

Abstract
Background:
Transient neonatal myasthenia gravis (TNMG) affects a proportion of infants born to mothers with myasthenia gravis (MG). Symptoms usually resolve completely within the first few months of life, but persistent myopathic features have been reported in a few isolated cases.
Methods:
Here we report 8 patients from 4 families born to mothers with clinically manifest MG or mothers who were asymptomatic but had elevated acetylcholine receptor (AChR) antibody levels.
Results:
Clinical features in affected infants ranged from a mild predominantly facial and bulbar myopathy to arthrogryposis multiplex congenita. Additional clinical findings included hearing impairment, pyloric stenosis, and mild CNS involvement. In all cases, antibodies against the AChR were markedly elevated, although not always specific for the fetal AChR γ subunit. There was a correlation between maternal symptoms; the timing, intensity, and frequency of maternal treatment; and neonatal outcome.
Conclusions:
These findings suggest that persistent myopathic features following TNMG may be more common than currently recognized. Fetal AChR inactivation syndrome should be considered in the differential diagnosis of infants presenting with unexplained myopathic features, in particular marked dysarthria and velopharyngeal incompetence. Correct diagnosis requires a high degree of suspicion if the mother is asymptomatic but is crucial considering the high recurrence risk for future pregnancies and the potentially treatable nature of this condition. Infants with a history of TNMG should be followed up for subtle myopathic signs and associated complications.
Transient neonatal myasthenia gravis (TNMG) is a rare complication of maternal myasthenia gravis (MG), affecting around 10%–15% of infants of mothers with antibodies to the acetylcholine receptor (AChR)1 and, less frequently, muscle-specific kinase (MuSK).2 In the only detailed study, the fetal AChR, a pentameric complex composed of 2 α, 1 β, 1 δ, and 1 γ subunit is present until around 30 weeks, at which time it is replaced by the adult ε subunit.3
Infants born to mothers with MG, which is due to antibodies that act on AChRs at the neuromuscular junction, are at risk of TNMG; these antibodies commonly bind to both adult and fetal AChR, which differ in one subunit. Antibodies that selectively inhibit the function of the fetal γ subunit can be associated with severe cases of fetal arthrogryposis.4 Recently, a persistent myopathy (“fetal AChR inactivation syndrome”) following exposure to maternal antibodies has been noted in 2 families5,6 and attributed to loss or inactivation of the fetal AChR during a critical period of fetal development.
Here we report 8 infants from 4 unrelated families presenting with highly variable features of fetal AChR inactivation syndrome, suggesting that this condition may be more common than previously recognized.
METHODS
Patients.
We included infants with persistent myopathic features following an initial presentation with neonatal MG, a maternal history of MG, and/or maternal AChR antibodies. Details regarding maternal history and treatments, presentation, and examination findings were recorded from the case notes.
Methods.
Techniques used routinely in the Oxford Neuroimmunology service were applied, including radioimmunoassay for AChR antibodies and cell-based assays to distinguish binding of serum antibodies to γ (adult) or ε (fetal) AChRs. For these, human embryonic kidney (HEK) cells were transfected with complementary DNA (cDNA) for either the fetal or adult AChR and for the clustering protein rapsyn. cDNA for enhanced green fluorescent protein was introduced into the cDNA for the γ and ε subunits to indicate transfected cells. Sera were tested at 1:20 and 1:250 dilution, and binding of immunoglobulin (Ig) G detected with a secondary antibody conjugated to Alexa Fluor 568 (red) anti-human IgG at 1:750 dilution.
Standard protocol approvals, registrations, and patient consents.
Informed consent was obtained from all families. The ethical approval for further antibody testing on referred samples was from the Oxfordshire Regional Ethical Committee A (07/Q1604/28).
RESULTS
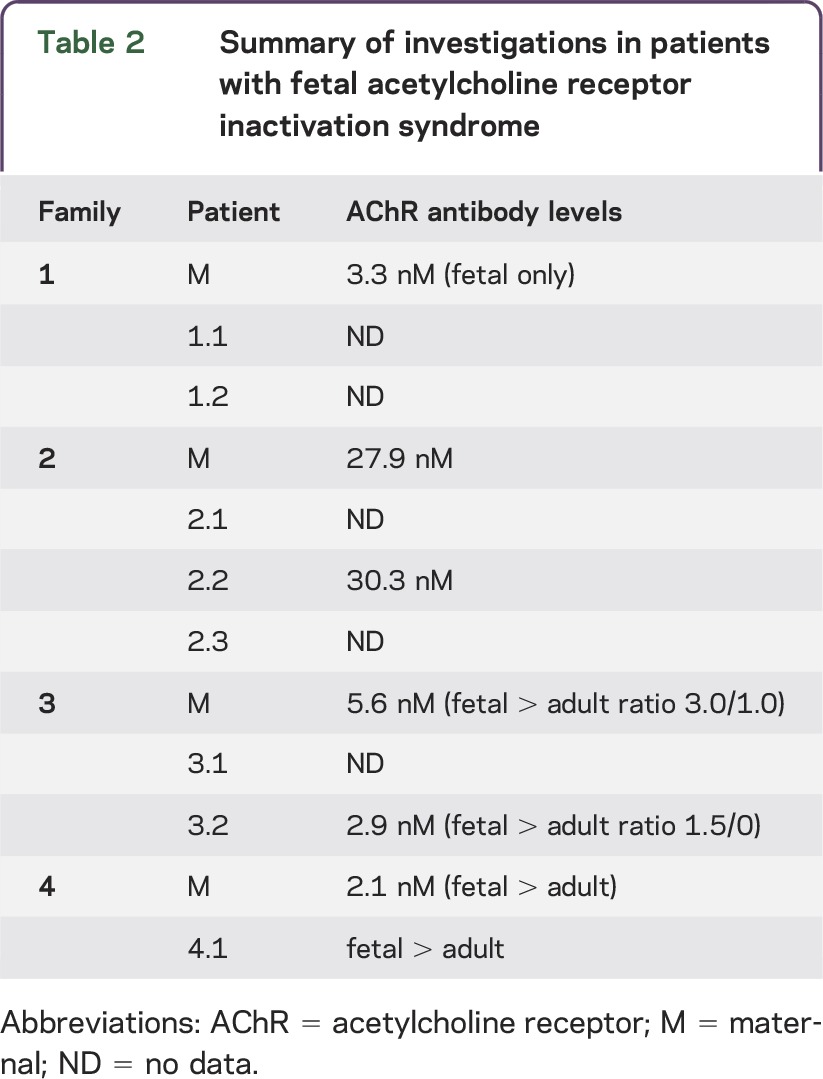
Clinical features from our patients, maternal history, and details of treatments are summarized in table 1 and outlined in more detail below. Key investigations (including maternal and patient AChR antibody levels) are summarized in table 2.
Table 1.
Clinical findings in patients with fetal acetylcholine receptor inactivation syndrome
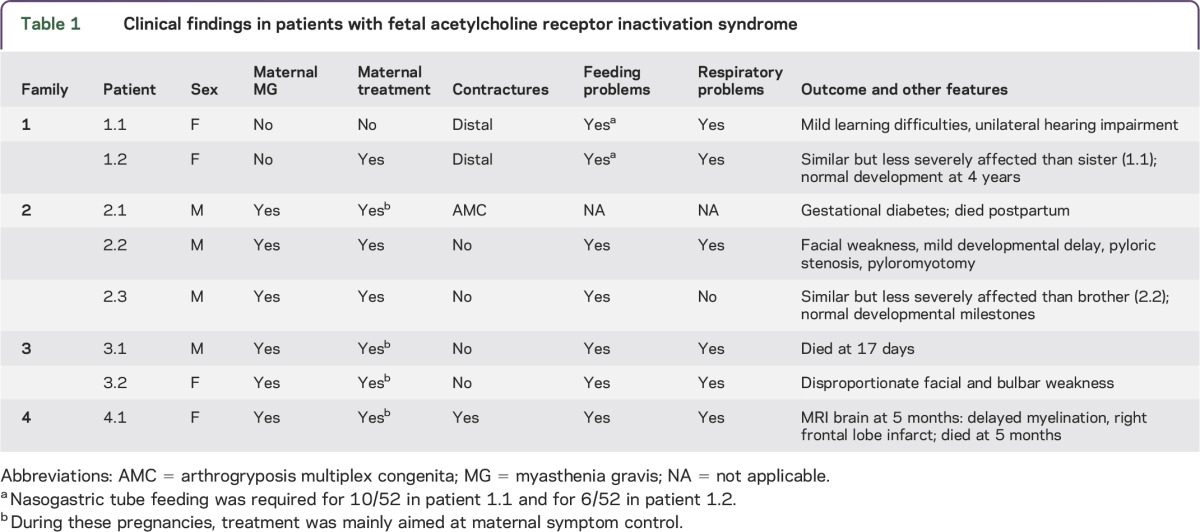
Table 2.
Summary of investigations in patients with fetal acetylcholine receptor inactivation syndrome
Family 1.
A female infant (patient 1.1) was born at 36 weeks to an asymptomatic mother following a pregnancy complicated by polyhydramnios from 33 weeks' gestation. She required ventilation from birth and tube feeding for 2.5 months. She had proximal and distal interphalangeal joint contractures. Creatine phosphokinase levels were normal. She made good developmental progress with some mild motor and speech delay. At 3 years, antibodies to the fetal AChR γ subunit were identified in her asymptomatic mother. The patient is currently 14 years old and has mild learning difficulties, residual facial weakness (figure 1, A–D) with poor palatal movement, and mild, presumably conductive, right hearing impairment. Contractures have completely resolved.
Figure 1. Clinical features in patients with fetal acetylcholine receptor inactivation syndrome.

In the index case from family 1 (A–D), there is a myopathic facial appearance with a horizontal smile but no ptosis. Her younger sister (E–G) was less severely affected and had only mild facial weakness as well as mild finger flexion deformities. The first surviving sibling in family 2 (H–K) had marked facial weakness with an inverted V-shaped mouth and bilateral ptosis. He made good developmental progress but had persistent axial weakness that slowly improved over time.
During her second pregnancy, the mother received alternate-day plasma exchange from 26 weeks' gestation. This was tolerated for 3 weeks with reduction of antibody levels but was then discontinued due to a fall in white blood cell count. A female infant (patient 1.2) was delivered via elective cesarean section at 34 weeks. She appeared similar to (figure 1, E–G) but less affected than her older sister, with milder facial weakness and only mild arthrogryposis affecting elbows, fingers, and knees. Feeding difficulties required tube feeding for 6 weeks. Motor developmental milestones were normal. She is currently 4 years old and does not have any difficulties other than slightly nasal speech.
Family 2.
A 24-year-old primigravida was diagnosed with MG at 20 weeks' gestation. Severe maternal myasthenic symptoms eventually responded to a combination of pyridostigmine, prednisolone, IV immunoglobulin (IVIg), and 8 cycles of plasma exchange, although AChR antibody titers remained high. A female infant (patient 2.1) was born at 34 weeks with severe and neonatally lethal arthrogryposis multiplex congenital (AMC). The mother had a thymectomy 5 weeks postpartum. Details of her antenatal and perinatal anesthetic management have been previously reported.7
During her second pregnancy, myasthenic symptoms were well-controlled by a combination of plasmapheresis on 3 occasions at the end of the first trimester and IVIg as required. At birth, the male infant (patient 2.2) had hypotonia with poor suck and swallow. He was ventilated for almost 3 weeks but subsequently established and maintained respiratory independence. He was initially tube fed but because of persistent difficulties required intermittent gastrostomy insertion. At 6 weeks corrected age he developed hypertrophic pyloric stenosis. He continues to make developmental progress. He has persistent but improving facial and axial weakness (figure 1, H–K).
During her third pregnancy, maternal myasthenic symptoms were controlled using IVIg until the fifth month. A male infant (patient 2.3) was born by elective cesarean section at 38 weeks and was noted to have paucity of movements and low tone but no contractures. He was transferred to the special care baby unit because of a supplemental oxygen requirement and an inadequate suck and swallow. He was started on pyridostigmine with moderate response. He required additional nasogastric tube feeds because of an inefficient, fatigable suck. He is currently 10 months old and his development is appropriate.
Family 3.
A 23-year-old woman was diagnosed with MG, and after initial medical treatment she underwent thymectomy followed by symptomatic improvement. At the time of her first pregnancy at 25 years, she was on prednisone 20 mg alternate days and pyridostigmine 60 mg 4 times daily. Reduced fetal movement and polyhydramnios were noted. She delivered a male infant at term (patient 3.1) who had severe hypotonia with respiratory insufficiency, requiring intubation and ventilation. He died at 17 days of age from respiratory failure.
Two years later she became pregnant again. Her myasthenic symptoms had improved on a reduced dose of prednisolone 10 mg alternate days. She had polyhydramnios but fetal movements were normal. A female infant (patient 3.2) was born at 38 weeks with hypotonia, respiratory distress, and generalized weakness. Following initial nasogastric tube feeding, she underwent gastrostomy placement at 7 months. Neonatal MG was diagnosed based on the detection of circulating antibodies that bound preferably to the fetal AChR (γ subunit) rather than the adult AChR (ε subunit) in both mother and infant (figure 2; table 2). The infant improved slowly but had persistent bulbar and facial weakness. No additional benefit was gained from immunotherapy and pyridostigmine treatment. A muscle biopsy taken at 7 months from the quadriceps was normal. Nerve conduction studies were normal but EMG showed myopathic changes, in particular in the facial muscles. At 20 months of age, she has made developmental progress and her bulbar function has improved, but her nasal speech and facial paresis remain severe.
Figure 2. Cell-based assay demonstrating differential binding of AChR antibodies to the adult and fetal receptors.
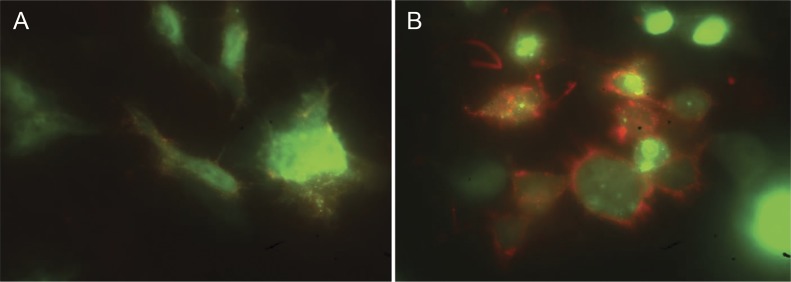
The fetal (gamma subunit specific) and adult (epsilon subunit specific) forms of the receptors were expressed in human embryonic kidney cells, identified with green fluorescence. Binding of immunoglobulin G was detected by anti-human IgG (red fluorescence). Results of a maternal serum from patient 3 that bound very weakly to the adult receptor (A), but strongly to the fetal receptor (B).
Family 4.
A 26-year-old primagravida with known MG was treated with high-dose pyridostigmine only during pregnancy. At 27 weeks' gestation, polyhydramnios was detected, requiring therapeutic amniocentesis. A course of plasmapheresis with 7 exchanges resulted in a decrease in maternal AchR antibody levels. Because of signs of fetal distress, a female infant (patient 4.1) was delivered by emergency cesarean section at 31 + 5 weeks' gestation. Birth weight was 1.545 kg. She was profoundly hypotonic and required intubation and ventilation. A trial of neostigmine showed no effect on respiration, but both muscle tone and antigravity movements improved. On examination, she had hypotonia, weakness, contractures of the third to fifth fingers bilaterally, and mild tightness of the hip adductors. At 8 weeks she was extubated but remained dependent on facial oxygen and/or nasal continuous positive airway pressure. She was never able to swallow saliva or fluids, but limb strength remained relatively good. Therapeutic trials with IVIg and pyridostigmine were ineffective. EMG studies including repetitive nerve stimulation in the extremities were normal. At 4 months, she required reintubation and 2 weeks of ventilator support. In accordance with parental wishes it was decided not to reintubate her in case of further deterioration, and she eventually died at 5 months of age from respiratory failure.
DISCUSSION
TNMG results from placental transfer of maternal antibodies and usually remits spontaneously over days to weeks. However, in rare cases the disorder persists and leaves the child with permanent disability, either multiple joint contractures (AMC) or myopathic features. Here we describe the first small case series of 8 children from 4 families with persistent myopathy and a history of either maternal MG or asymptomatically elevated maternal AChR antibody levels. These cases illustrate the persistent and potentially severe disability that can result from maternal AChR antibody exposure in utero and the possibility that better prenatal and perinatal surveillance and treatments may improve outcomes.
TNMG has been associated with antibodies to both the AChR1 and, very occasionally, MuSK.2 There is no correlation to maternal disease severity or absolute maternal antibody levels, but TNMG tends to recur in further pregnancies.8 Maternal thymectomy appears to have a protective effect.9 TNMG is usually transient, but persistent myopathic manifestations have been reported in a few isolated cases,5,6,10–13 previously thought to be due to coexisting pathology.
Within our cohort, at the most severe end of the spectrum infants presented with marked arthrogryposis and profound respiratory impairment resulting in early death, whereas the mildest cases had normal developmental milestones and only mild weakness, mainly affecting the facial and bulbar muscles. Severity of clinical symptoms in affected infants appeared to be related to the timing, intensity, and frequency of maternal treatment, as demonstrated in families 1, 2, and 3, in which fetal outcomes were better following optimization of maternal treatment and control of maternal symptoms, corresponding to earlier observations in isolated families.5 Control in subsequent pregnancies was easier to attain following maternal thymectomy in family 2. However, the relatively severe phenotype in family 4 suggests that fetal outcome may only be partly modified by optimal maternal treatment. With the exception of those who died early, the clinical course was characterized by an initial period of profound hypotonia, bulbar and respiratory impairment for several weeks, and continuous improvement but variable persistent myopathic features over subsequent years. The course in these patients suggests 2 distinct pathogenic mechanisms causing first an acute phase due to a transient neuromuscular transmission defect and second a chronic phase due to permanent myopathic damage; the latter observation would be consistent with an absent response to immunomodulatory/anticholinergic treatments and lack of evidence for a neuromuscular transmission defect in the chronic phase of the condition.
Marked dysarthria with or without velopharyngeal incompetence (VPI) was one of the most prominent persistent features observed in our series and also by others.5,13 It is interesting that TNMG has been reported as an identifiable factor in the medical history in a large cohort of patients with VPI,12 indicating a possible association. Pyloric stenosis was a feature in one of our patients and has previously been reported in an isolated case with TNMG14; it is uncertain whether this association reflects a shared disease mechanism and/or the effects of anticholinergic treatment. One patient each had mild learning difficulties and delayed myelination, but larger series will be required to identify whether mild CNS involvement is part of the phenotypical spectrum or reflective of dual pathology. Lastly, hearing impairment was present in the index case in family 1 and was previously reported in one family with fetal AChR inactivation syndrome5 but also in surviving children with AMC.4 The relevance of these observations and the pathophysiology of the hearing loss are not clear yet, bearing in mind that neuronal AChRs are structurally similar but not identical to muscle AChRs.
Maternal antibodies to AChRs were the defining feature in all 4 families, in 2 cases binding more strongly to the γ subunit containing fetal AChRs than to the ε subunit containing adult AChRs. It was suggested that, despite considerable variability in absolute AChR antibody levels, the anti-γ/anti-ε AChR antibody titer ratio remains relatively stable and can predict fetal outcome,15 but exceptions have been reported.1 Recently, a more appropriate cell-based assay for the detection of AChR antibodies binding to extracellular AChR epitopes on live HEK cells16 has been developed and can be used to measure the fetal and adult AChR antibodies independently. However, the binding of antibodies to fetal AChRs does not necessarily indicate the subgroup of fetal-specific antibodies that might cause paralysis in vivo, particularly those that inhibit AChR function.17 Moreover, it is possible that complement-mediated damage is also involved, as was demonstrated in one case of AMC,18 suggesting that different pathogenic mechanisms, perhaps acting at different times in gestation, may be relevant.
Our findings suggest a continuum between diverse disorders previously associated with maternal antibodies to the fetal AChR γ subunit, namely a subgroup of AMC and the more recently described “fetal AChR inactivation syndrome.”5 AMC is defined by multiple fixed joints, usually resulting from intrauterine lack of movement, and is caused by a wide range of different environmental or genetic factors, many of which are still unknown. High levels of maternal antibodies to the fetal AChR γ subunit were initially identified in rare cases of AMC,4 often recurring in successive pregnancies and not always associated with evidence of maternal MG, corresponding to family 1 in which the mother was also asymptomatic. High titers of antibodies specifically blocking the function of the fetal AChR γ subunit are rare in “classical” MG, explaining why AMC, although present in the most severe cases within our series, is generally uncommon in babies born to mothers with typical MG. At the milder end of the spectrum, our patients shared many findings with the “fetal AChR inactivation syndrome” described by Oskoui et al.5 in a family with 3 affected males, featuring facial diplegia but preserved strength in the limbs, high arched palate, VPI, hearing loss, and cryptorchidism. A similar isolated case has been reported more recently,6 indicating that long-term myopathic sequelae of TNMG are probably more common than currently recognized. We propose that “fetal AChR inactivation syndrome” comprises a wide range of manifestations, depending on both maternal antibodies to the fetal AChR γ subunit and modifying factors such as the timing, intensity, and frequency of maternal treatment.
The pivotal role of the AChR γ subunit in the pathogenesis of the phenotype reported in this article is supported by observations in various animal models and related inherited human disorders. During normal embryonic development, the fetal AChR helps to establish the primary connection between muscle and the nerve axon and is likely to be important not only for neuromuscular signal transduction but also for neuromuscular organogenesis,19 a hypothesis supported by the lethal phenotype in AChR γ subunit knockout mice.20 Moreover, patients with Escobar syndrome (Online Mendelian Inheritance in Man # 265000) secondary to mutations in the CHRNG gene encoding the AChR γ subunit21 typically present with joint contractures, excess webbing (“pterygia”), and respiratory distress. In contrast to congenital myasthenic syndromes secondary to mutations in genes encoding the α, β, and ε AChR subunits, which are expressed postnatally, patients with Escobar syndrome have no myasthenic symptoms later in life; this corresponds to observations in our own series, in which neurophysiologic assessments did not reveal any evidence for a persistent neuromuscular transmission defect and anticholinergic treatment was uniformly ineffective in the chronic stages of the condition. The persistent features observed in our patients are therefore most likely to reflect the effects of a prenatal exposure to antibodies that cause a fetal hypokinesia sequence, including an irreversible endplate myopathy corresponding to findings in antibody-mediated AMC,18 rather than an ongoing neuromuscular transmission defect.
While neonatal MG is mostly transient, maternal antibodies against the AChR γ subunit may cause persistent myopathic features in a subset of affected infants. Correct diagnosis requires a high degree of suspicion if the mother is asymptomatic but is crucial considering the high recurrence risk for future pregnancies and the essentially treatable nature of this condition. Fetal AChR inactivation syndrome should be considered in the differential diagnosis of infants presenting with unexplained myopathic features, in particular marked dysarthria and VPI, even in the absence of maternal symptoms. Lastly, infants with a history of TNMG should be followed up for signs of dysarthria or hearing impairment, as those may be subtle but at least partially amenable to supportive management.
GLOSSARY
- AChR
acetylcholine receptor
- AMC
arthrogryposis multiplex congenita
- cDNA
complementary DNA
- HEK
human embryonic kidney
- Ig
immunoglobulin
- IVIg
IV immunoglobulin
- MG
myasthenia gravis
- MuSK
muscle-specific kinase
- TNMG
transient neonatal myasthenia gravis
- VPI
velopharyngeal incompetence
AUTHOR CONTRIBUTIONS
Yael Hacohen: obtaining, analysis, and interpretation of the data, drafting the manuscript. Leslie W. Jacobson: obtaining, analysis, and interpretation of the data, drafting the manuscript. Susan Byrne: obtaining the data, drafting the manuscript. Fiona Norwood: obtaining, analysis, and interpretation of the data, revising the manuscript. Abhimanu Lall: obtaining the data, revising the manuscript. Stephanie Robb: obtaining, analysis, and interpretation of the data, revising the manuscript. Robertino Dilena: obtaining, analysis, and interpretation of the data, revising the manuscript. Monica Fumigalli: obtaining the data, revising the manuscript. Alfred Peter Born: obtaining, analysis, and interpretation of the data, revising the manuscript. Debbie Clarke: revising the manuscript. Ming Lim: design of the study, interpretation of the data, revising the manuscript. Angela Vincent: design of the study, interpretation of the data, revising the manuscript. Heinz Jungbluth: design of the study, interpretation of the data, revising the manuscript.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
Y. Hacohen received research support from NIHR. L.W. Jacobson, S. Byrne, F. Norwood, A. Lall, S. Robb, R. Dilena, and M. Fumagalli report no disclosures. A.P. Born is an associate editor for Acta Pediatrica and received research support from GlaxoSmithKline. D. Clarke reports no disclosures. M. Lim has received travel grants from Merck Serono, consulted for CSL Behring, and received research support from Action Medical Research, MS Society UK, SPARKS Charity, GOSH, and DES. A. Vincent has received honoraria from Baxter International Inc. and Biogen Inc.; is on the editorial board for Neurology; is an associate editor for Brain; holds a patent with Oxford University for VGKC complex antibodies; receives royalties from Athena Diagnostics, Euroimmun AG, Blackwell Publishing, and Mac Keith Press; consulted for Athena Diagnostics; and received research support from NIHR. H. Jungbluth is an editorial board member for Neuromuscular Disorder, MBC Paediatrics, and Journal of Neuromuscular Diseases and received research support from Guy's and St. Thomas' Hospital Charitable Foundation. Go to Neurology.org/nn for full disclosures.
REFERENCES
- 1.Vernet-der Garabedian B, Lacokova M, Eymard B, et al. Association of neonatal myasthenia gravis with antibodies against the fetal acetylcholine receptor. J Clin Invest 1994;94:555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Niks EH, Verrips A, Semmekrot BA, et al. A transient neonatal myasthenic syndrome with anti-musk antibodies. Neurology 2008;70:1215–1216. [DOI] [PubMed] [Google Scholar]
- 3.Hesselmans LF, Jennekens FG, Van den Oord CJ, Veldman H, Vincent A. Development of innervation of skeletal muscle fibers in man: relation to acetylcholine receptors. Anat Rec 1993;236:553–562. [DOI] [PubMed] [Google Scholar]
- 4.Vincent A, Newland C, Brueton L, et al. Arthrogryposis multiplex congenita with maternal autoantibodies specific for a fetal antigen. Lancet 1995;346:24–25. [DOI] [PubMed] [Google Scholar]
- 5.Oskoui M, Jacobson L, Chung WK, et al. Fetal acetylcholine receptor inactivation syndrome and maternal myasthenia gravis. Neurology 2008;71:2010–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D'Amico A, Bertini E, Bianco F, et al. Fetal acetylcholine receptor inactivation syndrome and maternal myasthenia gravis: a case report. Neuromuscul Disord 2012;22:546–548. [DOI] [PubMed] [Google Scholar]
- 7.Chieza JT, Fleming I, Parry N, Skelton VA. Maternal myasthenia gravis complicated by fetal arthrogryposis multiplex congenita. Int J Obstet Anesth 2011;20:79–82. [DOI] [PubMed] [Google Scholar]
- 8.Norwood F, Dhanjal M, Hill M, et al. Myasthenia in pregnancy: best practice guidelines from a UK multispecialty working group. J Neurol Neurosurg Psychiatry 2014;85:538–543. [DOI] [PubMed] [Google Scholar]
- 9.Hoff JM, Daltveit AK, Gilhus NE. Myasthenia gravis in pregnancy and birth: identifying risk factors, optimising care. Eur J Neurol 2007;14:38–43. [DOI] [PubMed] [Google Scholar]
- 10.Morel E, Eymard B, Vernet-der Garabedian B, Pannier C, Dulac O, Bach JF. Neonatal myasthenia gravis: a new clinical and immunologic appraisal on 30 cases. Neurology 1988;38:138–142. [DOI] [PubMed] [Google Scholar]
- 11.Ahlsten G, Lefvert AK, Osterman PO, Stalberg E, Safwenberg J. Follow-up study of muscle function in children of mothers with myasthenia gravis during pregnancy. J Child Neurol 1992;7:264–269. [DOI] [PubMed] [Google Scholar]
- 12.Rieder AA, Conley SF, Rowe L. Pediatric myasthenia gravis and velopharyngeal incompetence. Int J Pediatr Otorhinolaryngol 2004;68:747–752. [DOI] [PubMed] [Google Scholar]
- 13.Jeannet PY, Marcoz JP, Kuntzer T, Roulet-Perez E. Isolated facial and bulbar paresis: a persistent manifestation of neonatal myasthenia gravis. Neurology 2008;70:237–238. [DOI] [PubMed] [Google Scholar]
- 14.Regenbaum S, Sidhu K, Smith CE. Transient neonatal myasthenia gravis and pyloric stenosis. J Clin Anesth 1995;7:515–518. [DOI] [PubMed] [Google Scholar]
- 15.Gardnerova M, Eymard B, Morel E, et al. The fetal/adult acetylcholine receptor antibody ratio in mothers with myasthenia gravis as a marker for transfer of the disease to the newborn. Neurology 1997;48:50–54. [DOI] [PubMed] [Google Scholar]
- 16.Leite MI, Jacob S, Viegas S, et al. IgG1 antibodies to acetylcholine receptors in “seronegative” myasthenia gravis. Brain 2008;131:1940–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riemersma S, Vincent A, Beeson D, et al. Association of arthrogryposis multiplex congenita with maternal antibodies inhibiting fetal acetylcholine receptor function. J Clin Invest 1996;98:2358–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reimann J, Jacobson L, Vincent A, Kornblum C. Endplate destruction due to maternal antibodies in arthrogryposis multiplex congenita. Neurology 2009;73:1806–1808. [DOI] [PubMed] [Google Scholar]
- 19.Koenen M, Peter C, Villarroel A, Witzemann V, Sakmann B. Acetylcholine receptor channel subtype directs the innervation pattern of skeletal muscle. EMBO Rep 2005;6:570–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi M, Kubo T, Mizoguchi A, Carlson CG, Endo K, Ohnishi K. Spontaneous muscle action potentials fail to develop without fetal-type acetylcholine receptors. EMBO Rep 2002;3:674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffmann K, Muller JS, Stricker S, et al. Escobar syndrome is a prenatal myasthenia caused by disruption of the acetylcholine receptor fetal gamma subunit. Am J Hum Genet 2006;79:303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]