

Abstract
Duchenne Muscular Dystrophy is a chronic, progressive and ultimately fatal skeletal muscle wasting disease characterised by sarcolemmal fragility and intracellular Ca2+ dysregulation secondary to the absence of dystrophin. Mounting literature also suggests that the dysfunction of key energy systems within the muscle may contribute to pathological muscle wasting by reducing ATP availability to Ca2+ regulation and fibre regeneration. No study to date has biochemically quantified and contrasted mitochondrial ATP production capacity by dystrophic mitochondria isolated from their pathophysiological environment such to determine whether mitochondria are indeed capable of meeting this heightened cellular ATP demand, or examined the effects of an increasing extramitochondrial Ca2+ environment. Using isolated mitochondria from the diaphragm and tibialis anterior of 12 week-old dystrophin-deficient mdx and healthy control mice (C57BL10/ScSn) we have demonstrated severely depressed Complex I-mediated mitochondrial ATP production rate in mdx mitochondria that occurs irrespective of the macronutrient-derivative substrate combination fed into the Kreb’s cycle, and, which is partially, but significantly, ameliorated by inhibition of Complex I with rotenone and stimulation of Complex II-mediated ATP-production with succinate. There was no difference in the MAPR response of mdx mitochondria to increasing extramitochondrial Ca2+ load in comparison to controls, and 400 nM extramitochondrial Ca2+ was generally shown to be inhibitory to MAPR in both groups. Our data suggests that DMD pathology is exacerbated by a Complex I deficiency, which may contribute in part to the severe reductions in ATP production previously observed in dystrophic skeletal muscle.
Introduction
Duchenne Muscular Dystrophy (DMD) is a fatal neuromuscular disease characterised by progressive fibre necrosis secondary to the absence of the protein dystrophin from the sarcolemma [1]. This leads to severe muscle wasting and weakness, and eventually death in all patients afflicted with the disease, usually by the third decade of life [2]. A prominent yet commonly ignored feature of DMD is compromised bioenergetical status. A 50% deficit in resting ATP levels in dystrophic skeletal muscle has been reported [3], [4] which is likely reflective of both an increased demand for calcium (Ca2+) buffering, satellite cell cycling and muscle regeneration, alongside an inability of cellular energy systems to match this heightened demand with sufficient ATP production. Indeed, functional aberrations in key intracellular energy systems, including the mitochondria, have been consistently reported in the literature [4]–[8]. It is likely that these aberrations are strongly associated with the drastically increased intracellular [Ca2+] that is observed in dystrophin-deficient myofibres [9], and contribute significantly to the muscle wasting phenotype of DMD.
Mitochondria are important regulators of [Ca2+]i in skeletal muscle and work synchronously with the sarcoplasmic reticulum (SR) to maintain a resting [Ca2+]i of approximately 50 nM, and handle 100-fold functional oscillations of up to 5 µM during excitation-contraction coupling. Increasing [Ca2+]mit during activity is thought to provide functional benefits to the muscle whereby oxidative ATP production can be matched to demand at the cross-bridge level [10], [11]. Ca2+ is thus considered a positive stimulator of oxidative phosphorylation (OXPHOS) [10]–[13] and in dystrophic muscle where resting [Ca2+]i is demonstrably two-fold higher [14]–[16] the stimulus for mitochondrial ATP production should, theoretically, be elevated.
A reduced capacity for OXPHOS by dystrophic mitochondria has, however, been consistently reported in the literature [5]–[8], [17]–[20], Assessment of O2 consumption indicates a 30–35% decrease in mitochondrial respiration rates of both type I and type II fibres [8], [21] and mdx myoblasts [7] yet it is unclear as to whether reduced OXPHOS capacity is attributable to any specific metabolic pathway or intrinsic to the mitochondria. Chi et al. [22] has reported impaired glycolysis and hence carbohydrate metabolism, but speculate that increased fatty acid metabolism adequately compensates this deficit. Conversely, Kuznetsov et al. [6] report a 50% reduction in electron transport chain (ETC) enzyme activity but no change in glycolysis or TCA cycling rates, whilst Even et al. [5] report normal glycolytic but abnormal TCA activity.
A reason for these fundamentally different results might be that mitochondria are particularly adaptive to the environment in which they exist, and that the metabolic studies performed thus far have failed to ascertain the functional and/or pathophysiological status of the fibres/cells/myotubes that assays have been performed upon. In particular, mitochondrial function is highly dependent upon the calcium status of the cell such that highly necrotic fibres/cells are likely to have a large proportion of mitochondria that are overloaded with calcium and morphologically and/or functionally altered. As such, isolated mitochondria preparations are useful assay platforms for the investigation of disease states as the complex pathology-induced environment within the cell is effectively removed allowing intrinsic defects and/or persistent adaptations within the mitochondria to be determined. Removing mitochondria from the cellular environment allows dysfunction specific to the mitochondria to be determined, and for artificial environments around the mitochondria to be created to determine the impact of isolated variables on OXPHOS.
The present study is the first to directly quantify ATP production by dystrophic mdx mouse skeletal muscle mitochondria using a biochemical luciferin/luciferase reporter system. In particular, mitochondria isolated from diaphragm - which unlike hind limb muscle, parallels the progressive severity evident in human DMD [23] – was assessed. We aimed to characterise the effect of TCA substrate manipulation and increasing environmental [Ca2+] on mitochondrial ATP production rate (MAPR) to determine if mdx mitochondria respond normally to an artificially induced-dystrophinopathic (i.e. high [Ca2+]i) environment.
Materials and Methods
Ethics Statement
All experiments were approved by the Animal Ethics Experimentation Committee, Victoria University (AEETH 07/02), and conformed to the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (7th edition, National Health and Medical Research Council (NHMRC), 2004). Animals were housed in pairs and provided enrichment, and all procedures were conducted to maximise animal welfare by following the Guidelines to Promote the Wellbeing of Animals used for Scientific Purposes (NHMRC, 2008).
Animals
Age-matched normal C57BL/10ScSn (13.17±0.21 weeks) and dystrophic mdx (12±0.5 weeks) mice were sourced from the Animal Resources Centre (Western Australia, Australia), housed at the Victoria University Animal Facility (Werribee Campus, Victoria, Australia) on a 12∶12 hour light-dark cycle and were permitted ad libitum access to food and water. A total of 36 animals were used in this study: For MAPR, CS function, and mitochondrial protein experiments n = 10 for both control and mdx groups; for the mitochondrial swelling experiment n = 8 for both control and mdx groups.
The diaphragm was selected to study the full range of metabolic substrates in this study due to its human-DMD comparative phenotype [23], whereas a smaller subset of substrates (PPKM and S+R) was used to assess TA. On the day of experimentation, animals were anaesthetised (sodium pentobarbitone; 60 mg/kg; IP) and both muscle were excised for mitochondrial isolation.
Mitochondrial Isolation
For the MAPR assay, mitochondria were isolated according to adapted methods of Wibom et al. [24] and is described briefly. Muscles were dissected free of connective tissue, finely minced and ground-glass homogenised in Solution A (KCl 100 mM; TRIS 50 mM; MgCl2.6H2O 5 mM; ATP 1.8 mM; EDTA 1 mM; pH 7.2) (M1). M1 homogenate was centrifuged (650 G, 4°C, 3 mins) and the supernatant retained and re-centrifuged (15000 G, 4°C, 3 mins). The mitochondrial pellet was retained and re-suspended in Solution A with graded fine-bore glass pasteur pipettes and re-centrifuged (15000 G, 4°C, 3 mins). This step was repeated. The final pellet was re-suspended in 200 µL mitochondrial storage solution (200 µl, Solution B containing: sucrose 180 mM; KH2PO4 35 mM; Mg Acetate 5 mM; EDTA 1 mM; pH 7.5 with KOH) to yield the final mitochondrial suspension (M2). M2 suspension was diluted 1∶5 with Solution B and a volume portion was snap frozen for later determination of mitochondrial protein content, a volume retained on ice for determination of citrate synthase activity and the remaining volume further diluted 1∶5 with ATP monitoring reagent containing firefly luciferase (AMR; containing: sucrose 180 mM; KH2PO4, 35 mM; Mg acetate, 5 mM; EDTA, 1 mM; Na4P2O7 mM; and 1% BSA, pH 7.5 with KOH; reconstituted FL-AAM ATP assay mix (Sigma Chemical Company, MO, USA) containing luciferase and luciferin) to yield the working mitochondrial suspension (MS). MS was used immediately to measure ATP production rate.
Measurement of Mitochondrial ATP Production Rate (MAPR)
Substrate cocktails of pyruvate and malate (1 mM each; P+M); palmitoyl-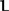 -carnitine (0.005 mM) and malate (10 mM) (PC+M); α-ketoglutarate (10 mM; α-KG); pyruvate (1 mM), palmitoyl--carnitine (0.005 mM), α-ketoglutarate (10 mM) and malate (10 mM) (PPKM); and succinate (20 mM and rotenone (0.1 mM) were prepared in AMR solution with a final [ADP] of 0.04 mM to stimulate the various entry points for substrate channelling into the TCA to fuel OXPHOS.
-carnitine (0.005 mM) and malate (10 mM) (PC+M); α-ketoglutarate (10 mM; α-KG); pyruvate (1 mM), palmitoyl--carnitine (0.005 mM), α-ketoglutarate (10 mM) and malate (10 mM) (PPKM); and succinate (20 mM and rotenone (0.1 mM) were prepared in AMR solution with a final [ADP] of 0.04 mM to stimulate the various entry points for substrate channelling into the TCA to fuel OXPHOS.
To test the effects of increasing extra-mitochondrial [Ca2+] on MAPR, a series of stock CaCl2 solutions were prepared to give final concentrations in the substrate cocktails of 50, 100, 200 and 400 nM, respectively. To determine the effects of [Ca2+] on MAPR for both diaphragm and TA, only PPKM and S+R substrates were utilised as these substrates give a good indication of how each of NADH- and FADH2-stimulated OXPHOS is affected by Ca2+, respectively.
MAPR was determined using firefly luciferase (Sigma, Australia) according to the methods of Wibom et al. [25] as previously used in our laboratory [26], [27]. Each well was calibrated with an internal ATP standard at the commencement of the assay. Basal ATP production was measured and ATP synthesis induced via the addition of ADP. [26], [27]. For all experiments, a background assay was performed containing mitochondria with no substrate to determine non-mitochondrial ATP production via the adenylate kinase and other non-specific reactions (see S1 Table). Linear, time-dependent ATP production was confirmed for all assays. Data is the average MAPR of triplicate readings and corrected for intact mitochondrial yield (%) as determined by sequential citrate synthase activity analysis. Control (n = 10) and mdx (n = 10) group sizes were utilised for these experiments.
Measurement of Citrate Synthase
CS synthase activity was measured spectrophotometrically (412 nm, 25°C), according to the methods of Srere [28]. CS activity was measured in the original mitochondrial extract on the day of experimentation post-MAPR assay (CSbefore), in the original mitochondrial extract containing 1% Triton-X after periodical snap-freeze and -thaw cycles to encourage fracture and disintegration of the mitochondrial membranes and CS liberation (CSafter), and in a separate snap-frozen sample of the same muscle on which MAPR was performed after mechanical homogenisation and complete disintegration of sub-cellular structures (CStotal).
CSafter and CStotal samples were stored and homogenised in homogenising buffer (KCl 175 mM and ethylenediaminetetra-acetic acid (EDTA) 2 mM; pH 7.4) and activity was calculated utilising the extinction coefficient for CS of 13.6 [28]. The proportion of CS activity in CSbefore, CSafter and CStotal samples was used to calculate intact mitochondrial yield for correction of MAPR.
Measurement of Mitochondrial Proteins
Mitochondrial protein content was analysed on snap frozen samples derived from MS using a Bradford Protein Assay kit in triplicate (Bio-Rad Protein Assay, Bio-Rad Laboratories, Hercules, CA, USA). Mitochondrial protein was used to correct for CS activity.
Measurement of Mitochondrial Swelling
Mitochondrial swelling was measured spectrophotometrically in a microplate assay adapted from [29]. 5 µL of diaphragmatic MS (∼10 µg mitochondrial protein) was added to each well and the assay was initiated via the addition of 90 uL of Solution B containing glutamate, malate and succinate (G+M+S; all 10 mM; [30]). A baseline reading was recorded for 4 mins (excitation wavelength = 490 nm; emission wavelength = 525 nm) before the addition of 5 µL of vehicle control (G+M+S in Solution B) or the appropriate [Ca2+]. An additional 30 µM [Ca2+] was utilised as a positive control as a known activator of the mitochondrial permeability transition pore inducing maximal mitochondrial swelling subsequent to bursting. Mitochondrial swelling was followed for 16 minutes, with a decrease in absorbance indicating increased size of the mitochondria (n = 8 for both groups for these experiments).
Statistics
Results are expressed as mean ± standard error of the mean. [Ca2+]-treated MAPR data is expressed relative to MAPR at 0 nM [Ca2+]. A three-way analysis of variance (ANOVA) was utilised to detect between group, substrate and muscle differences in MAPR at 0 nM [Ca2+], and between group, [Ca2+] and muscle differences in MAPR across the [Ca2+] range. A two-way ANOVA was utilised to detect between group and muscle differences for CS function, mitochondrial protein concentration and mitochondrial yield in both TA and diaphragm. A two-way ANOVA was utilised to detect between group and [Ca2+] difference in mitochondrial morphology. Subsequent post hoc analysis using Tukey’s test was utilised to identify interactions between the variables. An α value of 0.05 was considered significant.
Results
MAPR of mdx mitochondria from diaphragm and TA
This study notably demonstrates that NADH-dependent MAPR is severely depressed in mdx mitochondria isolated from diaphragm (Figs. 1 and 2; p<0.001) and TA (Fig. 2; p<0.05) compared to controls and that this depression occurs irrespective of the oxidizing substrate used to stimulate OXPHOS. Stimulation of various substrate entry points within the Kreb’s Cycle that mimic the entry points for glycolytic (P+M), β-fat oxidation (PC+M) and anaplerotic glutamate oxidation (α-KG) derivatives all produced chronically depressed MAPR in diaphragmatic mdx mitochondria that was 70% (p<0.005), 67% (p<0.01) and 60% (p<0.01) less than controls, respectively. A similar 75% depression in diaphragm MAPR was observed when Kreb’s cycle substrates were combined (PPKM) to stimulate OXPHOS (Fig. 2; p<0.01). This effects was also observed for TA MAPR, which was 62% less than controls following PPKM stimulation (p<0.05; Fig. 2). Succinate-induced FADH2-dependent OXPHOS, however, significantly ameliorated MAPR of mdx diaphragmatic and TA mitochondria when compared to the NADH-generating PPKM substrate combination (p<0.05), albeit was still 30% less than control levels (p<0.05). Interestingly, this effect of the S+R combination on MAPR was not observed in the control group (p>0.05).
Figure 1. MAPR of normal (control C57BL/10) and dystrophic mdx diaphragm at 0 nM extra-mitochondrial [Ca2+] following pyruvate and malate (P+M), palmitoyl carnitine and malate (PC+M) and α-ketoglutarate (α-KG) stimulation.

***p<0.005 from controls; **p<0.01 from controls; n = 10 control and n = 10 mdx.
Figure 2. MAPR of normal (control C57BL/10) and dystrophic mdx diaphragm and TA at 0 nM extra-mitochondrial [Ca2+] following pyruvate + palmitoyl carnitine + α-ketoglutarate + malate (PPKM) and succinate + rotenone (S+R) stimulation.

**p<0.01 from control group; *p<0.05 from control group; ∧p<0.05 from other substrates in mdx group; n = 10 control & n = 10 mdx.
Effects of a “resting” extramitochondrial [Ca2+] spectra on MAPR
Fig. 3 shows the effect of a 50–400 nM extra-mitochondrial [Ca2+]em (normalised to 0 nM [Ca2+]), on PPKM- and S+R-stimulated MAPR in control and mdx mitochondria extracted from the diaphragm and TA. Overall, there was no significant effect of [Ca2+] on MAPR of mdx mitochondria when compared to controls (p>0.05 group effect). There was a trend towards reduced stimulation of PPKM-fuelled MAPR in mdx compared to control mitochondria from both diaphragm (p = 0.141; Fig. 3A) and TA (p = 0.069; Fig. 3C) upon addition of Ca2+ em however this observation was not concentration-specific. Irrespective of group, 400 nM [Ca2+]em inhibited PPKM-stimulated MAPR of diaphragmatic mitochondria compared to all other [Ca2+]em (all p<0.05); Fig. 3A). A similar effect was observed for TA mitochondria, with 400 nM Ca2+ inhibiting PPKM-stimulated MAPR compared to 50 nM and 100 nM (both p<0.05) but not 200 nM [Ca2+]em (p>0.05; Fig. 3C).
Figure 3. Relative MAPR of normal (control C57BL/10) and dystrophic mdx diaphragm following (A) pyruvate, palmitoyl carnitine, α-ketoglutarate and malate (PPKM) stimulation; (B) succinate and rotenone (S+R) stimulation; and TA following (C) PPKM stimulation; and (D) S+R stimulation across a 50–400 nM [Ca2+] range.

Data is expressed relative to MAPR at 0 [Ca2+] as mean ± SEM. *p<0.05 400 nM different from other [Ca2+]; n = 10 control & n = 10 mdx.
For S+R-stimulated MAPR in diaphragm, there again was a trend towards relative Ca2+-induced MAPR depression (from 0 nM MAPR) in mdx compared to control mitochondria (p = 0.074; Fig. 3B). This trend was notably absent in TA mitochondria (p = 0.435 mdx versus control; Fig. 3D). There was no effect of extra-mitochondrial [Ca2+] on S+R-stimulated MAPR at any of the assayed concentrations for TA mitochondria, irrespective of group (Fig. 3D). In diaphragm, a 400 nM extramitochondrial [Ca2+] was inhibitory to S+R-induced MAPR compared to 50 nM and 100 nM [Ca2+]em (both p<0.05) but not to 200 nM (p>0.05; Fig. 3D).
Citrate Synthase activity
All data for CS function is described in Table 1. CSbefore activity (a measure of free CS availability from damaged mitochondria in the MS) was 40% lower in mdx mitochondria from TA and diaphragm compared to controls (p<0.01). CSafter activity (a measure of the total number of mitochondria that were brought through the mitochondrial extraction process whether intact or damaged), was reduced by ∼30% in mdx TA and diaphragm (p<0.01) from controls. No difference was observed between control and mdx groups in the CSbefore:CSafter ratio, a marker of the robustness of the extracted intact mitochondria that were available to OXPHOS and therefore MAPR measurement. In a separate homogenised sample of the same muscle from which mitochondria were isolated, CStotal activity was unchanged between control and mdx groups in both TA and diaphragm. A significant decrease in the CSafter:CStotal ratio (p<0.01), which represents the proportion of possible mitochondria that are able to withstand the extraction procedure, was observed in mdx muscles, in both diaphragm and TA. Mitochondrial yield (% of successfully extracted mitochondria in the MS compared to the total number of mitochondria available for extraction in the same mass of muscle) was markedly (35%) higher in control compared to mdx MS (p<0.01).
Table 1. Citrate synthase (CS) function and mitochondrial yield (% of total available) and protein content (% of total) of control (c57BL/10) and dystrophic mdx diaphragm and TA.
| CON | MDX | p values | ||||
| TA | DIA | TA | DIA | Strain | Muscle | |
| CSbefore (mmol.mg−1 mitochondrial protein.min−1) | 1.3±0.2 | 1.6±0.3 | 0.8±0.1 | 0.9±0.092 | 0.004** | 0.338 |
| CSafter (mmol.mg−1 mitochondrial protein.min−1) | 11.8±0.9 | 11.7±0.8 | 8.7±0.6 | 8.3±1.5 | 0.002** | 0.282 |
| CStotal (mmol.mg−1 mitochondrial protein.min−1) | 55.8±3.2 | 67.2±4.4 | 53.9±4.6 | 64.4±5.6 | 0.395 | 0.071 |
| CSafter:CStotal Ratio | 0.20±0.01 | 0.15±0.02 | 0.17±0.02 | 0.13±0.02 | 0.009** | 0.008** |
| CSbefore:CSafter Ratio | 0.10±0.02 | 0.12±0.02 | 0.09±0.01 | 0.13±0.02 | 0.900 | 0.063 |
| Mitochondrial Yield (%) | 18.0±1.5 | 13.9±1.1 | 15.5±2.1 | 10.1±1.3 | 0.012* | 0.009** |
| Mitochondrial Protein Content (% of total) | 21.6±2.2 | 22.1±0.9 | 17.5±0.8 | 31.3±2.6 | 0.499 | 0.269 |
*p<0.05 from control group, **p<0.01 from control group; n = 10 control & n = 10 mdx for TA and diaphragm.
Comparison of CS function between muscles was also made due to the marked differences in disease progression between muscles of the lower limb and diaphragm in mdx mice. Mitochondrial yield was ∼20% higher in TA than in diaphragm (p<0.05) as was the CSafter:CStotal ratio (p<0.01). There was a strong trend toward increased CStotal activity of the diaphragm compared to TA in both control and mdx muscle (20%; p = 0.07), however there were no significant differences in any of the other CS measures between muscles (CSbefore, CSafter and CSbefore:CSafter all p>0.05).
Mitochondrial Morphology
Mdx mitochondria were significantly larger (more swollen) as reflect by a reduced % change from baseline optical density at a 525 nm wavelength in both a Ca2+ free environment and when exposed to increasing [Ca2+]em (p<0.05; Fig. 4). Irrespective of strain, increasing [Ca2+] induced concomitant increases in mitochondrial size (p<0.05). Notably, there was large variation in the response of mdx mitochondria to increasing Ca2+ em load that was not evident in control mitochondria.
Figure 4. [Ca2+]-dependent swelling of normal (control C57BL/10) and dystrophic mdx diaphragm mitochondria respiring on glutamate, malate and succinate (GMS).

Data is expressed as the percentage change from baseline OD at 525 nm. *p<0.05 different from control at all [Ca2+]; **p<0.001 all different from 0 nM [Ca2+] n = 8 control & n = 8 mdx.
Discussion
This study confirms compromised ATP production by mdx mitochondria (MAPR), and in particular, in mitochondria respiring on optimal and continuous delivery of oxidising substrates. This is an important finding because mitochondria have been extracted from muscle at a relatively stable period of disease progression (12 weeks) and thus, the pathophysiology and concomitant inflammatory nature of the disease is less influential. This suggests an inherent deficit of the mitochondria, which is likely reflective of strong and persistent inhibition or structural abnormality of key mitochondrial machinery leading to deficient OXPHOS. While potentially induced by the extreme pathological environment in which the mitochondria reside in vivo, a mounting collection of evidence suggests a natural history of inherited metabolic impairment alongside dystrophin deficiency. Onopiuk et al. [7] has demonstrated that metabolic dysfunction is present in dystrophic myoblasts prior to the time of dystrophin expression [7]. This suggests that while dystrophin-deficiency induced pathophysiology may exacerbate mitochondrial dysfunction, metabolic impairment exists beforehand. That female carriers of DMD who do not express the disease exhibit abnormal muscle energy metabolism, especially when ATP demand is increased during exercise, lends further credence to this notion [31], [32].
Whilst several groups have demonstrated depressed oxygen consumption rate and isolated mitochondrial enzyme function in dystrophic skeletal muscle [5]–[8], [18], [21], [22], [33], we have highlighted the differential contributions of Complex I and II H+ flux into the ETC and resultant ATP production in dystrophic mouse mitochondria. Importantly, these impairments were shown in the ‘healthy’ mitochondria that survived the isolation process. MAPR was shown in this study to be severely depressed by up to 75% of control levels in both diaphragm (Figs. 1 and 2) and TA (Fig. 2) mitochondria from the mdx mouse. MAPR depression was more evident when stimulated by substrates that enter the TCA cycle (P+M, PC+M, α-KG and PPKM) and rely on NADH-mediated shuttling of H+ into the ETC through Complex I. In contrast, the complete inhibition of Complex I with rotenone and stimulation of Complex II-mediated MAPR with succinate (S+R) partially ameliorated mdx MAPR, albeit depression was still evident (Fig. 2). This suggests a problem with NADH flux into the ETC of mdx mitochondria, whereby NADH is either being sequestered away from, or is unable to be efficiently oxidised by, Complex I to establish proton motive force. In this instance, the accumulation of NADH at Complex I would be inhibitory to all dehydrogenases of the Krebs cycle except for succinate dehydrogenase (including pyruvate, isocitrate, α-KG dehydrogenase)(as reviewed in [34]), which would explain why succinate stimulation was able to partially restore MAPR of mdx mitochondria closer to control levels, whereas other Kreb’s substrates had no effect. Recent literature has demonstrated reduced Complex I activity in permeabilised skeletal muscle from mdx mice [33] and in mdx brain, in which dystrophin is normally expressed but is also notably absent in DMD [35]. The expression of various Complex I subunits is also evident in mdx skeletal muscle at the protein level (∼1.5 fold decrease), in human DMD skeletal muscle at the transcript level (3–6 fold decrease) [36] and in mdx cardiac muscle. Thus our data together with others [20], [33] suggests that a persistent impairment of Complex I function underpins dystrophic pathology, which strongly limits – but does not obliterate – the ATP-producing capacity of mitochondria. While Godin et al. [33] suggests that reduced mitochondrial biomass (i.e. Complex expression/activity) underscores loss of ETC function rather than specific respiratory impairment, our data would suggest the opposite as the stimulation of respiration with succinate following Complex I inhibition with rotenone partially restored MAPR in mdx mitochondria with equivalent biomass.
It has been widely documented that [Ca2+]mit is a positive stimulator of OXPHOS (reviewed in [37], [38]), but despite dramatic elevations of [Ca2+]mit within dystrophic skeletal [39] and cardiac [17] muscle, corresponding increases in OXPHOS do not appear to occur in dystrophic muscle. In our study, manipulating the [Ca2+]em across a 50–200 nM range (Fig. 3) had no effect on MAPR in either control or mdx mitochondria. With the exception of relative S+R-stimulated MAPR in TA, in which control and mdx MAPR were remarkably comparable, there was a general trend for reduced modulation of MAPR in mdx compared to control mitochondria (p values ranging from 0.069 to 0.141), however this what not statistically significant. Interestingly, in contrast to the lack of response a 50–200 nM [Ca2+] range effected, the addition of 400 nM [Ca2+ em] induced PPKM-stimulated MAPR depression in both control and mdx mitochondria from diaphragm and TA (Figs. 3A and 3C), and S+R-stimulated MAPR depression in both control and mdx mitochondria from diaphragm (Fig. 3B). While this demonstrates that mdx mitochondria are capable of responding to [Ca2+]em, and, indeed, that they respond in the same way to this “upper resting” load as per normal control mitochondria, general MAPR depression at this concentration is an anomalous finding. Several groups have demonstrated that 400 nM Ca2+ is sufficient to induce activation of key regulatory enzymes of OXPHOS including PDH and FAD-glycerol-3-phosphate dehydrogenase (reviewed in [38]), thus a stimulatory effect on MAPR would be expected. Nevertheless, we have shown the same inhibitory effect induced by 400 nM Ca2+ in control and mdx diaphragm mitochondria for PPKM- and S+R-stimulated MAPR, suggesting that this effect is not substrate specific and therefore, probably not a result of enzymatic PDH inhibition.
Remarkably, we were unable to show 400 nM [Ca2+]em-induced MAPR inhibition in either control or mdx mitochondria from TA, when stimulated with S+R. This would further suggest that diaphragm and TA respond very differently to extra-mitochondrial Ca2+ load, and that the target for MAPR inhibition by 400 nM Ca2+ may lie somewhere between Complex I and II, since in TA mitochondria MAPR inhibition was observed following PPKM- but not S+R-stimulation. It is interesting to speculate that the inhibitory effect of 400 nM [Ca2+]em on S+R-stimulated MAPR in mdx diaphragm, but not TA mitochondria may contribute to the more human DMD-like progressive wasting evident in this muscle, especially in the instance that Complex I function is impaired in DMD mitochondria which would increase reliance on FADH2-donated H+ and electrons through Complex II. A notable difference between mitochondria from the predominantly type I fibre-composed diaphragm and type-II fibre-composed TA is the absence of the mitochondrial calcium binding protein, calmitine. This protein is expressed only in type I fibres [40], although is notably quantitatively reduced in mdx mitochondria [41]. It is not known what, if any, role calmitine plays in directly regulating OXPHOS.
In this study, CS analysis of mdx muscle was performed to determine the quality of our isolated mitochondria preparations and subsequently normalise MAPR for comparative mitochondrial damage/loss caused by the extraction process and available functionality. While CStotal activity was unchanged in dystrophic mitochondria compared to controls in both TA and diaphragm, thus indicating comparable CS expression and mitochondrial content, it was interesting to observe highly significant reductions in the absolute (CSafter) and functional CS activity (CSbefore) of extracted MS. Taken together with a significant reduction in the CSafter/CStotal ratio and % mitochondrial yield of mdx muscle compared to controls, our data suggests that less mdx mitochondria are being successfully brought through the extraction process. This finding implicates that dystrophic mitochondria are more susceptible to damage (mechanical or biochemical) during the extraction process, than control mitochondria. Increased fragility of both the inner and outer mitochondrial membranes has been reported previously in mitochondrial fractions derived from gastrocnemius biopsies of DMD patients from as early as one year of age [42] – this is an age at which all muscles show relative stability and normality of function. We have confirmed the same fragility in mitochondria from 12 week old mdx mice (Table 1) – this is an age at which the severe cyclical degenerative episodes evident in hind limb muscles earlier in life have attenuated, and the diaphragm has yet to progress to its severe wasting phenotype. Mitochondrial membrane fragility following biochemical isolation is likely associated with the swollen morphology observed in mdx mitochondria comparative to controls (Fig. 4) which has also been observed by others [43], and, was apparent both prior to and following the addition of increasing [Ca2+] to the extramitochondrial environment. [Ca2+] load, however, did not induce any faster swelling of mdx mitochondria. This finding is in accordance with Reutenauer et al. [44] who demonstrated comparable responses to Ca2+ challenge between biochemically isolated control and mdx mitochondria, with the calcium retention capacity and Ca2+ overload concentration required to induce permeability transition following repeat 5 µM Ca2+ bursts, equivalent [39]. In contrast, Godin et al. [33] has shown a greater susceptibility (decreased time) to permeability transition pore opening and a reduced Ca2+_ retention capacity in isolated fibres from the mdx mouse, highlighting differences in the behaviour between in vivo and in vitro mitochondrial morphology. Indeed, it is interesting that swelling was persistent despite mitochondria bathing in a low Ca2+ em environment throughout the isolation procedure, storage and the Ca2+-free swelling assay in our study, which suggests that mdx mitochondria have a swollen morphological phenotype that is less responsive or unable to respond to subsequent reductions in the concentration of the Ca2+ em environment. Alternatively, this finding may reflect an artefact of our biochemical isolation technique as subsarcolemmal mitochondrial size is demonstrably comparable between mdx and control muscles in vivo, as detected by transmission electron microscopy [20]. As the subsarcolemmal mitochondrial pool accounts for only ∼10% of total skeletal muscle mitochondria and the comparative morphology of mdx intrafibrillar mitochondria (which account for ∼90% of total mitochondria) is yet to be assessed [20], further characterisation of mitochondrial swelling is clearly required.
In summary, we have demonstrated reduced MAPR in mitochondria extracted from diaphragm and TA from the mdx mouse model of DMD. As this reduction can be partially ameliorated by bypassing Complex I and directly stimulating Complex II, it suggests that Kreb’s-fuelled NADH-dependent Complex I function is deficient. We have also demonstrated a tendency for MAPR to be modulated differently by the addition of Ca2+ to the extra-mitochondrial environment in mdx compared to control mitochondria, though in a non-concentration-dependent manner. Further, we have shown depression of PPKM-stimulated MAPR in the presence of 400 nM [Ca2+]em in both diaphragm and TA, and S+R-stimulated MAPR in diaphragm in both control and mdx mitochondria. We cannot conclude if these findings are reflective of a pre-existing state either inherent to the DMD phenotype or induced by an in vivo pathophysiological environment that produces persistent morphological maladaptation. As such, further investigation is required to characterise the precise bioenergetical profile of mdx mitochondria and the fragility of mitochondrial membranes. Extension of our findings to determine whether Complex I deficiency worsens with age and/or repeat exposure to cyclical damage bouts, is present in other forms of muscular dystrophy characterised by intracellular Ca2+ dysregulation, and/or underscores the cardiac muscle pathology evident in DMD, would be of value to the current body of literature.
Supporting Information
Background ATP production (mmol.min−1.intact mitochondrial yield−1) of control (c57BL/10) and dystrophic mdx TA and diaphragm. *p<0.05 mdx different from control strain. There was no effect of muscle type (p = 0.323) or extramitochondrial [Ca2+] (p = 0.852).
(DOCX)
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its supporting information files.
Funding Statement
The authors have no support or funding to report.
References
- 1. Hoffman EP, Brown RH, Kunkel LM (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51:919–928. [DOI] [PubMed] [Google Scholar]
- 2. Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R, et al. (2002) Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscular Disorders 12:926–929. [DOI] [PubMed] [Google Scholar]
- 3. Austin L, De Niese M, McGregor A, Arthur H, Gurusinghe A, et al. (1992) Potential oxyradical damage and energy status in individual muscle fibres from degenerating muscle diseases. Neuromuscular Disorders 2:27–33. [DOI] [PubMed] [Google Scholar]
- 4. Cole M, Rafael J, Taylor D, Lodi R, Davies K, et al. (2002) A quantitative study of bioenergetics in skeletal muscle lacking utrophin and dystrophin. Neuromuscular Disorders 12:247–257. [DOI] [PubMed] [Google Scholar]
- 5. Even P, Decrouy A, Chinet A (1994) Defective regulation of energy metabolism in mdx-mouse skeletal muscles. Biochemical Journal 304:649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuznetsov AV, Winkler K, Wiedemann F, von Bossanyi P, Dietzmann K, et al. (1998) Impaired mitochondrial oxidative phosphorylation in skeletal muscle of the dystrophin-deficient mdx mouse. Molecular and cellular biochemistry 183:87–96. [DOI] [PubMed] [Google Scholar]
- 7. Onopiuk M, Brutkowski W, Wierzbicka K, Wojciechowska S, Szczepanowska J, et al. (2009) Mutation in dystrophin-encoding gene affects energy metabolism in mouse myoblasts. Biochemical and Biophysical Research Communications 386:463–466. [DOI] [PubMed] [Google Scholar]
- 8. Passaquin AC, Renard M, Kay L, Challet C, Mokhtarian A, et al. (2002) Creatine supplementation reduces skeletal muscle degeneration and enhances mitochondrial function in mdx mice. Neuromuscular Disorders 12:174–182. [DOI] [PubMed] [Google Scholar]
- 9. Dunn J, Radda G (1991) Total ion content of skeletal and cardiac muscle in the mdx mouse dystrophy: Ca2+ is elevated at all ages. Journal of the Neurological Sciences 103:226–231. [DOI] [PubMed] [Google Scholar]
- 10. McCormack J, Denton R (1993) Mitochondrial Ca2+ Transport and the Role of Intramitochondrial Ca2+ in the Regulation of Energy Metabolism. Developmental neuroscience 15:165–173. [DOI] [PubMed] [Google Scholar]
- 11. McCormack JG, Denton R (1993) The role of intramitochondrial Ca2+ in the regulation of oxidative phosphorylation in mammalian tissues. Biochemical Society Transactions 21:793–799. [DOI] [PubMed] [Google Scholar]
- 12. Das A, Harris DA (1990) Intracellular calcium as a regulator of the mitochondrial ATP synthase in cultured cardiomyocytes. Biochemical Society Transactions 18:554–555. [DOI] [PubMed] [Google Scholar]
- 13. Wernette M, Ochs R, Lardy H (1981) Ca2+ stimulation of rat liver mitochondrial glycerophosphate dehydrogenase. Journal of Biological Chemistry 256:12767–12771. [PubMed] [Google Scholar]
- 14. Fong P, Turner PR, Denetclaw WF, Steinhardt RA (1990) Increased activity of calcium leak channels in myotubes of Duchenne human and mdx mouse origin. Science (New York, NY) 250:673. [DOI] [PubMed] [Google Scholar]
- 15. Franco A, Lansman JB (1990) Calcium entry through stretch-inactivated ion channels in mdx myotubes. Nature 344:670–673. [DOI] [PubMed] [Google Scholar]
- 16. Mongini T, Ghigo D, Doriguzzi C, Bussolino F, Pescarmona G, et al. (1988) Free cytoplasmic Ca++ at rest and after cholinergic stimulus is increased in cultured muscle cells from Duchenne muscular dystrophy patients. Neurology 38:476–476. [DOI] [PubMed] [Google Scholar]
- 17. Bhattacharya SK, Johnson PL, Thakar JH (1993) Reversal of impaired oxidative phosphorylation and calcium overloading in the in vitro cardiac mitochondria of CHF-146 dystrophic hamsters with hereditary muscular dystrophy. Journal of the Neurological Sciences 120:180–186. [DOI] [PubMed] [Google Scholar]
- 18. Sperl W, Skladal D, Gnaiger E, Wyss M, Mayr U, et al. (1997) High resolution respirometry of permeabilized skeletal muscle fibers in the diagnosis of neuromuscular disorders. Molecular and cellular biochemistry 174:71–78. [PubMed] [Google Scholar]
- 19. Godin R, Daussin F, Matecki S, Li T, Petrof BJ, et al. (2012) Peroxisome proliferator-activated receptor γ coactivator 1-α gene transfer restores mitochondrial biomass and improves mitochondrial calcium handling in post-necrotic mdx mouse skeletal muscle. The Journal of physiology 590:5487–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Percival JM, Siegel MP, Knowels G, Marcinek DJ (2013) Defects in mitochondrial localization and ATP synthesis in the mdx mouse model of Duchenne muscular dystrophy are not alleviated by PDE5 inhibition. Human Molecular Genetics 22:153–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schuh RA, Jackson KC, Khairallah RJ, Ward CW, Spangenburg EE (2012) Measuring mitochondrial respiration in intact single muscle fibers. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology 302:R712–R719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chi MMY, Hintz CS, McKee D, Felder S, Grant N, et al. (1987) Effect of Duchenne muscular dystrophy on enzymes of energy metabolism in individual muscle fibers. Metabolism 36:761–767. [DOI] [PubMed] [Google Scholar]
- 23. Stedman H, Sweeney H, Shrager J, Maguire H, Panettieri R, et al. (1991) The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 352:536–539. [DOI] [PubMed] [Google Scholar]
- 24. Wibom R, Lundin A, Hultman E (1990) A sensitive method for measuring ATP-formation in rat muscle mitochondria. Scandinavian journal of clinical and laboratory investigation 50:143–152. [DOI] [PubMed] [Google Scholar]
- 25. Wibom R, Söderlund K, Lundin A, Hultman E (2005) A luminometric method for the determination of ATP and phosphocreatine in single human skeletal muscle fibres. Journal of Bioluminescence and Chemiluminescence 6:123–129. [DOI] [PubMed] [Google Scholar]
- 26. Williams AD, Selig SE, Hare DL, Hayes A, Krum H, et al. (2004) Reduced exercise tolerance in CHF may be related to factors other than impaired skeletal muscle oxidative capacity. Journal of cardiac failure 10:141–148. [DOI] [PubMed] [Google Scholar]
- 27. Williams AD, Carey MF, Selig S, Hayes A, Krum H, et al. (2007) Circuit resistance training in chronic heart failure improves skeletal muscle mitochondrial ATP production rate–a randomized controlled trial. Journal of cardiac failure 13:79–85. [DOI] [PubMed] [Google Scholar]
- 28. Srere P (1969) [1] Citrate synthase:[EC 4.1. 3.7. Citrate oxaloacetate-lyase (CoA-acetylating)]. Methods in enzymology 13:3–11. [Google Scholar]
- 29. Jürgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, et al. (1998) Bax directly induces release of cytochrome c from isolated mitochondria. Proceedings of the National Academy of Sciences 95:4997–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dlasková A, Hlavatá L, Ježek J, Ježek P (2008) Mitochondrial Complex I superoxide production is attenuated by uncoupling. The international journal of biochemistry & cell biology 40:2098–2109. [DOI] [PubMed] [Google Scholar]
- 31. Barbiroli B, Funicello R, Ferlini A, Montagna P, Zaniol P (1992) Muscle energy metabolism in female DMD/BMD carriers: A 31P-MR spectroscopy study. Muscle & Nerve 15:344–348. [DOI] [PubMed] [Google Scholar]
- 32. Barbiroli B, Funicello R, Iotti S, Montagna P, Ferlini A, et al. (1992) 31P-NMR spectroscopy of skeletal muscle in Becker dystrophy and DMD/BMD carriers: Altered rate of phosphate transport. Journal of the Neurological Sciences 109:188–195. [DOI] [PubMed] [Google Scholar]
- 33.Godin R, Daussin F, Matecki S, Li T, Petrof BJ, et al. (2012) PGC1α gene transfer restores mitochondrial biomass and improves mitochondrial calcium handling in post-necrotic mdx MOUSE skeletal muscle. The Journal of Physiology. [DOI] [PMC free article] [PubMed]
- 34. Adam-Vizi V, Chinopoulos C (2006) Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends in Pharmacological Sciences 27:639–645. [DOI] [PubMed] [Google Scholar]
- 35. Tuon L, Comim CM, Fraga DB, Scaini G, Rezin GT, et al. (2010) Mitochondrial respiratory chain and creatine kinase activities in mdx mouse brain. Muscle & Nerve 41:257–260. [DOI] [PubMed] [Google Scholar]
- 36. Tseng BS, Zhao P, Pattison JS, Gordon SE, Granchelli JA, et al. (2002) Regenerated mdx mouse skeletal muscle shows differential mRNA expression. Journal of Applied Physiology 93:537–545. [DOI] [PubMed] [Google Scholar]
- 37. Brookes PS, Yoon Y, Robotham JL, Anders M, Sheu SS (2004) Calcium, ATP, and ROS: a mitochondrial love-hate triangle. American Journal of Physiology-Cell Physiology 287:C817–C833. [DOI] [PubMed] [Google Scholar]
- 38. Gellerich FN, Gizatullina Z, Trumbeckaite S, Nguyen HP, Pallas T, et al. (2010) The regulation of OXPHOS by extra mitochondrial calcium. Biochimica et Biophysica Acta (BBA)-Bioenergetics 1797:1018–1027. [DOI] [PubMed] [Google Scholar]
- 39. Robert V, Massimino ML, Tosello V, Marsault R, Cantini M, et al. (2001) Alteration in Calcium Handling at the Subcellular Level inmdx Myotubes. Journal of Biological Chemistry 276:4647–4651. [DOI] [PubMed] [Google Scholar]
- 40. Lucas-Héron B, Schmitt N, Ollivier B (1990) Age-related calmitine distribution in mitochondria of normal and mdx mouse skeletal muscle. Journal of the Neurological Sciences 99:349–353. [DOI] [PubMed] [Google Scholar]
- 41. Lucas-Heron B, Schmitt N, Ollivier B (1990) Calmitine: a calcium-binding mitochondrial protein specific for fast-twitch muscle fibers. Neuroscience Letters 115:103–107. [DOI] [PubMed] [Google Scholar]
- 42. Scholte H, Busch H (1980) Early changes of muscle mitochondria in duchenne dystrophy: Partition and activity of mitochondrial enzymes in fractionated muscle of unaffected boys and adults and patients. Journal of the Neurological Sciences 45:217–234. [DOI] [PubMed] [Google Scholar]
- 43. Millay DP, Sargent MA, Osinska H, Baines CP, Barton ER, et al. (2008) Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nature medicine 14:442–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reutenauer J, Dorchies O, Patthey-Vuadens O, Vuagniaux G, Ruegg U (2008) Investigation of Debio 025, a cyclophilin inhibitor, in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. British journal of pharmacology 155:574–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Background ATP production (mmol.min−1.intact mitochondrial yield−1) of control (c57BL/10) and dystrophic mdx TA and diaphragm. *p<0.05 mdx different from control strain. There was no effect of muscle type (p = 0.323) or extramitochondrial [Ca2+] (p = 0.852).
(DOCX)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its supporting information files.