

Summary
Neural stem cells have been adopted to model a wide range of neuropsychiatric conditions in vitro. However, how well such models correspond to in vivo brain has not been evaluated in an unbiased, comprehensive manner. We used transcriptomic analyses to compare in vitro systems to developing human fetal brain and observed strong conservation of in vivo gene expression and network architecture in differentiating primary human neural progenitor cells (phNPCs). Conserved modules are enriched in genes associated with ASD, supporting the utility of phNPCs for studying neuropsychiatric disease. We also developed and validated a machine learning approach called CoNTExT that identifies the developmental maturity and regional identity of in vitro models. We observed strong differences between in vitro models, including hiPSC-derived neural progenitors from multiple laboratories. This work provides a systems biology framework for evaluating in vitro systems and supports their value in studying the molecular mechanisms of human neurodevelopmental disease.
Introduction
Human neural stem cells are poised to revolutionize our ability to make mechanistic inferences, bridging the gap between traditional model systems and human biology (Dolmetsch and Geschwind, 2011; Merkle and Eggan, 2013). Ideally, such a platform should maximize translational potential by recapitulating in vivo brain development and function as much as possible. Human embryonic stem (hES), induced pluripotent stem (hiPS), and primary human neural progenitor (phNPC) cells all have the ability to differentiate into functional neurons (Espuny-Camacho et al., 2013; Hansen et al., 2011; Palmer et al., 2001; Sandoe and Eggan, 2013). In each of these systems, disease related processes can be modeled and studied by either generating hiPSCs from patients with known mutations or introducing genetic modifications into normal neural stem cell lines (An et al., 2012; Brennand et al., 2011; Israel et al., 2012; Marchetto et al., 2010; Pasca et al., 2011; Rosen et al., 2011; Ryan et al., 2013; Soldner et al., 2011). Despite many options, there is neither a clear consensus as to which system or culture conditions are better suited to model aspects of neurodevelopment and disease, nor a rubric for answering this question. It has not been demonstrated using a rigorous genome-wide statistical framework: (1) how well in vitro models match in vivo development, (2) what level of developmental maturity is achieved after differentiation, (3) what neuroanatomical identity is modeled, (4) what specific neurodevelopmental processes and molecular mechanisms are preserved and (5) what specific aspects remain to be better modeled in vitro, providing a guide for future work optimizing in vitro systems.
Recent large-scale efforts to measure the transcriptome from post-mortem human brain provide an unbiased in vivo standard to which in vitro systems can be compared. These datasets measure gene expression at time points from embryonic to late adulthood and across several cortical and subcortical regions (Kang et al., 2011). In addition, gene expression in micro-dissected cortical laminae has been measured at mid to late fetal time periods (Miller et al., 2014) providing datasets with increased spatial resolution within a restricted developmental window.
Here, we develop and demonstrate genome-wide methods to quantify the similarity between in vitro neural stem cell models and brain development in vivo and apply them to a newly generated set of phNPC lines. We demonstrate remarkable preservation of neurodevelopmental processes spanning embryonic to fetal corticogenesis in phNPCs in vitro. But, even after months in a dish, neither phNPCs nor hiPSC-derived neurons mature beyond fetal stages. We further uncover gene expression networks driving these processes, show their preservation in vitro, and reveal they are enriched in ASD (Autism Spectrum Disorders) risk genes. Finally, we expand this analysis to hiPSC and hES-based neural stem cell models and find striking differences in overlap to in vivo development and preservation of network architecture. We have implemented this framework into a user-friendly website (http://context.semel.ucla.edu) as a resource for the community.
Results
phNPCs express telencephalic markers and undergo stereotypical neuronal morphogenesis upon differentiation
We generated phNPC lines from 15–18 post conception week (PCW) human fetal brains by a neurosphere isolation method (Konopka et al., 2012; Palmer et al., 2001; Rosen et al., 2011; Wexler et al., 2011) (Figure 1A; Experimental Procedures). All lines were genotyped to determine sex and to exclude samples with aneuploidy. Immunostaining of undifferentiated phNPCs was consistent with standard methods used to define dorsal telencephalic progenitors/radial glia of human cortex (Hansen et al., 2010) (Figures 1B and 1C). During differentiation, the typical progression of decrease in mitotic and neural stem cell markers (Ki-67, NES) with concomitant increase in neuronal markers (Tuj1, DCX, MAP2) during cortical maturation was observed (Figures 1D and 1E and data not shown). We also detected cells expressing TBR2, a marker of intermediate progenitor cells (Englund et al., 2005; Hansen et al., 2010), all of which co-stained with PAX6, as shown in the macaque (Betizeau et al., 2013), but has yet to be shown in the human cortex (data not shown). The in vitro expression profile of genes marking laminar potential and fate across differentiation was also consistent with sequential generation of lower and upper layer projection neurons during corticogenesis (Arlotta et al., 2005; McEvilly et al., 2002) (Figures S1A–C). In addition, phNPCs generate neurons with stereotypical morphologies similar to what is observed in vivo, first forming bipolar migrating cells, followed by axonogenesis and increases in dendritic arborization (Figures 1F and 1G).
Figure 1.

phNPCs express canonical telencephalic markers and undergo stereotypical neuronal morphogenesis. (A) Isolation, culture, and differentiation of phNPCs from human fetal cortex. (B,C) Isolated phNPCs express dorsal telencephalic and radial glia markers. Undifferentiated phNPCs were subjected to immunocytochemistry with the indicated antibodies and the DNA-binding dye 4′,6-diamidino-2-phenylindole (DAPI). Telencephalic (FOXG1, 100%, ASCL1, 19%; n=368 cells), dorsal telencephalic and radial glia (PAX6, SOX2; 97%, n=395 cells) and mitotic (Ki-67; 36%, n=395 cells) markers are expressed. (D, E) Differentiation of phNPCs into MAP2+ neurons with concomitant decrease in neural progenitors (Nestin, Ki-67) across the indicated timepoints (n=794 cells; Ki-67, n=1693 cells). (F) phNPCs were infected with a low titer of turboRFP-expressing lentivirus (pTRIPZ) and subjected to immunocytochemistry with MAP2 and turboRFP antibodies at 2, 5 and 12 weeks post-differentiation. Axo-dendritic polarization and increased dendritic complexity is observed over development in vitro. Arrows and arrowheads indicate dendrites and axons, respectively. (G) Quantification of dendrite morphogenesis in MAP2+ neurons treated as in F shows robust dendrite growth and branching over differentiation (n= 23 cells/timepoint in 5 lines from 3 donors; mean ± SE displayed in barplot). Scale bar = 50μm for all panels.
Few commonly used literature based markers are specific to a single region (Figure S2A). For instance, PAX6 is widely used as a dorsal telencephalic marker (Espuny-Camacho et al., 2013; Pasca et al., 2011), as it largely separates the dorsal pallium from ventral pallium, but it is also strongly expressed in other brain regions including the developing diencephalon and hindbrain (Manuel and Price, 2005) (Figure S2A). To overcome this limitation, we generated a list of genes enriched in different brain regions based on transcriptomic data from human brain (Kang et al., 2011) (Supplementary Experimental Procedures) and used these gene sets as an initial standard. Cortical regional markers were highly expressed in all phNPC lines, while expression of markers of other brain regions was much lower (Figure 2). Further, expression of the majority of literature-based markers was also very similar between in vivo cortex and phNPCs (Figure S2B and S2C), further validating the cortical identity of phNPC cultures using an unbiased approach.
Figure 2.

Expression of transcriptomically-defined regional markers in vitro and in vivo. Expression levels of 5 to 20 marker genes enriched in each indicated brain region during late mid fetal in vivo development (Period 6) along with 12 wk PD phNPCs is shown. Genes are shown on the x-axis with colors to indicate the region for which the gene is a marker. The y-axis indicates the in vivo dissected region or in vitro cell line, specified by donor identification number and region identification number. Relative expression is indicated by color within the heat map and normalized for each gene from 0 to 1. Abbreviations: Cortex: OFC, DFC, VFC, MFC, M1C, S1C, IPC, A1C, STC, ITC, V1C; Hippocampus: HIP; Amygdala; AMY; Striatum: STR; Medial Dorsal Nucleus of the Thalamus: MD; Cerebellar Cortex: CBC.
Mapping in vitro differentiation of phNPCs to in vivo cortical development
We next developed an unbiased genome-wide transition mapping approach to assess the extent of overlap between in vivo cortical development and in vitro differentiation. Transition mapping (TMAP) consists of serialized differential expression analysis between any two in vivo developmental periods defined from a spatiotemporal transcriptomic atlas (Kang et al., 2011) and any two in vitro differentiation timepoints, followed by quantification of overlap using the Rank-Rank Hypergeometic Overlap test (Plaisier et al., 2010) for each combination of time periods. This method has the advantage of allowing for a threshold-free (1) identification of which in vivo developmental transition best overlaps with differentiating phNPCs and (2) statistical assessment of the extent of overlap. This technique generates a map of the overlap between any two systems across development.
We applied TMAP, comparing a signed, p-value ranked list of differentially expressed (DE) genes over 12 weeks of differentiation in vitro to genes DE between the listed developmental periods from human cortex (Figure 3A), and observed strong overlap between phNPC and cortical developmental periods up to late mid-fetal periods (max -log10(P-value) > 460). The strong matching observed in transitions from period 1 or 2 is consistent with the differentiation of the progenitor-enriched wk 1 PD timepoint (Figure 1C), since the in vivo dataset only includes cortical germinal zones in periods 1 and 2 (Kang et al., 2011). Genes co-upregulated in vitro and in vivo driving the overlap are related to processes of normal neuronal differentiation, whereas co-down-regulated genes are related to mitosis, cell cycle arrest and carbohydrate utilization (Figures 3B and 3C).
Figure 3.

TMAP identifies in vivo developmental period and cortical laminae most similar to differentiating phNPCs. Rank-rank hypergeometric overlap (RRHO) maps (Plaisier et al., 2010) comparing the transitions between in vivo developmental periods (A–C) or laminae in the developing cortex (D–E) to differentiation of phNPCs from 1 to 12 wks. (A,D) Each pixel represents the overlap between in vivo and in vitro transcriptome, color-coded according to the -log10 p-value of a hypergeometric test (step size = 200). On each map, the extent of shared upregulated genes is displayed in the bottom left corner, whereas shared downregulated genes are displayed in the top right corners (see schematic on rightmost panel). (B,E) Venn diagrams display the extent of overlap between in vivo and in vitro transcriptomes at the best matched periods (1 vs 6; B) and between SZ and CPi (E). Gene ontology (GO Elite) analysis shows co-upregulated genes are related to synaptic transmission and nervous system development, whereas co-downregulated genes are involved in mitosis. (C) Selected genes co-upregulated and co-downregulated are graphed through time both in developing phNPCs and in vivo.
Examples of key candidate genes that have preserved rankings show remarkably similar trajectories of gene expression, when compared to in vivo development up to period 6 (Figure 3C). Mapping the in vitro transitions between periods 1 to 4, periods 4 to 8, and periods 8 to 12 wks PD to in vivo developmental periods allows for visualization of the progressive maturation of phNPCs cultures (Figure S3B). It is evident that later transitions in differentiation correspond to later periods of human development.
Next, we sought to apply this same TMAP tool to transcriptomes derived from laser capture microdissection of cortical laminae from post-mortem human fetal brain (15–21 PCW) (Miller et al., 2014), permitting a direct assessment of in vivo matching between cortical neural progenitors and their post-mitotic derivatives. The transition between 1 to 12 wk PD in differentiating phNPCs has strongest correspondence to the transition between the ventricular zone or subventricular zone to subplate and inner cortical plate (Figure 3D). As expected, this overlap is largely driven by synaptic genes co-upregulated and cell cycle genes co-downregulated during the transition between subventricular zone and inner cortical plate in vivo and the transition between 1 to 12 wk PD in vitro (Figure 3E).
Individual in vitro sample prediction of regional identity and developmental period
The above analyses perform group-based determination of regional identity and developmental period. We sought to develop an approach that would allow determination of these features in individual samples, thus precluding the need to pool samples into groups or to compare between specific stages or transitions. We developed a multi-label (developmental period, regional identity) multi-class (periods: 1–15; regions: Cortex, Hippocampus, Amygdala, Thalamus, Striatum, Cerebellum) machine-learning framework (Tsoumakas and Katakis, 2007), named CoNTExT (Classification of Neuroanatomical and Temporal Expression via Transcriptomics), for this purpose. We trained the algorithm on a spatiotemporal transcriptome atlas of the human brain (Kang et al., 2011) (Figure 4A). After feature selection and parameter optimization (Experimental Procedures), cross-validation accuracy in the training dataset was high (10-fold cross validation with 90% split percentage; exact accuracy for developmental period: 99.6%; accuracy for regional identification: 96.9%).
Figure 4.

CoNTExT predicts developmental period and regional identity in individual phNPC cultures. (A) The CoNTExT algorithm was trained on all samples from a spatio-temporal atlas of human brain expression (Kang et al., 2011) (1340 samples using Affymetrix Exon 1.0 ST Array), and validated in several post-mortem expression datasets. Probability (color in heat map) of each predicted class assignment (y-axis) is shown for each sample of known regional and temporal identity (x-axis). (B) Cross platform accuracy was evaluated in 49 samples spanning all postnatal developmental periods and both cerebellar and cortical regions (Liu et al., 2012) (Affymetrix Gene 1.0 ST Array). Developmental period was classified with 84% (+/− one period) and 100% accuracy in region. (C) The validated machine learning algorithm was applied to one line (Donor: 8 Region: 49) of differentiating phNPCs to predict developmental period and regional identity (Illumina HT-12 Beadchip). A clear maturation across differentiation weeks is seen at the individual sample level, with the culture reaching early to late fetal periods of development. In addition, the cultures are predicted to be cortical, consistent with immunocytochemical profile and expression of regional markers.
We then applied this recognition algorithm to four other datasets of transcriptional profiling from post-mortem human brain to provide independent validation (Hernandez et al., 2012; Israel et al., 2012; Johnson et al., 2009; Liu et al., 2012), attaining high levels of accuracy in each of these new datasets, despite differences in microarray platforms, processing of samples and laboratories (Figures 4A and 4B, Figure S4A–C). Importantly, in the same platform as the in vitro phNPCs data (Illumina HT-12 Beadchip) the algorithm was able to correctly classify both fetal (Figure S4B) (Israel et al., 2012) and adult brain (911 samples; Figure S4C) (Hernandez et al., 2012). Overall, CoNTExT is accurate at differentiating distinct in vivo developmental epochs from each other (e.g. between: early to mid to late fetal; fetal vs. newborn; childhood vs. adolescence; childhood vs. adulthood).
We next applied CoNTExT to phNPC cultures to predict the developmental period and regional identity of each phNPC sample (see Supplemental Experimental Procedures). A progressive maturation of the culture can be seen across multiple differentiation weeks (Figure 4C). Similar to results from TMAP, but now for each single sample individually, we predict that these cultures match most closely to embryonic development at 1 wk PD, and early to late mid-fetal development at 12 wks PD. As in TMAP, the strong prediction of embryonic development at 1 wk PD is likely due to the in vivo dataset containing proliferative laminae of the cortex for these time periods only and the culture containing predominantly proliferative cells. In addition, regional prediction is strongly cortical, consistent with high expression of cortical markers in the developing phNPCs (Figure 1 and Figure 2).
Network analysis identifies key preserved neurodevelopmental processes in differentiating phNPCs
Although TMAP and CoNTExT define a cohesive framework to determine neuroanatomical region and developmental maturity, these methods are not tailored to characterize the preservation of specific cellular or biochemical processes. To identify which specific neurodevelopmental processes were preserved in vitro, we employed weighted gene co-expression network analysis (WGCNA). WGCNA identifies modules of co-expressed genes that correspond to shared function, for example, organelles, cell types or biological processes. (Hawrylycz et al., 2012; Johnson et al., 2009; Kang et al., 2011; Miller et al., 2014; Oldham et al., 2008; Parikshak et al., 2013; Voineagu et al., 2011).
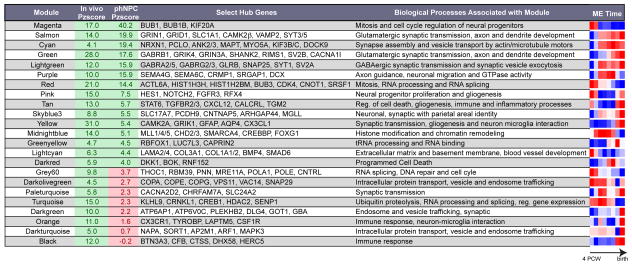
We constructed a network based on co-expression of genes in developing human cortex (Periods 1–8) (Kang et al., 2011) and assessed preservation of the network (Langfelder et al., 2011) (Experimental Procedures) in an independent in vivo dataset of developing human cortex (Colantuoni et al., 2011) to identify the most robust and generalizable modules, identifying 28 well preserved modules (Preservation Zscore 4) (Table 1; Figure S5A). This network, defined in developing fetal cortex, represents a rubric for comparing in vitro development to in vivo development and identifying shared processes and mechanisms. We found that the majority of our high-confidence modules were strongly correlated with developmental period, but not as strongly with areal identity or hemisphere, suggesting modules are largely reflective of processes related to differentiation and maturation (Figure S5B).
Table 1.
Summary of high-confidence modules defined in human fetal cortex. Module preservation Z scores are shown for an independent in vivo dataset of developing human cortex (Colantuoni et al., 2011) and in phNPCs. Modules not preserved in vitro are highlighted in pink with red font, whereas modules preserved in vitro and in vivo are highlighted in green. Select hub genes (from top 100 in each module) related to the biological processes of the module are shown. Modules with few genes (< 70) lacking clear or specific GO/KEGG enrichment and no over-representation of genes in manually curated lists comprising key neurodevelopmental processes, brain cell populations, and their subcellular compartments (white, steelblue, saddlebrown, yellowgreen, violet) are preserved in vivo but are not shown in the table.
|
We next assessed shared gene function within the module by enrichment of Gene Ontology (GO) terms, KEGG pathway analysis and over-representation of genes in manually curated lists comprising key neurodevelopmental processes, brain cell populations, and their subcellular compartments (Figure 5B; Table 1; Figure S5D). We identified modules related to major histogenetic and cellular processes driving cortical development including, exit from cell cycle (magenta, red and grey60 modules), neurogenesis and differentiation (pink and magenta modules), axon growth and guidance and neuronal migration (purple, salmon, and green modules), dendrite growth (salmon and green modules) and synaptogenesis (green, salmon, lightgreen, cyan, paleturquoise and yellow modules; Figures 5A and 5B; Table 1). The trajectory of gene expression over the developmental period (e.g. module eigengene), GO terms and key hub genes are illustrated for a subset of these modules in Figure 5B. For instance, modules related to neural progenitor proliferation (magenta, pink) have eigengenes that decrease over time and contain mitotic checkpoint kinases as hubs (BUB1, BUB1B; magenta) (Taylor and McKeon, 1997) as well as known markers of radial glia (PAX6, VIM, NES; magenta), intermediate progenitors (EOMES; magenta), members of the Notch/delta signaling pathway NOTCH1 (magenta), NOTCH2 and HES1 (pink) (Kageyama et al., 2008) and members of the SHH signaling pathway (SMO, GLI2 and GLI3; magenta) (Sousa and Fishell, 2010). Interestingly, the pink module is also enriched in genes related to gliogenesis and astrocytes, including SOX8 and SOX9 (Kang et al., 2012; Stolt et al., 2003), and its eigengene is upregulated in late fetal development consistent with the timing of gliogenesis in human cortex (Figure 5B; Figure S5D) (Kriegstein and Alvarez-Buylla, 2009; Rakic, 2003).
Figure 5.

Network analysis identifies major neurodevelopmental processes that are preserved in differentiating phNPCs. (A) A weighted gene co-expression network was formed using human cortex samples from embryonic (Period 1) to neonatal periods (Period 8). Timeline of known cellular and histogenetic processes in the developing human brain (Andersen, 2003; Kang et al., 2011) and modules enriched in genes related these processes are labeled. (B) A subset of modules related to key neurodevelopmental processes are highlighted with corresponding GO analysis, selected hub genes, and module eigengene trajectories (see Table 1 for full list). (C) Module preservation (Langfelder et al., 2011) in phNPCs differentiated over 12 weeks shows high preservation (Z 4) of 12 modules. (D,E) The green module, enriched in genes involved in synaptic function, shows a similar pattern of connectivity in vivo (D) and in vitro (E). The width and color of the edges are weighted by the strength of bi-correlation. (F,G,H) Module preservation varies over differentiation time. Early differentiation periods (F) show stronger preservation of chromatin modification genes (midnightblue module) while later time periods (G,H) show stronger preservation of gliogenic (pink, tan modules) and synaptogenic (salmon, green, lightgreen, cyan modules) genes.
A set of modules with trajectories of increasing expression over development mark different phases of neurogenesis. The purple module related to neuronal migration and axon guidance displays elevated expression during Periods 3–6, faithfully recapitulating the time period of these processes in vivo (Figure 5A) and contains the hub genes DCX and CRMP1, known to be involved in cytoskeleton regulation and neuronal migration (Gleeson et al., 1999; Goshima et al., 1995). Six additional modules are related to later phases of development including axon and dendrite growth and synaptogenesis (purple, salmon, green, lightgreen, cyan, paleturquoise). They contain hub genes involved in glutamatergic and GABAergic synapse function (CAMK2B, GRIN1, GRID1, VAMP2, SYT3/5 - Salmon; GRIA4, GRIK4, GRIN3A, SHANK2, RIMS1, SV2B - Green; GABRA2/5, GABRG2/3 Lightgreen and genes related to axon and dendrite growth (Table 1; Figures 5B and 5D). Consistent with a role in synaptic function, these modules are also enriched in genes found in biochemically isolated postsynaptic density (PSD) fractions and in genes known to interact with PSD-95 (Bayes et al., 2011) (Table 1; Figure S5D).
We also identified modules related to histone modification and chromatin remodeling (midnightblue) and RNA splicing and processing (red, grey60, turquoise). The midnightblue module peaks during cortical neurogenesis and contains several members of the chromodomain helicase family (CHD2/3) and the MLL (MLL1/4/5) and BAF (SMARCC2, SMARCA4) complexes involved in chromatin remodeling including BAF170/SMARCC2, which was recently implicated in regulation of cortical thickness (Tuoc et al., 2013). Notably, this module also contains transcription factors associated with generation of lower layer neurons including BCL11B and SOX5 and is expressed early in development when these layers are formed (Molyneaux et al., 2007). The red, grey60 and turquoise modules are also highly expressed early in development, but instead are enriched in genes related to RNA processing and splicing, including several proneural transcription factors (NEUROD1/2/6, NEUROG2; turquoise) and transcription factors expressed in cortical neural progenitors (FEZF2, EMX1; red) and early-generated neurons (TBR1; turquoise). Together these modules point to critical roles of RNA processing and epigenetic and transcriptional regulation in neural stem cell differentiation during corticogenesis.
Remarkably, we observed that 87% of genes in the network were present in phNPC-conserved modules (17 out of 28 modules; Zscore 4; Figure 5C; Table 1 and S1; (Langfelder et al., 2011)), including all modules related to the major cellular and histogenetic processes discussed above. The network architecture of many modules is also strongly preserved, as visually demonstrated by in vivo and in vitro network diagrams (Figures 5D and 5E). Of the modules that were not preserved, two of them, are related to immune response (orange and black) and neuron-microglia interactions (orange). This lack of preservation is consistent with the composition of phNPC cultures, which have no detectable microglia (absence of IBA1 staining, data not shown) or immune cells. We also examined module preservation at different time periods in vitro (Figures 5F–H) and found trends in preservation that are consistent with the module’s function - synaptic and gliogenic modules are better preserved at later time points (Figure 5A, Table 1). Half of the modules that were not preserved, including two modules related to vesicle and endosome trafficking have eigengenes upregulated late in development close to birth, consistent with the mid-fetal maturity of phNPC cultures. Taken together, these parallels in transcriptional network architecture reveal conservation of many biochemical and cellular processes between in vivo fetal brain and in vitro phNPC cultures.
Preserved modules in phNPCs are enriched in ASD genes
An important question facing the modeling of neurodevelopmental diseases in vitro is to what extent disease-related processes are conserved. Recently, we showed that ASD genes coalesce in several transcriptional modules during fetal human brain development (Parikshak et al., 2013). Similar to Parikshak et al. (2013), we started with a curated list of ASD associated candidate genes (ASD SFARI) (Banerjee-Basu and Packer, 2010), which showed strong enrichment with the highly preserved in vitro modules related to synaptic function and neurite outgrowth (salmon and yellow; Figure 6A). Importantly, genes co-expressed in ASD and related to synaptic function, previously found to be down-regulated in the ASD brain by unbiased transcriptomic analyses (asdM12) (Voineagu et al., 2011) were also enriched in phNPC modules with overlapping function (salmon, skyblue3, lightgreen, green, purple modules; Figure 6A).
Figure 6.

phNPC preserved modules are enriched in ASD associated genes. (A) In vivo defined modules preserved in differentiating phNPCs were tested for enrichment of disease associated genes from a curated list of autism-associated genes (ASD SFARI; (Banerjee-Basu and Packer, 2010)), rare de-novo protein disrupting mutations found in ASD and siblings (Iossifov et al., 2012; Neale et al., 2012; O’Roak et al., 2012; Sanders et al., 2012), two ASD associated modules defined in post-mortem brain (asdM12 and asdM16; (Voineagu et al., 2011)), a curated list of genes associated with intellectual disability (ID All) (Inlow and Restifo, 2004; Lubs et al., 2012; Parikshak et al., 2013; Ropers, 2008; van Bokhoven, 2011), the intersection of ASD SFARI with ID (ASD-ID overlap), the relative complement of these (ASD only, ID only), a list of genes disrupted by CNVs in schizophrenia (Levinson et al., 2011), genes near significantly associated loci in GWAS of schizophrenia (Ripke et al., 2013), and genes near significantly associated loci in GWAS of late onset Alzheimer’s disease (Naj et al., 2011). Enrichment was assessed using Fisher’s exact test and the FDR (Benjamini and Hochberg, 1995) correction for multiple comparisons P-value is displayed. (B) Protein disrupting de novo SNVs found in ASD are enriched in the preserved midnightblue module. The correlation of each gene to the module eigengene (kME) is shown for in vivo and phNPC data (1 and 4 wk PD). ASD-associated genes are highlighted in red. Genes close to the solid red line (y=x) have the same kME in vivo and in vitro. The dotted red line represents an in vivo kME value 3 times less than observed in vivo. (C) Several genes that contain ASD-associated de novo SNVs have similar expression patterns in vivo and in vivo. (D) kME is shown for in vivo and phNPC data (1, 4, 8, and 12 wk PD) for the preserved salmon module that is enriched in asdM12 genes (Voineagu et al., 2011). ASD-associated genes in asdM12 are in red and are labeled. (E) Several asdM12 genes show similar expression patterns in vivo and in vitro.
We next tested if rare de novo protein disrupting mutations found in ASD (Iossifov et al., 2012; Neale et al., 2012; O’Roak et al., 2012; Sanders et al., 2012) were similarly enriched in preserved modules. Similar to Parikshak et al. (2013), we found that the conserved phNPC midnightblue module, which contains transcriptional and chromatin regulatory genes, is enriched for these variants (Figure 6A). Remarkably, this module and both synaptic modules are enriched in mRNAs known to interact with FMRP (Figure S5D), suggesting that activity dependent protein translation dysregulated in ASD can be modeled in vitro. Finally, a group of upregulated genes that are co-expressed in ASD (asdM16; Voineagu et al. 2011) and enriched in astrocyte and microglial markers, is enriched in the preserved yellow, pink, tan, magenta, and red modules. As microglia are not observed in the culture, this implies that the astrocyte component of asdM16 shows enrichment in these modules.
Interestingly, genes implicated in other neurodevelopmental diseases including intellectual disability (Inlow and Restifo, 2004; Lubs et al., 2012; Parikshak et al., 2013; Ropers, 2008; van Bokhoven, 2011) and schizophrenia (Levinson et al., 2011) showed enrichment with preserved modules related to basic cellular processes (ubiquitylation, RNA splicing and gene expression) and synaptic transmission, respectively (Figure 6A). No enrichment was observed with genes implicated in neurodegenerative disorders, such as late onset Alzheimer’s disease (Naj et al., 2011), though this could be due to the relatively small number of genes identified (Figure 6A). Together these findings suggest that different disorders affect distinct neurodevelopmental processes, many of which can be effectively modeled in vitro using phNPCs.
We further assessed if the co-expression relationships of specific disease associated genes within our in vivo defined modules were also preserved in vitro. Many ASD-associated genes had generally similar hub status and expression pattern (Figures 6B–E) in prenatal human cortex and in phNPCs, marking these genes as high-priority candidates for modeling ASD in vitro. These findings link genetic and functional evidence of ASD to known developmental processes that can be modeled in vitro, providing a powerful platform for therapeutic discovery.
Heterogeneity of in vivo modeling by different human neural stem cell systems
Advances in stem cell biology in the last decade have facilitated the use of human pluripotent cells including hES and hiPS cells to study development and disease (Brennand et al., 2011; Kim et al., 2002; Marchetto et al., 2010; Millar et al., 2005; Pasca et al., 2011; Rosen et al., 2011; Ryan et al., 2013; Soldner et al., 2011). However, it is still not known to what degree they recapitulate in vivo development and most importantly, which specific processes are conserved in which system.
We evaluated the degree of overlap between developing human cortex in vivo and these different systems using the same functional genomics framework developed above. Datasets of differentiating hiPSC (three datasets including one previously published (Pasca et al., 2011) and two unpublished datasets from two separate labs), hESC (Fathi et al., 2011) and SY5Y neuroblastoma (Nishida et al., 2008) cells were compared to the human cortex spatiotemporal transcriptome (Kang et al., 2011) (Figure 7A) or the human LCM transcriptome (Miller et al., 2014) (Figure 7B) to map the transitions between developmental periods or cortical laminae in these alternate in vitro systems. These datasets, from different laboratories comprise a representative cohort of the differences in culture and differentiation protocols currently available (Supplementary Experimental Procedures). Just as with phNPCs, we found the best overlap in all systems when comparing in vivo period 1 or 2, which contain the germinal zones, to periods of mid-fetal development. As expected, neuroblastoma-derived SY5Y cells had the lowest degree of matching, while hES-derived cells matched relatively well to in vivo cortical development (Figure 7). When differentiated for the same length of time (3 weeks) phNPCs are roughly similar to hES-derived progenitors (Figure S6B). A great degree of heterogeneity of in vivo matching was observed in hiPSCs cultured in different labs using different protocols. Interestingly, hiPSC-derived progenitors from dataset 1, even when differentiated longer than hES-derived NPCs or phNPCs, still matched in vivo transcriptional profiles significantly less than phNPCs (Figure S6B,C). These differences were not driven by number of samples in the datasets as hES-derived cells (n=6) display better matching than hiPSC-derived cells from dataset 1 (n=29) (Supplemental Experimental Procedures). phNPCs from 1 to 8 weeks of differentiation show significantly greater overlap with in vivo development compared to all other in vitro datasets except for hiPSC dataset 3 which was roughly equivalent to phNPCs (Figure 3A; Figure 6B; Figure S6C). hiPSC dataset 3 showed strong matching even at 4 weeks of differentiation, implying that it reaches developmental maturity quicker than phNPCs.
Figure 7.

The extent of in vivo overlap observed in multiple in vitro neural stem cell models. The transitions between in vivo developmental periods of neocortex (Kang et al., 2011) (A) or laminae in the developing cortex (Miller et al., 2014) (B) are compared to the transition between proliferative and differentiated neuronal cultures derived from 3 datasets of hiPSC cells, hES (Fathi et al., 2011), and SY5Y neuroblastoma (Nishida et al., 2008) cells. The color bars are on the same scale as Figure 3. Comparisons of in vivo overlap between in vitro systems are found in Figure S6. (C) The machine learning framework CoNTExT was used to predict the regional and temporal identity of each sample. The identity of each sample as progenitor or differentiated is labeled below the heatmaps. The accuracy of CoNTExT predictions is related to the degree of in vitro matching, so low matching systems may not have accurate predictions (Figure S4). (D) Module preservation was used to test which processes were conserved in different in vivo systems on the same scale as Figure 5.
TMAP in laminar data (Miller et al., 2014) showed that all systems except hiPSC dataset 3 have significantly less overlap with in vivo laminar transitions than 8 wk PD phNPCs (Figure 3D; Figure 7B; Figure S6E). Overall, when differentiating for short periods of time, laminar matching is low (Figure S3D; Figure 7B), suggesting that more differentiation in vitro is required to better match in vivo development in most systems. In all systems, more overlap was detected in transitions between proliferative layers (VZ, SVZ) and postmitotic layers consistent with the differentiation of dividing neural progenitors into post mitotic neurons (Figure 3D; Figure 7B).
We next applied CoNTExT to predict individual sample developmental maturity and regional identity (Figure 7C). When CoNTExT predicted a greater difference in culture maturity from progenitor to differentiated cells, stronger in vivo matching was observed in TMAP. This implies that TMAP is mainly measuring similarity to in vivo neural differentiation. This can be seen in hiPSC dataset 1 where CoNTExT detected little difference in progenitor versus differentiated cultures and TMAP showed only a low degree of overlap. CoNTExT was also used to predict regional identity on each individual in vitro sample. Interestingly, several of the hiPSC cultures (dataset 1 and 3) showed mixed cortical and cerebellar regional identity. The two samples in dataset 3 classified as cortex were derived from the same line and had greater expression of cortical markers, and lower expression of caudal markers than other samples from the same dataset (data not shown).
To assess which biological processes were preserved in these systems, we applied the WGCNA framework. We first observed poor preservation of network architecture in SY5Y cells. Only the magenta module related to the cessation of proliferation was strongly preserved (Zscore>10), while synaptic modules were weakly preserved or not preserved, consistent with the restricted overlap to down-regulated genes in laminar transitions in SY5Y cells (Figures 7D). Across all other in vitro systems (phNPCs, hiPSCs, and hES-derived neural cells), a high degree of preservation (Z>10) was found for modules relating to neurogenesis and mitosis (magenta, red), neurite outgrowth and synaptogenesis (salmon, lightgreen, green), and RNA processing and splicing (red). Despite these similarities, differences in developmental processes were apparent across the hiPSC and phNPC systems. The chromatin remodeling midnightblue module (enriched for ASD associated de novo variation; Figure 6), while preserved in phNPCs was not preserved in the hES and most hiPSC-derived datasets (Figure 7D; this module was preserved in hiPSC dataset 2 when subsetting to 1 and 4 wks PD or 4 and 8 wks PD; Table S1). Similarly the pink module related to neural progenitor proliferation and gliogenesis and the darkred module related to programmed cell death were better or uniquely preserved in phNPCs (Figure 7D). Conversely the turquoise module was better preserved in hiPSC-derived cells. Nonetheless, preservation scores for all highly preserved modules common to all systems were generally higher in phNPCs.
Discussion
Understanding how in vitro systems model in vivo brain development is becoming more important as human neural in vitro systems are becoming more widely used. Benchmarks have long been sought in the stem cell field (Andrews et al., 2005); however, there currently exists no unbiased rubric on which to base such a comparison. We reasoned that transcriptional profiles would provide a comprehensive, unbiased foundation on which to build such a quantitative structure at a genome-wide level. Here, we demonstrate through multiple analytical approaches and validate via multiple independent datasets that phNPCs strongly match in vivo development and produce cortical neurons reaching late mid-fetal levels of maturity after 12 weeks of differentiation. Most, but not all neurodevelopmental processes of normal corticogenesis are highly preserved in phNPCs. Risk genes for ASD, ID, and schizophrenia are enriched in modules related to these processes. In addition, we find that phNPCs have a stronger overlap and matching with in vivo cortex than other in vitro systems in common use, including some hiPSCs. Furthermore, we identify specific neurodevelopmental processes related to ASD pathophysiology preserved exclusively in phNPCs and not in other in vitro models. Overall, we provide a basic experimental framework for assessing the preservation in vitro of specific biological processes and gene networks underlying cortical development in vivo.
One expectation is that this framework will allow comprehensive evaluation of in vitro systems as they are developed and refined. A variety of protocols exist to generate hiPSCs, including variations in the type of cells used to generate hiPSCs, the standards for characterizing resulting lines and neural induction and differentiation protocols (Haase et al., 2009; Obokata et al., 2014; Takahashi et al., 2007). Going forward, the comparative framework demonstrated here can be used to optimize protocols to best model in vivo development, including three-dimensional cultures (Kadoshima et al., 2013; Lancaster et al., 2013).
All four parts of our analytic framework are complementary, but provide different information. TMAP provides a global view of transcriptomic matching to in vivo development. TMAP is mainly driven by differentiation processes that are not regionally specific, so should not be considered a regional identity tool. Using human in vivo cerebellar or striatal data as input to TMAP, for example, still shows a high degree of in vitro matching with the cortical phNPCs (data not shown). CoNTExT is a useful framework for individual sample prediction of temporal and regional identity. One caveat to its use is that the algorithm must classify a sample into one of the defined regions or time periods, even if it matches none of them. As such, we defined through simulation how the level of in vivo matching affects the accuracy of classification (Figure S4D–E). Finally, CoNTExT and TMAP do not specify which molecular pathways are conserved. WGCNA module preservation is a well-validated tool to determine the specific functional processes that are preserved in vitro. For those processes not conserved, modifying the expression of key hub genes in the module using exogenous factors like small molecules or by targeted gene expression, may provide leverage for developing more faithful in vivo modeling in the future.
The observed differences between in vitro systems could be due to inherent properties of the system, reprogramming, culture conditions, or even culturing technique. While with the current data we cannot pinpoint the source of variance, it is interesting to note that hiPSC datasets generated using more recent protocols, which include addition of morphogens (SMAD inhibitors) for neural induction, match better to in vivo brain. Similarly, because the time spent in the neural induction stage differs between systems, the initial (NPC) timepoint may reflect different starting points of maturity. Nonetheless, significant heterogeneity exists even within hiPSCs cultured in identical conditions. In the highest in vivo matching hiPSC dataset by TMAP, we observed one clone with strongly different regional identity compared to another clone derived from the same donor (Figure 7; Dataset 3). The in vivo transcriptomic framework provided here can be used to systemically test how these and other factors can optimize matching to in vivo developmental transitions or regional identify.
Differences between pluripotent-derived and primary neural progenitors have been identified in previous studies, including low or undetectable expression of the radial glia marker GFAP and overall higher production of neurons upon differentiation (Falk et al, 2012). GFAP is expressed in radial glia in vivo (Wilkinson et al., 1990) and in undifferentiated phNPCs and its absence in hiPSC-derived progenitors may be related to their more immature neuroepithelial nature (Falk et al., 2012).
Growing evidence implicates abnormalities in the development and architecture of the human cerebral cortex in neuropsychiatric illness (Akbarian et al., 1996; Anderson et al., 1996; Ebert and Greenberg, 2013; Gulsuner et al., 2013; Hutsler and Zhang, 2010; Parikshak et al., 2013; Penzes et al., 2011; Strauss et al., 2006; Wegiel et al., 2012; Willsey et al., 2013). We identified a module related to chromatin remodeling (midnightblue) that is largely not preserved in hiPS-derived neural stem cells and contains key genes involved in the generation of lower layer neurons and that regulate cortical thickness in rodent (Tuoc et al., 2013). Chromatin remodeling and transcriptional regulation are key biological processes that are enriched for protein disrupting and missense rare de novo mutations in ASD (Parikshak et al., 2013). These findings underscore critical differences in widely used in vitro systems that may compromise their utility for the study of development and neuropsychiatric disease. At the same time, because they can be derived from well-characterized patients and carry their full genetic background, hiPSC-derived NPCs have clear value. Our approach provides a template to refine these systems to better match in vivo brain development.
In this regard, our analysis also highlights processes poorly preserved in all of the vitro models, including modules related to immune response and neuron-microglia interactions (orange and black). A hub in the orange module, CX3CR1, is specifically expressed in microglia and mediates neuron-microglia signaling implicated in regulating neuronal death during neuroinflammation (Cardona et al., 2006; Fuhrmann et al., 2010). The neuronal ligand of CX3CR1, the fractalkine CX3CL1, is in the weakly preserved neuronal yellow module; this signaling pair regulates developmental synaptic pruning (Paolicelli et al., 2011). Given the emerging role for microglia in neurodevelopmental and neurodegenerative diseases (Paolicelli et al., 2011; Vargas et al., 2005) it will be important to evaluate if addition of microglia to differentiating phNPCs will be sufficient to recapitulate these processes.
These results also stress that current human neural stem cell systems, including hiPSCs, represent an immature, fetal state, consistent with other published work (Brennand et al., 2014; Espuny-Camacho et al., 2013; Kadoshima et al., 2013; Mariani et al., 2012; Nicholas et al., 2013). This is a key issue that may present challenges for modeling late-onset brain disorders including Alzheimer’s disease. It will be important to evaluate to what extent phenotypes in these immature neurons can be translated effectively into relevant pathophysiological insights (Sandoe and Eggan, 2013). Transcriptomic data can be leveraged to identify key processes and genes that may drive the generation of specific populations of cells or a desired level of maturity.
The methods presented here represent a first step in determining the degree of matching of in vitro systems to in vivo development. We realize that the ultimate understanding of how well in vitro cultures match in vivo development will be from matching individual cell transcriptomes from different regions, laminae, and developmental time periods to individual cell transcriptomes grown in culture. Importantly, in the future as comprehensive datasets of additional genomic data across cell populations in the developing brain become available, our approach can integrate different data modalities to provide a robust picture of the matching of neural stem cell models to in vivo brain. This approach can also provide mechanistic insight into differences between systems, which can be leveraged to improve the fidelity of in vitro models. This is especially important if our goal is to understand and develop treatments for human disease.
Experimental Procedures
Tissue Acquisition and Cell Culture
Human fetal brain tissue was obtained from the UCLA Gene and Cell Therapy Core following IRB regulations. Primary human neural progenitor cultures were prepared from PCW 15–18 human fetal brain as described (Konopka et al., 2012; Wexler et al., 2011) and detailed in Supplemental Experimental Procedures.
Genotyping
DNA was acquired from phNPCs cultures or fetal brain tissue and donors were genotyped with Illumina HumanOmni2.5 chips. Sex was determined based on homozygosity in X chromosome SNPs (2 male, 1 female). High confidence CNVs called with CNVision (Sanders et al., 2011) were not found in autism-associated regions.
Immunocytochemistry
Immunostaining was performed on PFA fixed cells with the indicated antibodies and the DNA-binding dye 4′,6-diamidino-2-phenylindole (DAPI) to quantify total cell number and cell viability. Detailed protocol and a list of antibodies used in this study is provided in Supplemental Experimental Procedures. Images were captured using a Zeiss Axio Imager D1 (Thornwood, NY, USA) epifluorescence microscope and analyzed using ImageJ software.
RNA Isolation, Processing, and Microarray Hybridization
For each of 5 lines generated from 3 donors (15–16 PCW), two independent differentiation experiments each containing two replicates were performed and harvested at four time points (1, 4, 8, 12 wks PD; ~16 samples per line; 77 total samples). RNA was isolated using TRIzol reagent (Invitrogen) according to standard protocols. We confirmed RNA integrity by RIN score with the Agilent 2100 Bioanalyzer (mean +/− sd: 9.16 +/− 0.78). Further details are found in Supplemental Experimental Procedures. All RNA expression profiles acquired as part of this project are publicly available (GSE57595).
Transition Mapping
A spatiotemporal atlas of human brain gene expression (Kang et al., 2011) and laminar expression data dissected via Laser Capture Microdissection from fetal human brain (Miller et al., 2014) were downloaded. For TMAP, differential expression between time points was calculated using a mixed effects model implemented in the nlme package (Pinheiro and Bates, 2009), with a random effect representing cell line (in vitro) or donor (in vivo), to appropriately account for the repeated measures of multiple non-independent samples (replicates of lines in phNPCs; multiple regions from the same donor in vivo). The Rank Rank Hypergeometric Overlap test (Plaisier et al., 2010) was implemented using custom R scripts now available as a Bioconductor package (RRHO) to evaluate overlap between in vivo and in vitro datasets. Further details are found in Supplemental Experimental Procedures.
CoNTExT
A multi-class multi-label machine learning algorithm was implemented in the MEKA toolbox (http://meka.sourceforge.net/) (Read, 2010) and trained on all 1340 samples of the Kang et al., 2011 data using all regions and all time points. Classification accuracy of developmental period was evaluated for each downloaded dataset within a range of +/− one from the true value to account for edge effects and ambiguities in the determination of conception date. For each sample, the probability of belonging to each class was output and displayed in Figure 4 and Figure S4. A simulation was conducted to judge the accuracy of the classifier at different levels on in vivo matching Figure S4. Further details are found in Supplemental Experimental Procedures.
Weighted Gene Co-expression Network Analysis
WGCNA was conducted using the R package WGCNA (Langfelder and Horvath, 2008) on the neocortical samples from Kang et al. (2011) dataset from time periods 1 through 8. Module preservation analysis was conducted to determine if density and connectivity based measures were preserved in vitro (Langfelder et al., 2011). Further details are found in Supplemental Experimental Procedures.
GO ontologies
Gene ontology analysis was completed using GO-Elite v1.2.5 with default settings (Zambon et al., 2012). Relevant biological process and molecular function categories were plotted selected from the top 10 terms ranked by Z-score.
Supplementary Material
Highlights.
Quantitative framework permits comparisons of in vitro models to in vivo brain
phNPCs recapitulate cortical development up to late mid-fetal periods
In vivo cortical gene networks implicated in ASD are preserved in phNPCs
Highlights key differences between widely-used stem cell models and in vivo brain
Acknowledgments
Fetal tissue was acquired in collaboration with CFAR grant 5P30 AI028697. Expression and genotype array data were generated by the UCLA Neuroscience Genomics Core. This work was supported by NIH grants to DHG (5R37 MH060233, 5R01 MH094714), JLS (K99MH102357) and LTU (T32MH073526) the Broad Stem Cell Research Center (BSCRC) (DHG, JLS, LTU), Autism Speaks (JLS), the UCLA BSCRC-California Institute for Regenerative Medicine (LTU), and the Dr. Miriam and Sheldon Adelson Medical Foundation (KSK). We thank Yadong Huang and Hyung-Seok Kim for assistance with reprogramming of patient fibroblasts. We thank Yining Zhao for website development. We thank members of the Geschwind laboratory for helpful discussions and critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akbarian S, Kim JJ, Potkin SG, Hetrick WP, Bunney WE, Jr, Jones EG. Maldistribution of interstitial neurons in prefrontal white matter of the brains of schizophrenic patients. Arch Gen Psychiatry. 1996;53:425–436. doi: 10.1001/archpsyc.1996.01830050061010. [DOI] [PubMed] [Google Scholar]
- An MC, Zhang N, Scott G, Montoro D, Wittkop T, Mooney S, Melov S, Ellerby LM. Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell stem cell. 2012;11:253–263. doi: 10.1016/j.stem.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen SL. Trajectories of brain development: point of vulnerability or window of opportunity? Neuroscience and biobehavioral reviews. 2003;27:3–18. doi: 10.1016/s0149-7634(03)00005-8. [DOI] [PubMed] [Google Scholar]
- Anderson SA, Volk DW, Lewis DA. Increased density of microtubule associated protein 2-immunoreactive neurons in the prefrontal white matter of schizophrenic subjects. Schizophr Res. 1996;19:111–119. doi: 10.1016/0920-9964(96)88521-5. [DOI] [PubMed] [Google Scholar]
- Andrews PW, Benvenisty N, McKay R, Pera MF, Rossant J, Semb H, Stacey GN Steering Committee of the International Stem Cell I. The International Stem Cell Initiative: toward benchmarks for human embryonic stem cell research. Nat Biotechnol. 2005;23:795–797. doi: 10.1038/nbt0705-795. [DOI] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Banerjee-Basu S, Packer A. SFARI Gene: an evolving database for the autism research community. Disease models & mechanisms. 2010;3:133–135. doi: 10.1242/dmm.005439. [DOI] [PubMed] [Google Scholar]
- Bayes A, van de Lagemaat LN, Collins MO, Croning MD, Whittle IR, Choudhary JS, Grant SG. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nature neuroscience. 2011;14:19–21. doi: 10.1038/nn.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc B Met. 1995;57:289–300. [Google Scholar]
- Betizeau M, Cortay V, Patti D, Pfister S, Gautier E, Bellemin-Menard A, Afanassieff M, Huissoud C, Douglas RJ, Kennedy H, et al. Precursor diversity and complexity of lineage relationships in the outer subventricular zone of the primate. Neuron. 2013;80:442–457. doi: 10.1016/j.neuron.2013.09.032. [DOI] [PubMed] [Google Scholar]
- Brennand K, Savas JN, Kim Y, Tran N, Simone A, Hashimoto-Torii K, Beaumont KG, Kim HJ, Topol A, Ladran I, et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol Psychiatry. 2014 doi: 10.1038/mp.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473:221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nature neuroscience. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, Colantuoni EA, Elkahloun AG, Herman MM, Weinberger DR, et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature. 2011;478:519–523. doi: 10.1038/nature10524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch R, Geschwind DH. The human brain in a dish: the promise of iPSC-derived neurons. Cell. 2011;145:831–834. doi: 10.1016/j.cell.2011.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, Greenberg ME. Activity-dependent neuronal signalling and autism spectrum disorder. Nature. 2013;493:327–337. doi: 10.1038/nature11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25:247–251. doi: 10.1523/JNEUROSCI.2899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espuny-Camacho I, Michelsen KA, Gall D, Linaro D, Hasche A, Bonnefont J, Bali C, Orduz D, Bilheu A, Herpoel A, et al. Pyramidal neurons derived from human pluripotent stem cells integrate efficiently into mouse brain circuits in vivo. Neuron. 2013;77:440–456. doi: 10.1016/j.neuron.2012.12.011. [DOI] [PubMed] [Google Scholar]
- Falk A, Koch P, Kesavan J, Takashima Y, Ladewig J, Alexander M, Wiskow O, Tailor J, Trotter M, Pollard S, et al. Capture of neuroepithelial-like stem cells from pluripotent stem cells provides a versatile system for in vitro production of human neurons. PLoS One. 2012;7:e29597. doi: 10.1371/journal.pone.0029597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fathi A, Hatami M, Hajihosseini V, Fattahi F, Kiani S, Baharvand H, Salekdeh GH. Comprehensive gene expression analysis of human embryonic stem cells during differentiation into neural cells. PLoS One. 2011;6:e22856. doi: 10.1371/journal.pone.0022856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, Haass C, LaFerla FM, Kretzschmar H, Herms J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nature neuroscience. 2010;13:411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson JG, Lin PT, Flanagan LA, Walsh CA. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999;23:257–271. doi: 10.1016/s0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- Goshima Y, Nakamura F, Strittmatter P, Strittmatter SM. Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature. 1995;376:509–514. doi: 10.1038/376509a0. [DOI] [PubMed] [Google Scholar]
- Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, Rippey C, Shahin H, Nimgaonkar VL, Go RC, et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell. 2013;154:518–529. doi: 10.1016/j.cell.2013.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase A, Olmer R, Schwanke K, Wunderlich S, Merkert S, Hess C, Zweigerdt R, Gruh I, Meyer J, Wagner S, et al. Generation of induced pluripotent stem cells from human cord blood. Cell stem cell. 2009;5:434–441. doi: 10.1016/j.stem.2009.08.021. [DOI] [PubMed] [Google Scholar]
- Hansen DV, Lui JH, Parker PR, Kriegstein AR. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature. 2010;464:554–561. doi: 10.1038/nature08845. [DOI] [PubMed] [Google Scholar]
- Hansen DV, Rubenstein JL, Kriegstein AR. Deriving excitatory neurons of the neocortex from pluripotent stem cells. Neuron. 2011;70:645–660. doi: 10.1016/j.neuron.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, van de Lagemaat LN, Smith KA, Ebbert A, Riley ZL, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489:391–399. doi: 10.1038/nature11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez DG, Nalls MA, Moore M, Chong S, Dillman A, Trabzuni D, Gibbs JR, Ryten M, Arepalli S, Weale ME, et al. Integration of GWAS SNPs and tissue specific expression profiling reveal discrete eQTLs for human traits in blood and brain. Neurobiology of disease. 2012;47:20–28. doi: 10.1016/j.nbd.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutsler JJ, Zhang H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain research. 2010;1309:83–94. doi: 10.1016/j.brainres.2009.09.120. [DOI] [PubMed] [Google Scholar]
- Inlow JK, Restifo LL. Molecular and comparative genetics of mental retardation. Genetics. 2004;166:835–881. doi: 10.1093/genetics/166.2.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482:216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MB, Kawasawa YI, Mason CE, Krsnik Z, Coppola G, Bogdanovic D, Geschwind DH, Mane SM, State MW, Sestan N. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron. 2009;62:494–509. doi: 10.1016/j.neuron.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoshima T, Sakaguchi H, Nakano T, Soen M, Ando S, Eiraku M, Sasai Y. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc Natl Acad Sci U S A. 2013;110:20284–20289. doi: 10.1073/pnas.1315710110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama R, Ohtsuka T, Shimojo H, Imayoshi I. Dynamic Notch signaling in neural progenitor cells and a revised view of lateral inhibition. Nature neuroscience. 2008;11:1247–1251. doi: 10.1038/nn.2208. [DOI] [PubMed] [Google Scholar]
- Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, Sousa AM, Pletikos M, Meyer KA, Sedmak G, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang P, Lee HK, Glasgow SM, Finley M, Donti T, Gaber ZB, Graham BH, Foster AE, Novitch BG, Gronostajski RM, et al. Sox9 and NFIA coordinate a transcriptional regulatory cascade during the initiation of gliogenesis. Neuron. 2012;74:79–94. doi: 10.1016/j.neuron.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Auerbach JM, Rodriguez-Gomez JA, Velasco I, Gavin D, Lumelsky N, Lee SH, Nguyen J, Sanchez-Pernaute R, Bankiewicz K, et al. Dopamine neurons derived from embryonic stem cells function in an animal model of Parkinson’s disease. Nature. 2002;418:50–56. doi: 10.1038/nature00900. [DOI] [PubMed] [Google Scholar]
- Konopka G, Wexler E, Rosen E, Mukamel Z, Osborn GE, Chen L, Lu D, Gao F, Gao K, Lowe JK, et al. Modeling the functional genomics of autism using human neurons. Mol Psychiatry. 2012;17:202–214. doi: 10.1038/mp.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annual review of neuroscience. 2009;32:149–184. doi: 10.1146/annurev.neuro.051508.135600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? PLoS computational biology. 2011;7:e1001057. doi: 10.1371/journal.pcbi.1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinson DF, Duan J, Oh S, Wang K, Sanders AR, Shi J, Zhang N, Mowry BJ, Olincy A, Amin F, et al. Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am J Psychiatry. 2011;168:302–316. doi: 10.1176/appi.ajp.2010.10060876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Somel M, Tang L, Yan Z, Jiang X, Guo S, Yuan Y, He L, Oleksiak A, Zhang Y, et al. Extension of cortical synaptic development distinguishes humans from chimpanzees and macaques. Genome Res. 2012;22:611–622. doi: 10.1101/gr.127324.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubs HA, Stevenson RE, Schwartz CE. Fragile X and X-linked intellectual disability: four decades of discovery. Am J Hum Genet. 2012;90:579–590. doi: 10.1016/j.ajhg.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuel M, Price DJ. Role of Pax6 in forebrain regionalization. Brain research bulletin. 2005;66:387–393. doi: 10.1016/j.brainresbull.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, Chen G, Gage FH, Muotri AR. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani J, Simonini MV, Palejev D, Tomasini L, Coppola G, Szekely AM, Horvath TL, Vaccarino FM. Modeling human cortical development in vitro using induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2012;109:12770–12775. doi: 10.1073/pnas.1202944109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvilly RJ, de Diaz MO, Schonemann MD, Hooshmand F, Rosenfeld MG. Transcriptional regulation of cortical neuron migration by POU domain factors. Science. 2002;295:1528–1532. doi: 10.1126/science.1067132. [DOI] [PubMed] [Google Scholar]
- Merkle FT, Eggan K. Modeling human disease with pluripotent stem cells: from genome association to function. Cell stem cell. 2013;12:656–668. doi: 10.1016/j.stem.2013.05.016. [DOI] [PubMed] [Google Scholar]
- Millar JK, Pickard BS, Mackie S, James R, Christie S, Buchanan SR, Malloy MP, Chubb JE, Huston E, Baillie GS, et al. DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science. 2005;310:1187–1191. doi: 10.1126/science.1112915. [DOI] [PubMed] [Google Scholar]
- Miller JA, Ding SL, Sunkin SM, Smith KA, Ng L, Szager A, Ebbert A, Riley ZL, Aiona K, Arnold JM, et al. Transcriptional Landscape of the Prenatal Human Brain. Nature. 2014;508:199–206. doi: 10.1038/nature13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8:427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas CR, Chen J, Tang Y, Southwell DG, Chalmers N, Vogt D, Arnold CM, Chen YJ, Stanley EG, Elefanty AG, et al. Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell stem cell. 2013;12:573–586. doi: 10.1016/j.stem.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida Y, Adati N, Ozawa R, Maeda A, Sakaki Y, Takeda T. Identification and classification of genes regulated by phosphatidylinositol 3-kinase- and TRKB-mediated signalling pathways during neuronal differentiation in two subtypes of the human neuroblastoma cell line SH-SY5Y. BMC research notes. 2008;1:95. doi: 10.1186/1756-0500-1-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obokata H, Wakayama T, Sasai Y, Kojima K, Vacanti MP, Niwa H, Yamato M, Vacanti CA. Stimulus-triggered fate conversion of somatic cells into pluripotency. Nature. 2014;505:641–647. doi: 10.1038/nature12968. [DOI] [PubMed] [Google Scholar]
- Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, Geschwind DH. Functional organization of the transcriptome in human brain. Nature neuroscience. 2008;11:1271–1282. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer TD, Schwartz PH, Taupin P, Kaspar B, Stein SA, Gage FH. Cell culture. Progenitor cells from human brain after death. Nature. 2001;411:42–43. doi: 10.1038/35075141. [DOI] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, Geschwind DH. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155:1008–1021. doi: 10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Pasca AM, Cord B, Palmer TD, Chikahisa S, Nishino S, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nature medicine. 2011;17:1657–1662. doi: 10.1038/nm.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nature neuroscience. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro J, Bates D. Mixed-Effects Models in S and S-PLUS. 2. Springer; 2009. [Google Scholar]
- Plaisier SB, Taschereau R, Wong JA, Graeber TG. Rank-rank hypergeometric overlap: identification of statistically significant overlap between gene-expression signatures. Nucleic Acids Res. 2010;38:e169. doi: 10.1093/nar/gkq636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P. Developmental and evolutionary adaptations of cortical radial glia. Cerebral cortex. 2003;13:541–549. doi: 10.1093/cercor/13.6.541. [DOI] [PubMed] [Google Scholar]
- Read J. Scalable Multi-label Classification. Hamilton, New Zealand: University of Waikato; 2010. [Google Scholar]
- Ripke S, O’Dushlaine C, Chambert K, Moran JL, Kahler AK, Akterin S, Bergen SE, Collins AL, Crowley JJ, Fromer M, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. 2013;45:1150–1159. doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropers HH. Genetics of intellectual disability. Current opinion in genetics & development. 2008;18:241–250. doi: 10.1016/j.gde.2008.07.008. [DOI] [PubMed] [Google Scholar]
- Rosen EY, Wexler EM, Versano R, Coppola G, Gao F, Winden KD, Oldham MC, Martens LH, Zhou P, Farese RV, Jr, et al. Functional genomic analyses identify pathways dysregulated by progranulin deficiency, implicating Wnt signaling. Neuron. 2011;71:1030–1042. doi: 10.1016/j.neuron.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan SD, Dolatabadi N, Chan SF, Zhang X, Akhtar MW, Parker J, Soldner F, Sunico CR, Nagar S, Talantova M, et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell. 2013;155:1351–1364. doi: 10.1016/j.cell.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoe J, Eggan K. Opportunities and challenges of pluripotent stem cell neurodegenerative disease models. Nature neuroscience. 2013;16:780–789. doi: 10.1038/nn.3425. [DOI] [PubMed] [Google Scholar]
- Soldner F, Laganiere J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R, Khurana V, Golbe LI, Myers RH, Lindquist S, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146:318–331. doi: 10.1016/j.cell.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa VH, Fishell G. Sonic hedgehog functions through dynamic changes in temporal competence in the developing forebrain. Current opinion in genetics & development. 2010;20:391–399. doi: 10.1016/j.gde.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolt CC, Lommes P, Sock E, Chaboissier MC, Schedl A, Wegner M. The Sox9 transcription factor determines glial fate choice in the developing spinal cord. Genes & development. 2003;17:1677–1689. doi: 10.1101/gad.259003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss KA, Puffenberger EG, Huentelman MJ, Gottlieb S, Dobrin SE, Parod JM, Stephan DA, Morton DH. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med. 2006;354:1370–1377. doi: 10.1056/NEJMoa052773. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Taylor SS, McKeon F. Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell. 1997;89:727–735. doi: 10.1016/s0092-8674(00)80255-x. [DOI] [PubMed] [Google Scholar]
- Tsoumakas G, Katakis I. Multi-Label Classification: An Overview. International Journal of Data Warehousing & Mining. 2007;3:1–13. [Google Scholar]
- Tuoc TC, Boretius S, Sansom SN, Pitulescu ME, Frahm J, Livesey FJ, Stoykova A. Chromatin regulation by BAF170 controls cerebral cortical size and thickness. Developmental cell. 2013;25:256–269. doi: 10.1016/j.devcel.2013.04.005. [DOI] [PubMed] [Google Scholar]
- van Bokhoven H. Genetic and epigenetic networks in intellectual disabilities. Annual review of genetics. 2011;45:81–104. doi: 10.1146/annurev-genet-110410-132512. [DOI] [PubMed] [Google Scholar]
- Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Annals of neurology. 2005;57:67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegiel J, Schanen NC, Cook EH, Sigman M, Brown WT, Kuchna I, Nowicki K, Wegiel J, Imaki H, Ma SY, et al. Differences between the pattern of developmental abnormalities in autism associated with duplications 15q11.2-q13 and idiopathic autism. Journal of neuropathology and experimental neurology. 2012;71:382–397. doi: 10.1097/NEN.0b013e318251f537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler EM, Rosen E, Lu D, Osborn GE, Martin E, Raybould H, Geschwind DH. Genome-wide analysis of a Wnt1-regulated transcriptional network implicates neurodegenerative pathways. Science signaling. 2011;4:ra65. doi: 10.1126/scisignal.2002282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson M, Hume R, Strange R, Bell JE. Glial and neuronal differentiation in the human fetal brain 9–23 weeks of gestation. Neuropathology and applied neurobiology. 1990;16:193–204. doi: 10.1111/j.1365-2990.1990.tb01156.x. [DOI] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155:997–1007. doi: 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambon AC, Gaj S, Ho I, Hanspers K, Vranizan K, Evelo CT, Conklin BR, Pico AR, Salomonis N. GO-Elite: a flexible solution for pathway and ontology over-representation. Bioinformatics. 2012;28:2209–2210. doi: 10.1093/bioinformatics/bts366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.