[4, 5] However, recent studies, including those from our laboratory, have shown that it is possible to design highly selective and potent D3 ligands.[6–8] For example, based upon pramipexole (1), a potent D3 agonist with only a modest selectivity over the D2 receptor, we have developed CJ-1368 (2) and CJ-1639 (3) as potent and selective D3 agonists (Figure 1).[8] More recently, we reported the design of highly selective D3 antagonists based upon the structure of tranylcypromine.[9]
Figure 1.
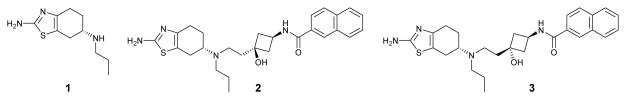
Chemical structures of pramipexole (1) and two potent and selective D3 agonists.
Both compounds 2 and 3 display a high affinity for the D3 receptor and excellent selectivity over the D2 and D1 receptors, and thus are promising lead compounds. Toward identification of highly selective D3 agonists that can be used for investigation in vivo, we have performed further evaluations of compounds 2 and 3. It was found that these compounds have moderate or poor human liver microsomal stability, a shortcoming for their use in vivo. In the present study, we have performed modifications of these compounds with the objective of improving their metabolic stability, as well as further enhancing their selectivity for the D3 over the D2 receptor, while maintaining high affinity to the D3 receptor.
Results and Discussion
Microsomal stability testing showed that compound 2 has a t1/2 of 26 minutes and compound 3 has a t1/2 of 9.5 minutes in human liver microsomes (Table 1). In order to improve the metabolic stability of the compounds, we analyzed the metabolites of compound 2 in human liver microsomes. Our results suggest that hydroxylation of the electron-rich naphthalene group is the major metabolic biotransformation in human liver microsomes (Figure 2 and Supporting Information- SI). Accordingly, in this study we focused our modifications on the naphthalene group.
Table 1.
Human liver microsomal stability of representative compounds. Percentage remaining of compounds in human liver microsomes at selected time points.a
| Time (min) | Compounds | ||||
|---|---|---|---|---|---|
| 2 | 3 | 20 | 21 | 23 | |
| 0 | 100 | 100 | 100 | 100 | 100 |
| 5 | 84.6 | 79.2 | 97.7 | 99.7 | 96.6 |
| 10 | 72.2 | 49.9 | 94.1 | 98.5 | 99.2 |
| 15 | 63.6 | 34.9 | 94.0 | 92.5 | 99.0 |
| 30 | 41.1 | 11.7 | 85.5 | 83.0 | 90.0 |
| 60 | 20.1 | 2.1 | 76.7 | 74.1 | 65.5 |
| t1/2 (min) | 26.1 | 9.48 | >60 | >60 | >60 |
Data represent a single determination.
Figure 2.
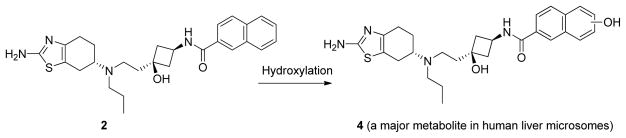
Major biotransformation of compound 2 in human liver microsomes.
We hypothesized that replacement of the naphthyl group in compound 2 with less electron-rich groups may improve the metabolic stability and accordingly, a series of new compounds (11-23) with groups less electron-rich than the naphthyl group, such as unsubstituted or substituted phenyl rings, were synthesized and evaluated.
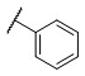
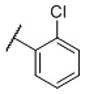
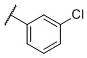
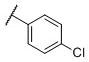
These compounds were first evaluated for their binding affinities to the rat D1-like, D2-like and D3 receptors using a rat brain preparation (Table 2). Our binding data showed that all compounds with a phenyl ring, substituted or unsubstituted, have high affinities for the D3 receptor, with Ki values ranging from 0.43–1.9 nM. Compound 11, which has a phenyl group in place of the naphthyl group, binds to the D3 receptor with a Ki value of 0.74 nM and is 301 times more selective for the D3 receptor over the D2 receptor. Chlorine substitution at the o-, m- and p-positions on the phenyl group results in compounds 12, 13 and 14, respectively. These have similar affinities to the D3 receptor and also similar selectivities over the D2 receptor, as compound 11. Fluorine substitution at the three different positions on the phenyl ring yields compounds 15-17. While compounds 16 and 17 with a m-fluoro or a p-fluoro substituent have similar affinities for the D3 receptor and also similar selectivities over the D2 receptor, as compound 11, compound 15 with the o-fluoro substituent has a high affinity for the D3 receptor and a selectivity of >30,000-fold over the D2 receptor. The m-chloro p-fluoro compound (18) also has a high affinity for the D3 receptor (Ki = 0.43 nM) and an outstanding selectivity (>15,000-fold) over the D2 receptor. Methoxyl substitution at three different positions on the phenyl ring generates compounds 19-21 which all have similar binding affinities to the D3 receptor (0.70–1.0 nM). The m-methoxy compound (21) has a selectivity of >12,000-fold over the D2 receptor, highest among these three compounds. Hence, it appears that while substitution on the phenyl ring has modest effect on the affinity for the D3 receptor, it can have a major effect on the selectivity for the D3 receptor over the D2 receptor.
Table 2.
Binding affinities of original lead compound 2 and new compounds 11-23 at the D1-like, D2-like, and D3 receptors in binding assays using rat brain preparation.
| ||||||
|---|---|---|---|---|---|---|
| ligand | R | Ki±SEM (nM) | Selectivity | |||
| D3 | D2-like | D1-like | D2-like/D3 | D1-like/D3 | ||
| 2 |
|
0.40±0.087 | 725±45 | 1,610±167 | 1,827 | 4,025 |
| 11 |
|
0.74±0.038 | 224±9 | 1,970±179 | 301 | 2,662 |
| 12 |
|
0.62±0.068 | 72±11 | 49,700±2,807 | 117 | 80,161 |
| 13 |
|
0.58±0.062 | 101±12 | 28,700±2,640 | 175 | 49,482 |
| 14 |
|
1.6±0.19 | 244±46 | 15,113±780 | 157 | 9,751 |
| 15 |
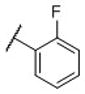
|
0.46±0.06 | 17,000±4,092 | 53,600±8,380 | 36,956 | 116,521 |
| 16 |
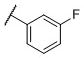
|
1.9±0.087 | 264±46 | 56,600±2,730 | 138 | 29,789 |
| 17 |
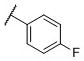
|
0.89±0.17 | 367±27 | 50,000±4,890 | 412 | 56,180 |
| 18 |
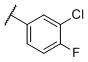
|
0.43±0.06 | 6,950±1,095 | 28,200±5,308 | 16,272 | 65,581 |
| 19 |
|
1.0±0.094 | 158±25 | 43,500±711 | 160 | 44,500 |
| 20 |
|
0.70±0.055 | 2,660±461 | 42,800±3,279 | 3,779 | 61,143 |
| 21 |
|
0.76±0.036 | 9,790±1,500 | 64,200±1,760 | 12,836 | 84,474 |
| 22 |
|
0.96±0.10 | 14,600±1,280 | 19,200±2,550 | 15,208 | 20,000 |
| 23 |
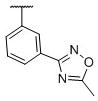
|
0.53±0.07 | 15,800±5,910 | 15,000±2,920 | 29,800 | 28,300 |
D3 receptor binding was determined using [3H]7-OH-DPAT and membranes prepared from ventral striatum.
D2-like receptor binding (D2, D3, and D4) was determined using [3H]spiperone and striatal membranes. D2-like receptor binding (D2, D3, and D4) was determined using [3H]spiperone and striatal membranes. For those compounds that produced a competition curve consistent with inhibition at 2-sites in this assay, which is consistent with the behavior of agonists in this assay, the Ki value for the high affinity component is reported. D1-like receptor binding (D1 and D5) was determined using [3H]SCH23390 and striatal membranes.
Data are the mean ± SEM of 3–6 independent determinations.
In addition to these simple substituents, we also synthesized compounds 22 and 23 containing a 5-methyl-1,2,4-oxadiazol-3-yl substituent at the m- or p-position respectively, of the phenyl ring. Compounds 22 and 23 bind to the D3 receptor with Ki values of 0.96 nM and 0.53 nM, respectively, and both show very high selectivities (>15,000) over the D2 receptor.
In addition to their high selectivity for the D3 receptor over the D2 receptor, compounds 15, 18, 21, 22 and 23 are also highly selective for the D3 receptor over the D1 receptor.
We next synthesized a series of compounds (24-35) in which the amide group in compounds 11-23 was replaced by a sulfonamide group (Table 3). These compounds retain high affinities to the D3 receptor, but they are much less selective than the most selective compounds with the amide group. Compound 34 is the most selective compound among compounds 24-35 containing a sulfonamide group but has a selectivity of only 1052-fold for the D3 receptor over the D2 receptor and consequently, this series of compounds was not pursued.
Table 3.
Binding affinities of new compounds 24-35 at the D1-like, D2-like, and D3 receptors in binding assays using rat brain preparation.
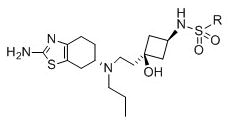
| ||||||
|---|---|---|---|---|---|---|
| Ki±SEM (nM) | Selectivity | |||||
|
|
||||||
| ligand | R | D3 | D2-like | D1-like | D2-like/D3 | D1-like/D3 |
| 24 |
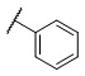
|
1.9±0.17 | 81±11 | 57,000±2,830 | 42 | 30,000 |
| 25 |
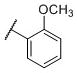
|
2.3±0.22 | 170±18 | 1,810±93 | 75 | 800 |
| 26 |
|
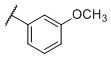
1.6±0.16 | 160±8.5 | 36,500±2,243 | 98 | 22,812 |
| 27 |
|
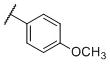
1.6±0.090 | 102±15 | 2,610±255 | 65 | 1,631 |
| 28 |
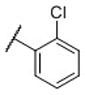
|
0.59±0.057 | 286±49 | 94,900±7,620 | 485 | 160,847 |
| 29 |
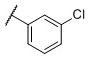
|
2.5±0.15 | 71±4.8 | 68,000±2,300 | 28 | 27,200 |
| 30 |
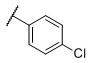
|
2.6±0.20 | 542±79 | 45,900±2,490 | 205 | 17,654 |
| 31 |
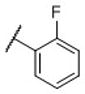
|
1.0±0.072 | 619±68 | 109,000±2,500 | 614 | 109,000 |
| 32 |
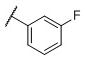
|
1.2±0.091 | 110±14 | 117,000±10,170 | 91 | 97,500 |
| 33 |
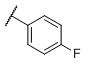
|
1.9±0.25 | 789±36 | 102,000±5,390 | 414 | 53,684 |
| 34 |
|
2.3±0.25 | 2,420±118 | 41,900±2,764 | 1,052 | 18,217 |
| 35 |
|
3.1±0.26 | 57±7.4 | 76,000±6,810 | 19 | 24,516 |
D3 receptor binding was determined using [3H]7-OH-DPAT and membranes prepared from ventral striatum.
D2-like receptor binding (D2, D3, and D4) was determined using [3H]spiperone and striatal membranes. D2-like receptor binding (D2, D3, and D4) was determined using [3H]spiperone and striatal membranes. For those compounds that produced a competition curve consistent with inhibition at 2-sites in this assay, which is consistent with the behavior of agonists in this assay, the Ki value for the high affinity component is reported. D1-like receptor binding (D1 and D5) was determined using [3H]SCH23390 and striatal membranes.
Data are the mean ±SEM of 3–6 independent determinations.
We next evaluated the human liver microsomal stability for compounds 20, 21 and 23, three potent and highly selective D3 ligands, in direct comparison with compounds 2 and 3. The results are provided in Table 1. Our data showed that compounds 20, 21 and 23 all have t1/2 >60 minutes, indicating that the microsomal stability of these compounds is indeed much improved over that of compounds 2 or 3.
We next evaluated compounds 15, 18, 21, 23, 33 and 34 for their functional activity at the human D3 receptor in a quinpirole-induced mitogenesis assay in hD3-transfected CHOp cells. The results, presented in Table 4 show that all these compounds behave as highly potent full D3 agonists.
Table 4.
Functional activity of representative D3 ligands in the quinpirole-induced mitogenesis assay in CHO cells transfected with the human D3 receptor.a
| Ligands | Agonist activity: IC50 (nM) | % of Stimulation compared to Quinpirole as the standard agonist |
|---|---|---|
| 15 | 2.0±0.49 | 100 |
| 18 | 2.0±0.34 | 100 |
| 21 | 18±2.7 | 100 |
| 23 | 19±1.5 | 100 |
| 33 | 17±4.4 | 100 |
| 34 | 26±4.1 | 100 |
Data are the mean ± SEM of 3–6 independent determinations.
Synthesis of designed compounds
The synthetic route to compounds 11-23 is shown in Scheme 1. The commercially available cyclobutanone (5) was reacted with allymagnesium bromide at −78 °C to give cis-allycyclobutanol (6). The stereochemistry of 6 was confirmed by transformation to a known compound 7, whose stereochemistry was determined by x-ray crystallographic analysis.[8] The aldehyde (8) was obtained by oxidation of 6 with NaIO4 and a catalytic amount of OsO4. Pramipexole (1) was allowed to react with 8 to give the key intermediate 9 which, treated with trifluoroacetic acid afforded the amine 10. This amine (10) was reacted with commercially available acid chlorides in the presence of N,N-diisopropylethylamine (DIPEA) and the resulting crude amides 11-23 were purified by preparative HPLC.
Scheme 1.

Synthesis of compounds 11-23. Conditions and reagents: a) Allylmagnesium bromide, THF, −78 °C, 4 h, 68%; b) 1. TFA, DCM, RT, 12 h, 2. 2-naphthoyl chloride, DIPEA, DCM, RT, 2 h, 72%; c) OsO4, NalO4, THF-H2O, RT, 30 min, 60%; d) 8, NaBH(OAc)3, HOAc, DCM, 27%; e) TFA, DCM, RT, 12 h, 90%; f) appropriate acid chlorides, DIPEA, DCM, RT, 2 h, 36–66%.
The synthesis of the sulfonamide-containing compounds 24-35 is shown in Scheme 2. Briefly, these compounds were synthesized by reaction of the intermediate amine 10 at room temperature with the appropriate sulfonyl chorides in N,N-diisopropyl-ethylamine and dichloromethane with 40–56% yield.
Scheme 2.
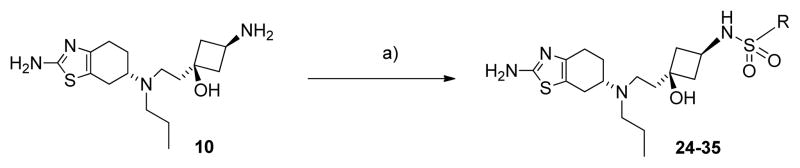
Synthesis of compounds 24-35. Conditions and reagents: a) appropriate sulfonyl chlorides, DIPEA, DCM, RT, 2 h, 40–56%.
Summary
Modifications of the naphthyl group in our previously reported D3 ligand (2) have yielded a series of new compounds with high binding affinities to the D3 receptor and high selectivity over the D1 and D2 receptors and significantly, with improved microsomal stability. Compound 23, for example, binds to the D3 receptor with a Ki value of 0.53 nM, shows a selectivity of >25,000 over the D1 and D2 receptors and superior microsomal stability to that of compound 2 in human liver microsomes. Compound 23 is a full agonist to the human D3 receptor in the quinpirole-induced mitogenesis assay and represents a potent and highly selective D3 agonist.
Experimental Section
Biology
In vitro dopamine receptor binding assays
The binding affinities of all the synthetic compounds were determined at the D1-like, D2-like and D3 receptors in membranes prepared from the brains of adult, male Sprague-Dawley rats (Pel-Freez, Rogers, AR). For these assays, all compounds were dissolved in 100% EtOH at a concentration of 5 mM.
[3H]R-(+)-7-OH-DPAT binding assays
[3H]R-(+)-7-OH-DPAT binding assays for the D3 dopamine receptors were performed as described previously under conditions that yield selective labeling of the D3 receptor.[10] Rat ventral striatal (nucleus accumbens and olfactory tubercles) membrane was prepared in an assay buffer (50 mM Tris, 1 mM EDTA; pH 7.4 at 23 °C) to yield a final concentration of 10 mg original wet weight (o.w.w.)/ml. Membranes were incubated with [3H]R-(+)-7-OH-DPAT (0.15 nM, SA = 163 Ci/mmol, GE Healthcare or SA = 143 Ci/mmol, Perkin-Elmer) and different concentrations of the test compounds (10−10 to 10−4 M). Nonspecific binding was defined by 1 μM spiperone. Assay tubes were incubated at 23 °C for 90 min. The reaction was terminated by rapid vacuum filtration. Data were analyzed using SigmaPlot 8.0.2. using KD = 0.15 nM for [3H]7-OH-DPAT.[10] Ki values are expressed as the mean ± SEM of 3–6 independent determinations.
[3H]Spiperone binding assays
[3H]Spiperone binding assays for D2-like receptors were performed as described previously[11] for [3H] 7-OH-DPAT with the following exception. Assays were performed using membranes prepared from rat caudate-putamen, which expresses D2 receptors in high density but with very low levels of D3 receptors, and the final membrane homogenate concentration was 1.5 mg o.w.w./mL. The assay buffer, 50 mM Tris-HCl, 5 mM KCl, 2 mM MgCl2, and 2 mM CaCl2, pH 7.4 at 23 °C, was used to optimize conditions for agonist binding.[12] The concentration of [3H]spiperone (24 Ci/mmol; SA = 105/mmol, GE Healthcare; SA= 60, American Radiolabeled Chemicals; or SA = 83.4; Perkin-Elmer) was 0.2 nM and the incubation time was 90 min at 23 °C. Nonspecific binding was determined in the presence of 1 μM (+)-butaclamol. Ki values were calculated using the experimentally determined KD value for [3H]spiperone of 0.4 nM. For those compounds that produced a competition curve consistent with inhibition at 2 sites, which is consistent with the behavior of agonists in this assay, the Ki value for the high affinity component is reported.
[3H]SCH 23390 binding assays
[3H]SCH 23390 (7-chloro-3-methyl-1-phenyl-1,2,4,5-tetrahydro-3-benxazepin-8-ol) binding assays for D1-like dopamine receptors were performed as described previously[13] for [3H]spiperone binding except the concentration of [3H]SCH 23390 (SA = 73 Ci/mmol, GE Healthcare or SA = 60 Ci/mmol, Americal Radiolabeled Chemicals) used was 0.3 nM. Ki values were calculated using the KD value for [3H]SCH 23390 of 0.3 nM.
DA D3 Mitogenesis Functional Assay
CHOp-D3 cells were maintained in alpha-MEM with 10% fetal bovine serum (FBS, Atlas Biologicals), 0.05% pen-strep, and 400 μg/ml of G418. To measure D3 stimulation of mitogenesis (agonist assay) or inhibition of quinpirole stimulation of mitogenesis (antagonist assay), CHOp-D3 cells were seeded in a 96-well plate at a concentration of 5,000 cells/well. The cells were incubated at 37 °C in alpha-MEM with 10% FBS. After 48–72 h, the cells were rinsed twice with serum-free alpha-MEM then incubated for 24 h at 37 °C. Serial dilutions of test compounds were made by the Biomek robotics system in serum-free alpha-MEM. In the functional assay for agonists, the medium was removed and replaced with 100 μl of a solution of the test compound in serum-free alpha-MEM. In the antagonist assay, the serial dilution of the putative antagonist test compound was added in 90 μl of the solution (1.1X of final concentration) and 300 nM quinpirole (30 nM final) was added in 10 μl of the solution. After a further 16-h incubation at 37 °C, 0.25 μCi of [3H]thymidine in alpha-MEM supplemented with 10% FBS was added to each well and the plates were further incubated for 2 h at 37 °C. The cells were trypsinized by addition of 10X trypsin solution (1% trypsin in calcium-magnesium-free phosphate-buffered saline) and the plates were filtered and counted. Quinpirole is assayed daily as an internal control and dopamine is included for comparative purposes.
Data analysis
For functional assays, GraphPAD Prism was used to calculate either EC50 (agonists) or IC50 (antagonists) values using data expressed as pg cAMP for adenylate cyclase activity and % quinpirole-stimulation for mitogenesis.
Human Liver Microsomal Stability Assay
1 μM of each test compound was metabolized at 37°C by incubating the test compound with 0.5 mg/mL human liver microsomes and 1 mM cofactor NADPH in a total volume of 400 μL of 100 mM potassium phosphate buffer (pH 7.4 containing 3.3 mM MgCl2). The reactions were stopped at 0, 5, 10, 15, 30, and 60 min, by adding 3 fold the volume of CH3CN containing 100 nM of internal standard. The collected fractions were centrifuged to collect the supernatant for LC-MS/MS analyses, from which the amount of parent compound remaining was determined. The natural log of the amount of parent compound remaining was plotted against time to calculate the rate of disappearance of the tested compounds and the half-life of the tested compounds. The intrinsic clearance of each compound was calculated using the equation:
Hydrolysis of 2, 3, 20, 21, and 23 in human liver microsomes and a PBS-only solution was monitored under similar conditions. 1 μM of each compound was incubated with 0.5 mg/mL human liver microsomes in PBS solution or with PBS only for 60 min. No change in the concentration of three compounds at 60 min compared to that at time zero suggests little hydrolysis had occurred for the three compounds in PBS solution with or without human liver microsomes.
Chemistry
General Methods
Solvents and reagents were purchased and used without further purification. Reactions were monitored by TLC carried out on 250 μm E. Merck silica gel plates (60F-254) using UV light as visualizing agent. E. Merck silica gel (60, particle size 15–40 μm) was used for flash column chromatography. NMR spectra were recorded on a Bruker Avance300 spectrometer (300 MHz). Chemical shifts (δ) are reported as δ values (ppm) downfield relative to TMS as an internal standard, with multiplicities reported in the standard form. All final compounds have purities > 95%, as determined by HPLC (UV detection at 254 nm).
t-Butyl (3-allyl-3-hydroxycyclobutyl)carbamate (6)
1M Allylmagnesium bromide solution in ether (64.8 mL, 64.8 mmol) was added dropwise to a solution of t-butyl (3-oxocyclobutyl)-carbamate 29 (6.0 g, 32.4 mmol) in anhydrous THF at −78 °C and the reaction mixture was stirred at −78 °C for 2 h. Then the mixture was allowed to warm up slowly to room temperature. The reaction was quenched by slow addition of aqueous NH4Cl solution. The mixture was extracted with EtOAc (3×40 mL) and organic layers were combined. The organic solvents were removed under vacuum and the residue was purified by column chromatography (SiO2, hexane/EtOAc 2:1) to give 6 (5.0 g, 68%) as a colorless oil. 1H NMR (CDCl3, 300 MHz) δ 5.90-5.70 (m, 1H), 5.25-5.10 (m, 2H), 4.70 (broad, 1H), 3.80-3.70 (m, 1H), 2.35 (d, J=3.2 Hz, 2H), 2.20 (s, 1H), 2.00-1.80 (m, 2H), 1.44 (s, 9H).
N-(cis-3-Allyl-3-hydroxycyclobutyl)-2-naphthamide (7)
TFA (1 mL) was added to a solution of 30 (500 mg, 2.20 mmol) in CH2Cl2 (5 mL) and the mixture was stirred at room temperature for 12 h. Solvent and TFA were removed under vacuum and the residue was dissolved in DCM (5 mL). Diisopropylethylamine (568 mg, 4.40 mmol) and 2-naphthoyl chloride (502 mg, 2.64 mmol) were added and the mixture was stirred at room temperature for 2 h. The reaction was quenched with H2O and the pH was adjusted to 9–10 by addition of aqueous Na2CO3. The mixture was extracted with CH2Cl2 (30 mLx3). The organic layer was separated, combined, dried, and evaporated. The residue was purified by chromatography (SiO2, hexane/EtOAc 1:1) to give 7 as a colorless solid (445 mg, 72% over two steps). 1H NMR (CDCl3, 300 MHz) δ 8.27 (s, 1H), 7.95-7.75 (m, 4H), 7.65-7.50 (m, 2H), 6.51 (d, J = 6.5 Hz, 1H), 6.00-5.80 (m, 1H), 5.30-5.20 (m, 2H), 4.45-4.27 (m, 1H), 2.80-2.70 (m, 2H), 2.43 (d, J = 7.1 Hz, 2H), 2.36 (s, 1H), 2.25-2.15 (m, 2H).
t-Butyl (3-hydroxy-3-(2-oxoethyl)cyclobutyl)carbamate (8)
OsO4 (223 mg, 0.881 mmol) was added to a solution of 30 (2.0 g, 8.81 mmol) in THF-H2O (80 mL, 1:1 ratio) at room temperature and the mixture was stirred at room temperature for 30 min. Then, NaIO4 (4.71 g, 22 mmol) was added and the mixture was stirred for 30 min. The mixture was extracted with EtOAc (40 mLx3) and organic layers were combined. Evaporation of organic solvents under vacuum gave crude product 8 (1.2 g, crude yield 60%) as a slightly yellow oil. This crude product was used directly for the next step without further purification. Purification of this crude product by silica gel column chromatography led to its decomposition.
t-Butyl (3-(2-(((S)-2-amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)ethyl)-3-hydroxycyclobutyl)carbamate (9)
Compound 8 (8.93 g, 39.0 mmol), acetic acid (3.51 g, 58.5 mmol) and sodium triacetoxyborohydride (12.4 g, 58.5 mmol) were added to a solution of pramipexole (8.3 g, 39.0 mmol) in CH2Cl2 (60 mL) and the reaction mixture was stirred at room temperature for 6 h. The reaction was quenched with H2O and the pH was adjusted to 9–10 by addition of aqueous Na2CO3 solution. The mixture was then extracted 3 times with CH2Cl2. The organic layers were separated, combined, and evaporated. The residue was chromatographed (SiO2, ethyl acetate/methanol 95:5) to give compound 9 (4.5 g, 27% yield). 1H NMR (CDCl3, 300 MHz) δ 4.90 (s, 2H), 4.77 (d, J=7.8 Hz, 1H), 3.80-3.60 (m, 1H), 3.25-3.10 (m, 1H), 2.80-2.35 (m, 10H), 2.00-1.45 (m, 8H), 1.43 (s, 9H), 0.89 (t, J=7.2 Hz, 3H).
3-Amino-1-(2-(((S)-2-amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)ethyl)-cyclobutanol (10)
TFA (5 mL) was added to a solution of 9 (4.5 g, 10.6 mmol) in CH2Cl2 (30 mL) and the mixture was stirred at room temperature for 12 h. Solvent and TFA were removed under vacuum. Water was added to the residue and pH was adjusted to 9–10 by addition of aqueous Na2CO3 solution. The mixture was extracted with CH2Cl2 (30 mLx3). The organic layers were separated, combined, dried, and evaporated. The residue was chromatographed (SiO2, CH2Cl2/MeOH 80:20) to give compound 10 as a colorless oil (3.1 g, 90% yield). 1H NMR (CD3OD, 300 MHz) δ 4.02-3.83 (m, 1H), 3.60-2.60 (m, 11H), 2.40-1.75 (m, 8H), 1.06 (t, J=7.2 Hz, 3H).
General procedure for the synthesis of compounds 11-23
Diisopropylethylamine (52 mg, 0.4 mmol) and the appropriate acid chloride (0.24 mmol) were added to a suspension of 10 (65 mg, 0.2 mmol) in CH2Cl2 (10 mL) and the mixture was stirred at room temperature for 2 h. The reaction was quenched with H2O and pH was adjusted to 9–10 by addition of aqueous Na2CO3. The mixture was extracted three times with CH2Cl2 (30 mLx3). The organic layer was separated, combined, dried, and evaporated. The residue was purified by preparative HPLC to give the product. This procedure was used to prepare compounds 11-23, as detailed below.
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)benzamide (11)
Colorless solid (47 mg, 55%). 1H NMR (CD3OD, 300 MHz) δ 7.83 (d, J=7.1 Hz, 2H), 7.60-7.40 (m, 3H), 4.20-3.90 (m, 2H), 3.60-2.60 (m, 10 H), 2.40-1.80 (m, 8H), 1.08 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.76, 170.03, 135.43, 134.63, 132.77, 129.57, 128.35, 112.93, 69.74, 60.02, 54.44, 44.10, 38.12, 34.42, 23.87, 23.39, 22.94, 19.82, 11.17; m/z 429 [M+H]+.
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-2-chlorobenzamide (12)
Colorless solid (42 mg, 45%). 1H NMR (CD3OD, 300 MHz) δ 7.50-7.37 (m, 4H), 4.18-3.82 (m, 2H), 3.60-2.60 (m, 10H), 2.40-1.70 (m, 8H), 1.07 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.77, 169.62, 137.46, 134.36, 132.20, 131.94, 130.99, 129.79, 128.15, 112.92, 69.64, 59.98, 54.42, 44.00, 37.88, 34.34, 23.83, 23.34, 22.85, 19.79, 11.16; m/z 463 [M+H]+.
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)-amino)ethyl)-3-hydroxycyclobutyl)-3-chlorobenzamide (13)
Colorless solid (52 mg, 56%). 1H NMR (CD3OD, 300 MHz) δ 7.83-7.70 (m, 2H), 7.60-7.40 (m, 2H), 4.18-3.83 (m, 2H), 3.60-2.60 (m, 10H), 2.40-1.72 (m, 8H), 1.08 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.76, 168.32, 137.37, 135.60, 134.46, 132.64, 131.22, 128.50, 126.75, 112.92, 69.71, 59.99, 54.45, 44.01, 38.22, 34.41, 23.85, 23.36, 22.88, 19.81, 11.16; m/z 463 [M+H]+.
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-4-chlorobenzamide (14)
Colorless solid (54 mg, 58%). 1H NMR (CD3OD, 300 MHz) δ 7.81 (d, J=8.6 Hz, 2H), 7.47 (d, J=8.6 Hz, 2H), 4.16-3.83 (m, 2H), 3.60-2.60 (m, 10H), 2.40-1.72 (m, 8H), 1.06 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.77, 168.73, 138.81, 134.41, 134.05, 130.08, 129.72, 112.92, 69.69, 59.99, 54.42, 44.03, 38.17, 34.41, 23.84, 23.35, 22.86, 19.80, 11.16; m/z 463 [M+H]+.
N-(cis-3-(2-(((S)-2-amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-2-fluorobenzamide (15)
Colorless solid (40 mg, 45%). 1H NMR (CD3OD, 300 MHz) δ 7.81-7.77 (m, 1H), 7.67-7.65(m, 1H), 7.42-7.30 (m, 2H), 4.25-4.08 (m, 2H), 3.55-2.83 (m, 10H), 2.46-1.96 (m, 8H), 1.19 (t, J=7.1 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.88, 166.72, 163.01 (d, JF-C=247.9 Hz), 134.49, 134.25 (d, JF-C=8.6 Hz), 131.37 (d, JF-C=2.5 Hz), 125.76 (d, JF-C=3.5 Hz), 124.54 (d, JF-C=13.8 Hz), 117.47 (d, JF-C=22.7 Hz), 113.04, 69.82, 60.10, 54.58, 44.30, 38.09, 34.46, 23.95, 23.45, 22.96, 19.92, 11.24; m/z 447 [M+H]+.
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-3-fluorobenzamide (16)
Colorless solid (32 mg, 36%). 1H NMR (CD3OD, 300 MHz) δ 7.70-7.43 (m, 3H), 7.35-7.24 (m, 1H), 4.16-3.85 (m, 2H), 3.60-2.62 (m, 10H), 2.42-1.74 (m, 8H), 1.08 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.78, 168.37, 164.07 (d, JF-C=244.0 Hz), 137.78 (d, JF-C=6.8 Hz), 134.35, 131.54 (d, JF-C=7.9 Hz), 124.22 (d, JF-C=2.7 Hz), 119.48 (d, JF-C=21.5 Hz), 115.29 (d, JF-C=23.1 Hz), 112.91, 69.71, 59.98, 54.46, 44.01, 38.19, 34.41, 23.83, 23.34, 22.84, 19.78, 11.16; m/z 447 [M+H]+.
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-4-fluorobenzamide (17)
Colorless solid (59 mg, 66%). 1H NMR (CD3OD, 300 MHz) δ 7.90-7.80 (m, 2H), 7.18 (t, J=8.8 Hz, 2H), 4.16-3.83 (m, 2H), 3.60-2.62 (m, 10H), 2.42-1.70 (m, 8H), 1.06 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.77, 168.75, 166.21 (d, JF-C=248.8 Hz), 134.34, 131.75 (d, JF-C=2.9 Hz), 130.97 (d, JF-C=9.0 Hz), 116.36 (d, JF-C=22.1 Hz), 112.90, 69.69, 59.97, 54.43, 44.05, 38.14, 34.41, 23.83, 23.33, 22.84, 19.79, 11.16; m/z 447 [M+H]+.
N-(cis-3-(2-(((S)-2-amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-3-chloro-4-fluorobenzamide (18)
Colorless solid (39 mg, 40%). 1H NMR (CD3OD, 300 MHz) δ 8.01-7.98 (m, 1H), 7.86-7.81(m, 1H), 7.40-7.34 (m, 1H), 4.30-3.80 (m, 2H), 3.65-2.63 (m, 10H), 2.46-1.76 (m, 8H), 1.08 (t, J=7.4 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 170.35, 165.97, 161.60 (d, JF-C=251.3 Hz), 133.16, 131.51 (d, JF-C=3.7 Hz), 129.83, 127.89 (d, JF-C=8.2 Hz), 120.83 (d, JF-C=18.2 Hz), 116.56 (d, JF-C=21.8 Hz), 111.53, 68.28, 58.62, 53.02, 42.60, 36.87, 33.03, 22.46, 21.99, 21.52, 18.42, 9.76; m/z 481 [M+H]+.
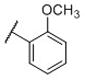
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-2-methoxybenzamide (19)
Colorless solid (50 mg, 54%). 1H NMR (CD3OD, 300 MHz) δ 7.84 (dd, J=1.7, 7.7 Hz, 2H), 7.53-7.45 (m, 1H), 7.14 (d, J=8.3 Hz, 1H), 7.08-7.00 (m, 1H), 4.18-3.83 (m, 2H), 3.92 (s, 3H), 3.60-2.60 (m, 10H), 2.40-1.70 (m, 8H), 1.06 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.77, 167.81, 159.00, 134.33, 134.18, 131.81, 122.99, 121.92, 112.89, 69.86, 59.97, 56.49, 54.43, 44.54, 37.68, 34.39, 23.82, 23.33, 22.83, 19.79, 11.16; m/z 459 [M+H]+.
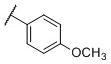
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-4-methoxybenzamide (20)
Colorless solid (41 mg, 45%). 1H NMR (CD3OD, 300 MHz) δ 7.80 (d, J=8.8 Hz, 2H), 6.97 (d, J=8.8 Hz, 2H), 4.18-3.85 (m, 2H), 3.84 (s, 3H), 3.60-2.60 (m, 10H), 2.40-1.70 (m, 8H), 1.05 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.76, 169.49, 163.96, 134.26, 130.23, 127.39, 114.69, 112.87, 69.72, 59.94, 55.92, 54.41, 44.12, 38.01, 34.39, 23.81, 23.30, 22.80, 19.77, 11.16; m/z 459 [M+H]+.
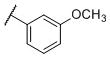
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-3-methoxybenzamide (21)
Colorless solid (53 mg, 58%). 1H NMR (CD3OD, 300 MHz) δ 7.42-7.30 (m, 3H), 7.13-7.05 (m, 1H), 4.18-3.81 (m, 2H), 3.84 (s, 3H), 3.60-2.60 (m, 10H), 2.40-1.70 (m, 8H), 1.06 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.77, 169.76, 161.22, 136.77, 134.32, 130.66, 120.45, 118.46, 113.70, 112.89, 69.72, 59.96, 55.87, 54.39, 44.05, 38.11, 34.40, 23.83, 23.32, 22.83, 19.78, 11.16; m/z 459 [M+H]+.
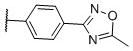
N-(cis-3-(2-(((S)-2-amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-4-(5-methyl-1,2,4-oxadiazol-3-yl)benzamide (22)
Colorless solid (46 mg, 45%). 1H NMR (CD3OD, 300 MHz) δ 8.17 (d, J=8.1 Hz, 2H), 7.98 (d, J=8.1 Hz, 2H), 4.20-3.85 (m, 2H), 3.60-2.70 (m, 10H), 2.69 (s, 3H), 2.45-1.82 (m, 8H), 1.10 (t, J=7.5 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 179.21, 171.75, 169.06, 168.87, 137.90, 134.70, 131.09, 129.05, 128.37, 112.97, 69.74, 60.05, 54.46, 44.08, 38.26, 34.43, 23.89, 23.41, 22.98, 19.85, 12.08, 11.17; m/z 511 [M+H]+.
N-(cis-3-(2-(((S)-2-amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-3-(5-methyl-1,2,4-oxadiazol-3-yl)benzamide (23)
Colorless solid (58 mg, 57%). 1H NMR (CD3OD, 300 MHz) δ 8.52 (m, 1H), 8.23-8.21 (m, 1H), 8.02-7.99 (m, 1H), 7.67-7.62 (m, 1H), 4.25-3.85 (m, 2H), 3.60-2.70 (m, 10H), 2.69 (s, 3H), 2.45-1.75 (m, 8H), 1.09 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 179.13, 171.77, 169.05, 168.99, 136.43, 134.55, 131.20, 131.00, 130.39, 128.63, 127.28, 112.94, 69.75, 60.03, 54.46, 44.07, 38.25, 34.45, 23.86, 23.40, 22.92, 19.82, 12.09, 11.17; m/z 511 [M+H]+.
General procedure for the synthesis of compounds 24-35
Diisopropylethylamine (52 mg, 0.4 mmol) and appropriate sulfonyl chlorides (0.24 mmol) were added to a suspension of 10 (65 mg, 0.2 mmol) in CH2Cl2 (10 mL) and the mixture was stirred at room temperature for 2 h. The reaction was quenched with H2O and pH was adjusted to 9–10 by addition of aqueous Na2CO3. The mixture was extracted 3 times with CH2Cl2 (30 mLx3). The organic lawyer was separated, combined, dried, and evaporated. The residue was purified by preparative HPLC to give the product. This procedure was used to prepare compounds 24-35 as described below.
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)benzenesulfonamide (24)
Colorless solid (37 mg, 40%).1H NMR (CD3OD, 300 MHz) δ 7.86 (d, J=6.9 Hz, 2H), 7.80-7.50 (m, 3H), 4.00-3.80 (m, 1H), 3.50-2.65 (m, 9H), 2.50-1.60 (m, 10H), 1.03 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.70, 142.43, 134.48, 133.74, 130.27, 127.97, 112.87, 69.33, 59.90, 54.31, 44.76, 40.50, 34.15, 23.76, 23.29, 22.85, 19.71, 11.11; m/z 465 [M+H]+.
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-2-methoxybenzenesulfonamide (25)
Colorless solid (42 mg, 42%).1H NMR (CD3OD, 300 MHz) δ 7.80 (d, J=8.3 Hz, 1H), 7.62 (t, J=7.6 Hz, 1H), 7.21 (d, J=8.3 Hz, 1H), 7.08 (t, J=7.6 Hz, 1H), 3.96 (s, 3H), 3.94-3.80 (m, 1H), 3.40-2.60 (m, 9H), 2.35-1.65 (m, 10H), 1.02 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.71, 158.14, 136.04, 134.27, 131.06, 129.28, 121.40, 113.59, 112.87, 69.28, 59.87, 56.57, 54.26, 44.51, 40.68, 34.07, 23.73, 23.25, 22.78, 19.73, 11.10; m/z 495 [M+H]+.
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-3-methoxybenzenesulfonamide (26)
Colorless solid (49 mg, 50%).1H NMR (CD3OD, 300 MHz) δ 7.50-7.13 (m, 4H), 3.95-3.80 (m, 1H), 3.83 (s, 3H), 3.50-2.60 (m, 9H), 2.42-1.62 (m, 10H), 1.03 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.70, 161.49, 143.56, 134.39, 131.42, 120.01, 119.36, 113.15, 112.86, 69.33, 59.89, 56.17, 44.72, 40.53, 34.16, 23.74, 23.27, 22.82, 19.73, 11.11; m/z 495 [M+H]+.
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-4-methoxybenzenesulfonamide (27)
Colorless solid (44 mg, 44%).1H NMR (CD3OD, 300 MHz) δ 7.78 (d, J=8.9 Hz, 2H), 7.07 (d, J=8.9 Hz, 2H), 3.97-3.83 (m, 1H), 3.85 (s, 3H), 3.42-2.60 (m, 9H), 2.42-1.65 (m, 10H), 1.03 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.71, 164.49, 134.35, 133.76, 130.16, 115.33, 112.87, 69.39, 59.89, 56.20, 44.75, 40.47, 34.19, 23.74, 23.27, 22.82, 19.70, 11.12; m/z 495 [M+H]+.
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-2-chlorobenzenesulfonamide (28)
Colorless solid (56 mg, 56%).1H NMR (CD3OD, 300 MHz) δ 8.04 (d, J=7.8 Hz, 1H), 7.62-7.42 (m, 3H), 3.98-3.80 (m, 1H), 3.48-2.63 (m, 9H), 2.40-1.70 (m, 10H), 1.02 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.73, 139.49, 135.13, 134.19, 132.94, 132.83, 132.17, 128.52, 112.85, 69.18, 59.87, 54.32, 44.57, 40.52, 34.06, 23.73, 23.24, 22.76, 19.72, 11.10; m/z 499 [M+H]+.
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-3-chlorobenzenesulfonamide (29)
Colorless solid (47 mg, 47%). 1H NMR (CD3OD, 300 MHz) δ 7.81-7.66 (m, 2H), 7.64-7.43 (m, 2H), 3.95-3.78 (m, 1H), 3.40-2.60 (m, 9H), 2.44-1.65 (m, 10H), 1.01 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.74, 144.51, 136.14, 134.22, 133.69, 131.99, 127.88, 126.38, 112.88, 69.29, 59.89, 54.36, 44.75, 40.51, 34.16, 23.76, 23.26, 22.79, 19.74, 11.12; m/z 499 [M+H]+.
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-4-chlorobenzenesulfonamide (30)
Colorless solid (49 mg, 49%). 1H NMR (CD3OD, 300 MHz) δ 7.84 (d, J=8.6 Hz, 2H), 7.58 (d, J=8.6 Hz, 2H), 3.95-3.80 (m, 1H), 3.50-2.65 (m, 9H), 2.49-1.70 (m, 10H), 1.02 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.73, 141.25, 139.93,134.19, 130.47, 129.75, 112.87, 69.33, 59.89, 54.32, 44.70, 40.50, 34.14, 23.74, 23.25, 22.77, 19.76, 11.11; m/z 499 [M+H]+.
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-2-fluorobenzenesulfonamide (31)
Colorless solid (39 mg, 40%). 1H NMR (CD3OD, 300 MHz) δ 7.87-7.80 (m, 1H), 7.70-7.60 (m, 1H), 7.40-7.25 (m, 2H), 3.95-3.80 (m, 1H), 3.52-2.63 (m, 9H), 2.45-1.70 (m, 10H), 1.01 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.72, 160.25 (d, JF-C=252.3 Hz), 136.42 (d, JF-C=8.4 Hz), 134.37, 131.21, 130.23 (d, JF-C=13.7 Hz), 125.79 (d, JF-C=3.8 Hz), 118.12 (d, JF-C=24.2 Hz), 112.87, 69.22, 59.90, 54.29, 44.71, 40.52, 34.11, 23.75, 23.27, 22.82, 19.73, 11.11; m/z 483 [M+H]+.
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-3-fluorobenzenesulfonamide (32)
Colorless solid (42 mg, 43%). 1H NMR (CD3OD, 300 MHz) δ 7.70-7.50 (m, 3H), 7.42-7.30 (m, 1H), 3.95-3.82 (m, 1H), 3.50-2.60 (m, 9H), 2.45-1.70 (m, 10H), 1.01 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.74, 163.86 (d, JF-C=147.9 Hz), 144.73 (d, JF-C=6.5 Hz), 134.21, 132.47 (d, JF-C=7.9 Hz), 123.95, 120.67 (d, JF-C=21.4 Hz), 115.04 (d, JF-C=24.5 Hz), 112.87, 69.29, 59.88, 54.31, 44.72, 40.53, 34.15, 23.74, 23.26, 22.77, 19.73, 11.12; m/z 483 [M+H]+.
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-4-fluorobenzenesulfonamide (33)
Colorless solid (49 mg, 51%). 1H NMR (CD3OD, 300 MHz) δ 7.89 (dd, J=5.1, 8.9 Hz, 2H), 7.29 (dd, J=8.9, 8.9 Hz, 2H), 3.95-3.80 (m, 1H), 3.48-2.65 (m, 9H), 2.49-1.70 (m, 10H), 1.01 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.72, 166.44 (d, JF-C=250.8 Hz), 138.73 (d, JF-C=3.2 Hz), 134.35, 130.94 (d, JF-C=9.3 Hz), 117.26 (d, JF-C=22.8 Hz),112.88, 69.33, 60.03, 59.89, 54.35, 44.73, 40.49, 34.16, 23.75, 23.27, 22.82, 19.73, 11.12; m/z 483 [M+H]+.
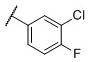
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-3-chloro-4-fluorobenzenesulfonamide (34)
Colorless solid (50 mg, 48%). 1H NMR (CD3OD, 300 MHz) δ 7.98 (dd, J=2.2, 6.8 Hz, 1H), 7.83-7.75 (m, 1H), 7.47 (t, J=8.8 Hz, 1H), 3.96-3.80 (m, 1H), 3.50-2.63 (m, 9H), 2.45-1.70 (m, 10H), 1.03 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.70, 161.66 (d, JF-C=254.7 Hz), 140.06 (d, JF-C=3.8 Hz), 134.52, 130.83, 129.09 (d, JF-C=8.6 Hz), 122.89 (d, JF-C=18.8 Hz), 118.68 (d, JF-C=22.4 Hz), 112.88, 69.29, 59.93, 54.32, 44.72, 40.49, 34.15, 23.77, 23.30, 22.88, 19.73, 11.13; m/z 517 [M+H]+.
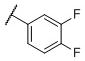
N-(cis-3-(2-(((S)-2-Amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)(propyl)amino)-ethyl)-3-hydroxycyclobutyl)-3,4-difluorobenzenesulfonamide (35)
Colorless solid (40 mg, 40%). 1H NMR (CD3OD, 300 MHz) δ 7.80-7.70 (m, 2H), 7.55-7.43 (m, 1H), 3.95-3.80 (m, 1H), 3.50-2.60 (m, 9H), 2.45-1.70 (m, 10H), 1.02 (t, J=7.3 Hz, 3H); 13C NMR (CD3OD, 75 MHz) δ 171.73, 154.41 (dd, JF-C=12.5, 212.0 Hz), 151.05 (dd, JF-C=12.5, 209.8 Hz), 139.75 (t, JF-C=4.2 Hz), 134.31, 125.58 (dd, JF-C=3.9, 7.6 Hz), 119.45 (d, JF-C=18.5 Hz), 117.79 (d, JF-C=19.9 Hz), 112.88, 69.29, 60.34, 54.30, 44.69, 40.50, 34.15, 23.75, 23.26, 22.81, 19.72, 11.12; m/z 501[M+H]+.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institute on Drug Abuse, National Institutes of Health (R01DA020669). We are grateful to the Addiction Treatment Discovery Programs at the NIDA and NIH for evaluation of ligands for their functional activity in cells transfected with cloned human dopamine receptors under the contract NIDA Y1-DA-0101-02, performed by Dr. Aaron Janowsky at the Oregon Health & Science University (Portland, Oregon).
Contributor Information
Dr. Jianyong Chen, Email: jiachen@umich.edu.
Prof. Shaomeng Wang, Email: Shaomeng@umich.edu.
References
- 1.Joyce JN. Pharmacol Ther. 2001;90:231–259. doi: 10.1016/s0163-7258(01)00139-5. [DOI] [PubMed] [Google Scholar]
- 2.Volkow ND, Fowler JS, Wang GJ. Behav Pharmacol. 2002;13:355–366. doi: 10.1097/00008877-200209000-00008. [DOI] [PubMed] [Google Scholar]
- 3.Shimohama S, Sawada H, Kitamura Y, Taniguchi T. Trends Mol Med. 2003;9:360–365. doi: 10.1016/s1471-4914(03)00117-5. [DOI] [PubMed] [Google Scholar]
- 4.Newman AH, Grundt P, Nader MA. J Med Chem. 2005;48:3663–3679. doi: 10.1021/jm040190e. [DOI] [PubMed] [Google Scholar]
- 5.Heidbreder CA, Newman AH. Ann N Y Acad Sci. 2010;1187:4–34. doi: 10.1111/j.1749-6632.2009.05149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newman AH, Beuming T, Banala AK, Donthamsetti P, Pongetti K, LaBounty A, Levy B, Cao J, Michino M, Luedtke RR, Javitch JA, Shi L. J Med Chem. 2012;55:6689–6699. doi: 10.1021/jm300482h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keck TM, Burzynski C, Shi L, Newman AH. Adv Pharmacol. 2014;69:267–300. doi: 10.1016/B978-0-12-420118-7.00007-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J, Collins GT, Levant B, Woods J, Deschamps JR, Wang S. ACS Med Chem Lett. 2011;2:620–625. doi: 10.1021/ml200100t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J, Levant B, Jiang C, Keck TM, Newman AH, Wang S. J Med Chem. 2014;57:4962–4968. doi: 10.1021/jm401798r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bancroft GN, Morgan KA, Flietstra RJ, Levant B. Neuropsychopharmacology. 1998;18:305–316. doi: 10.1016/S0893-133X(97)00162-0. [DOI] [PubMed] [Google Scholar]
- 11.Levant B, Grigoriadis DE, DeSouza EB. J Pharmacol Exp Ther. 1992;262:929–935. [PubMed] [Google Scholar]
- 12.Grigoriadis D, Seeman P. J Neurochem. 1985;44:1925–1935. doi: 10.1111/j.1471-4159.1985.tb07189.x. [DOI] [PubMed] [Google Scholar]
- 13.Levant B. Current Protocols in Pharmacology. John Wiley & Sons, Inc; 2001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.