

Summary
The bHLH transcription factor Hand2 plays critical roles during cardiac morphogenesis via expression and function within myocardial, neural crest, and epicardial cell populations. Here we show that Hand2 plays two essential Notch-dependent roles within the endocardium. Endocardial ablation of Hand2 results in failure to develop a patent tricuspid valve, intraventricular septum defects, and hypotrabeculated ventricles, which collectively resemble the human congenital defect tricuspid atresia. We show endocardial Hand2 to be an integral downstream component of a Notch endocardium-to-myocardium signaling pathway, and a direct transcriptional regulator of Neuregulin1. Additionally, Hand2 participates in endocardiumto-endocardium based cell-signaling, with Hand2 mutant hearts displaying an increased density of coronary lumens. Molecular analyses further reveal dysregulation of several crucial components of Vegf signaling, including VegfA, VegfR2, Nrp1, and VegfR3. Thus, Hand2 functions as a crucial downstream transcriptional effector of endocardial Notch signaling during both cardiogenesis and coronary vasculogenesis.
Keywords: Hand2, Nrg1, EfnB2, Tricuspid atresia, endocardium, Notch signaling
Introduction
In the primitive heart, communication between the endocardium, which is the endothelium-like tissue that lines the heart, and the myocardium, which is the muscular heart tissue, is essential for processes central to normal cardiac morphogenesis, including trabeculation, chamber septation, and coronary vasculogenesis (Bruneau, 2003; Brutsaert, 2003). The transmembrane receptor Notch1 is integral to this communication, as endocardial Notch activates endocardial EphrinB2, which through-unknown mechanisms, activates expression of the secreted factor Neurgulin1 (Nrg1). Nrg1 initiates the trabeculation process, and Notch signaling subsequently activates expression of Bmp10 (Chen et al., 2004) through a separate pathway (Grego-Bessa et al., 2007). Bmp10 expression stimulates proper proliferation of trabecular, and possibly septal, myocardium. Disruption of Notch-mediated endocardialmyocardial communication results in congenital heart defects (CHD), which are the most frequent human developmental anomalies (Hoffman, 1995).
Tricuspid atresia (TA) is a CHD characterized by the lack of a direct connection between the right atria (RA) and the right ventricle (RV), requiring both ventricular (VSD) and atrial (ASD) septal defects for embryonic survival (Anderson et al.). Conditional ablation of the bHLH factor Hand2 within the second heart field (SHF) via Mef2c-Cre results in TA (Tsuchihashi et al., 2011). Mef2c-Cre marks a pool of SHF progenitor cells that contribute to both the myocardium and endocardium (Tsuchihashi et al., 2011; Verzi et al., 2005), and SHF ablation of Hand2 causes TA via unknown mechanisms. As cardiomyocyte-specific ablation of Hand2 does not result in TA (Tsuchihashi et al., 2011), we hypothesized that loss of Hand2 function within the endocardium is causative of TA. Indeed, our data reveals that either endothelial or endocardial-specific deletion of Hand2 (H2CKO) using either Tie2-Cre or Nfatc1Cre, respectively, results in TA. These data show that Hand2 functions downstream of endocardial Notch to mediate endocardium-to-myocardium signaling via direct transcriptional regulation of the growth factor Neuregulin1 (Nrg1).
As cardiogenesis proceeds, development of the coronary vasculature allows for oxygenation of the thickening ventricular compact zone. Cardiac endothelial cells form the primitive coronary network by angiogenesis (Red-Horse et al., 2010; Wu et al., 2012), a process that extra-cardiac models demonstrate to be Notch-dependent. Early embryonic lethality in Notch pathway mutants has precluded robust analysis of Notch signaling in coronary development. Significantly, H2CKO mice survive long enough to assess the initiation of coronary vascularization, and our findings indicate that Hand2 modulates coronary development through regulation of multiple Vegf signaling components within the developing heart. Collectively, these data support a model whereby Notch signaling, via endocardial Hand2 function, regulates trabeculation, interventricular septum (IVS) formation, and coronary development.
Results
Endothelial/endocardial loss of Hand2 causes trabeculation and septation defects that resemble tricuspid atresia
To test the hypothesis that loss of endocardial Hand2 results in TA, we intercrossed the endothelial cell-specific Tie2-Cre allele (Kisanuki et al., 2001) with mice carrying the Hand2 conditional allele (Hand2fx). Tie2-Cre is expressed in all endothelial cells, including cells of the ventricular endocardium, coronary endothelium, and atrioventricular cushions (Kisanuki et al., 2001). Tie2-Cre expression initiates at E8.5 (Kisanuki et al., 2001), which is concurrent with the onset of endocardial Hand2 expression (Barnes et al., 2011). Hand2fx/fx;R26RlacZ females were mated with Tie2-Cre(+);Hand2+/− males, and neonates were genotyped. No H2CKO mice were observed (n=58, Table S1). We next set up timed matings and observed that Tie2-Cre(+);Hand2fx/− embryos die by E14.5 (Table S1). Hand2 deletion within the endocardium is evident by in situ hybridization (ISH) at E10.5 (Fig. 1A, B arrows). Examination of Tie2-Cre H2CKOs in whole mount (Fig. 1D, E) and in section histology (Fig. 1G, H) reveals hypotrabeculation (arrow in E) and a highly penetrant TA phenotype (Fig. 1H; Table S2) comparable to that reported in the SHF Mef2c-CreH2CKOs. At E12.5, we observe hypotrabeculation, VSDs, rightward septal displacement, and RV hypoplasticity in Tie2-Cre H2CKO hearts. Occasionally (~20%), we observe a patent tricuspid valve that makes a direct connection with the LV, resulting in a double inlet left ventricle (DILV), a CHD that functionally resembles TA (Fig. 1J arrow).
Figure 1. Endocardial deletion of Hand2 results in a VSD, hypotrabeculation, hypoplastic RV, IVS defects and TA.

Hand2 ISH of RV section from E10.5 control, Tie2-Cre H2CKO, and Nfatc1CreH2CKO, respectively (A–C). Wholemount view of E12.5 control heart, Tie2-Cre H2CKO, and Nfatc1Cre H2CKO, respectively (D–F). R26RlacZ stained sections from E12.5 Tie2-Cre(+) control embryo (G), Tie2-Cre H2CKO with TA (H). R26RlacZ stained E12.5 Tie2-Cre(+) control embryo (I), and Tie2-Cre H2CKO with DILV (J). R26RlacZ stained sections from E12.5 Nfatc1Cre control embryo (M), and Nfatc1Cre H2CKO (N; arrows denote multiple IVSs). Alcian blue staining of E12.5 Tie2-Cre(+) control AV cushions (K), and Tie2-Cre H2CKO (L). Asterisk denotes abnormalities in AV cushion shape and position. SAVC, superior atrioventricular cushion; IAVC, inferior atrioventricular cushion; tv, tricuspid valve; mv, mitral valve. Scale bars in A – C represent 100μm; scale bars in G – N represent 250μm.
To examine atrioventricular (AV) cushion formation, cell density, and extracellular matrix (ECM) deposition, we stained sections with alcian blue. Although comparison of H2CKO cushions with those of controls revealed no difference in size, (Fig 1K, L), there appears to be a cushion-positioning defect (asterisk in Fig. 1L). Despite normal AV cushion ECM deposition, no direct connection between the RA and RV is detected in any plane of section, resulting in a single left sided AV canal in most H2CKOs, thus meeting the clinical definition of TA (Fig. 1H). However, OFT morphogenesis occurs normally in H2CKOs (Fig. S1A–C).
While we also observe no defects in the yolk sac vasculature of Tie2-CreH2CKOs (Fig. S1D, E), Hand2 may play critical roles in vascular endothelium that could contribute to the observed embryonic lethality. Therefore, we used the endocardial-specific Nfatc1Cre allele (Wu et al., 2012) to generate H2CKOs (Fig. 1C, F, N). This Nfatc1Cre allele initiates expression throughout the endocardium at E9.0 (Wu et al., 2012), as opposed to the Nfatc1enCre allele, which labels only a subset of valve endothelial cells. Nfatc1Cre H2CKOs also show dramatic defects in trabeculation, malformed IVS, and atresia of the tricuspid valve (~70%; Fig. 1F, N). Interestingly, in some cases, IVS malformations include large protrusions of myocardium that appear to indicate the formation of multiple IVSs (Fig. 1N arrows; Table S2). Robust expression of the septal/compact zone marker Hey2 and the septal marker Irx2 in these large protrusions confirm this observation, while expression of the trabecular marker Anf is excluded (Fig. S2A–H). However, expression of the LV marker Hand1 is not observed beyond the left-most septum (Fig. S2I, J). Tbx5, which has been suggested to induce the IVS (Takeuchi et al., 2003), is also unchanged (Fig. S2K, L). Together, these data confirm that conditional deletion of Hand2 within the endocardium is critical for normal trabeculation and septation. Given that these phenotypes are myocardial in nature, this suggests that endocardial Hand2 plays a crucial role in endocardium-to-myocardium signaling.
Hand2 regulates Nrg1 expression within the endocardium
We next investigated changes in gene expression within E10.5 Tie2-CreH2CKOs using ISH. Expression analysis of Fog2 was of interest, as Fog2 deficient mice also display TA (Svensson et al., 2000); however, no change in Fog2 expression was observed (Fig. S3A, B). Deletion of Tgfbr2 from the endothelium results in DILV (Jiao et al., 2006). Analysis of Tgfbr2 expression revealed no differences between H2CKO and controls (Fig. S3C, D).
It has been directly demonstrated that endocardial Notch signaling plays an essential role in orchestrating morphogenic changes within the underlying myocardium, particularly during the process of trabeculation (Grego-Bessa et al., 2007). Therefore, we examined Notch pathway gene expression. We first analyzed expression of the Notch-regulated bHLH transcriptional repressors, and potential Hand2 dimer partners, Hey1 and Hey2 (Fig. S3E–H). Endocardial expression is not significantly changed. Next, we examined expression of the direct Notch1 target EphrinB2 (EfnB2; Grego-Bessa et al., 2007). EfnB2 ISH also shows no change in expression between H2CKOs and control embryos (Fig. 2A–D). However, EGF family member Neuregulin1 (Nrg1) is markedly down-regulated at E10.5 within the endocardium of H2CKOs (Fig. 2F, arrow H) compared to control embryos (Fig. 2E, arrow G). Nrg1 is known to be an essential mediator of trabeculation in the developing ventricles and is down-regulated in Tie2-Cre Notch1CKOs, and EfnB2 systemic null embryos (Grego-Bessa et al., 2007). The maintenance of EfnB2 and loss of Nrg1 in H2CKOs suggests that Hand2 acts downstream of EfnB2, but upstream of Nrg1, representing a novel step in Notch1 trabeculation and septation signaling.
Figure 2. H2CKOs exhibit down-regulation of Nrg1, and loss of Bmp10 expressing trabecular myocardium.

Expression of the Notch1 target EfnB2 is comparable between H2CKOs at E10.5 with controls (A–D). Nrg1 expression at E10.5 is markedly decreased in H2CKOs when compared to controls (E–H arrows in G and H denote endocardium). H2CKOs have less Bmp10 expressing trabecular myocardium in the RV at E12.5 (I, J arrows denote trabecular tissue). qPCR on isolated ventricles confirms the significant reduction (* p≤0.05) of Nrg1 in E10.5 H2CKOs. Scale bars in A, B, E, F, I, and J represent 200μm; scale bars in C, D, G, and H represent 100μm.
We next analyzed Bmp10 expression, as myocardially-expressed Bmp10 is crucial for proper trabeculation and is independently downstream of Notch1 signaling (Chen et al., 2004; Grego-Bessa et al., 2007). BMP10 ISH at E10.5 suggests a subtle down-regulation in H2CKOs (Fig. S3I, J) while E12.5 ISH shows a near complete loss of Bmp10 expressing trabecular tissue within the RV but not LV (Fig. 2I, J arrows). To quantitatively assess Bmp10 expression, we dissected ventricles from E10.5 hearts, and isolated RNA for qRT-PCR (qPCR). As expected, qPCR analysis confirms that both Hand2 and Nrg1 are significantly down-regulated in H2CKOs (Fig. 2K) but Bmp10 myocardial expression is not altered (Fig. 2K). Thus, the reduction of Bmp10 within the RV of E12.5 H2CKOs reflects the absence of RV trabeculation, as LV trabeculae express Bmp10 (Fig. 2J).
Endocardial Hand2 functions downstream of Notch1 and the direct Notch1 target EfnB2
Upon interaction with one of its transmembrane ligands, the transmembrane receptor Notch1 is proteolytically cleaved to generate the Notch1 intracellular domain (N1ICD). N1ICD translocates to the nucleus where it dimerizes with its partner RBPJk to activate transcription of target genes. Previous studies have shown that deletion of Notch1 or RBPJk results in hypotrabeculation due to loss of EfnB2 and Nrg1 (Grego-Bessa et al., 2007). To confirm that Hand2 lies within the Notch1 signaling pathway, we assayed Hand2 expression in E9.5 RBPJk−/− embryos. Whole mount analyses reveal a loss of endocardial Hand2, while expression within the OFT and pharyngeal mesenchyme is unaffected (Fig. 3A, B). Sectioned embryos confirm a specific loss of Hand2 within mutant endocardium (Fig. 3C, D), definitively establishing Hand2 as a Notch1 signaling effector.
Figure 3. Hand2 functions downstream of Notch1 and the direct Notch1 target EphrinB2 during trabeculation.
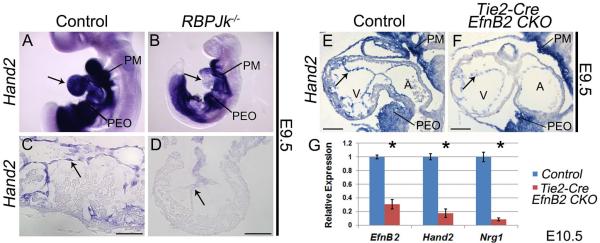
Wholemount Hand2 ISH in wild type (A) and RBPJk knockout embryos (B). Arrows indicate endocardial Hand2 expression in control (A) and loss of expression in RBPJk knockouts (B). Hand2 ISH (C) shows robust endocardial Hand2 expression that is downregulated within RBPJk−/− endocardium (D). Hand2 ISH (E) revealed a similar downregulation of Hand2 within the endocardium of Tie2-Cre EfnB2CKO hearts (F Arrows denote endocardium). PM, pharyngeal mesenchyme; PEO, proepicardial organ. qPCR on RNA isolated from ventricles confirms significant (* p≤0.05) downregulation of Hand2 and Nrg1 in Tie2-Cre EfnB2CKO hearts (G). Scale bars in C and D represent 50μm; scale bars in E and F represent 100μm.
During trabeculation N1ICD/RBP Jk directly trans-activates EfnB2, which acts through its EphB2/EphB4 tyrosine kinase receptors to upregulate Nrg1 (Grego-Bessa et al., 2007). While our data clearly show Hand2 to be upstream of Nrg1, it was not clear if Hand2 lies downstream of EphrinB2 signaling, or if Hand2 represents a parallel EfnB2-independent Notch signaling pathway. To address this question, we assayed Hand2 expression in E9.5 Tie2-Cre(+);EfnB2fx/fx embryos (Gerety and Anderson, 2002). ISH reveals that Hand2 is down-regulated in endocardial cells of Tie2-Cre(+);EfnB2fx/fx embryos (Fig. 3F arrow), whereas Hand2 expression in pharyngeal mesenchyme and the proepicardial organ is unaffected when compared to control hearts (Fig. 3E). qPCR analysis at E10.5 confirms Hand2 down-regulation within Tie2-Cre(+);EfnB2fx/fx isolated ventricles (Fig. 3G). Together, these data show that Hand2 is a Notch-dependent endocardial factor positioned between EfnB2 and Nrg1.
Hand2 regulation of Nrg1 is direct via interaction with the Nrg1 promoter and upstream enhancer sequences
As Hand2 encodes a transcription factor with a similar expression profile to Nrg1, we sought to determine if Hand2 regulation of Nrg1 is direct. An 850bp region of the Nrg1 promoter has been identified as necessary for high Nrg1 transcriptional activity in vitro (Frensing et al., 2008). As Hand2 binds the consensus sequence CANNTG, termed an E-box, or alternatively CGNNTG, a D-box (Firulli et al., 2007), we searched this aligned promoter region for these conserved cis-elements. Three were found within the 500bp directly upstream of the Nrg1 translation start site (Fig. 4A). To assess Hand2 interaction with this region of the Nrg1 promoter, we conducted ChIP assays in HeLa cells, employing a Myc-tagged Hand2 co-transfected with a plasmid encoding an untagged Hand2 dimerization partner E12. Negative controls included Myc-Hand2 + E12 immuno-precipitated without αMyc, and pCS2+Myc samples immuno-precipitated with αMyc. Enrichment of the Nrg1 promoter region was assessed by PCR using primers corresponding to a 103bp region of the human Nrg1 promoter. This amplicon shows robust enrichment within Myc-Hand2 + E12 immuno-precipitated samples (Fig. 4B). To confirm that Hand2-E12 heterodimers are capable of binding the conserved consensus E/D-box sequences, double-stranded oligos corresponding to these sites were used in electrophoretic mobility shift assays (EMSA) with in vitro translated Hand2 and E12 (Fig. 4C). Hand2-E12 heterodimers specifically bind oligos corresponding to sites 1 and 3 (Fig. 4D, E). No DNA binding is observed between Hand2-E12 complexes and site 2. To test if Hand2 is capable of trans-activating the Nrg1 promoter, Hand2 and E12 expression constructs were transfected into HeLa cells along with a Nrg1 luciferase reporter containing the 1000bp directly upstream of the murine Nrg1 translation start site. This region contains multiple transcription start sites located between −395 and −425 (Frensing et al., 2008). Co-transfection of Hand2 and E12 resulted in 7-fold reporter activation. Subsequent assays utilizing truncated promoters (−500/0bp and −500/−250bp) demonstrated more robust activation (~18 and 16 fold respectively; Fig. 4F). Transactivation data from the 500bp promoter revealed that E12 alone is not sufficient for activation, while Hand2 alone results in only modest (but significant) 3.5 fold transactivation. Consistent with EMSA results, co-transfection of Hand2 and E12 results in 18-fold transactivation, while a DNA binding deficient Hand2 construct (Hand2Δbasic) is incapable of Nrg1trans-activation alone or when co-transfected with E12 (Fig. 4G).
Figure 4. Hand2 directly regulates Nrg1.

Sequence alignments reveal three Hand2 consensus sites that are highly conserved among mammals (A). ChIP of the NRG1 promoter (B). In-vitro transcribed and translated Hand2 and E12 used in EMSAs (C). EMSAs for D-box 1 (D) and E-box 3 (E) show robust and specific binding of Hand2-E12 heterodimers. No binding is observed for site 2. Luciferase reporter assays with a deletion series of the Nrg1 promoter. Red numbers denote positions of the Hand2 consensus sites; green arrows denote positions of the transcription start sites; black arrows, location of ChIP primers (F). Transactivation of 500bp Nrg1 promoter reporter (G; *p≤0.05).
Replacement of Hand2 within EfnB2 deficient endocardium results in an improvement of cardiac trabeculation
EfnB2 mutant embryos display hypotrabeculation accompanied by loss of Hand2 and Nrg1 expression (Fig. 4). Therefore, if Hand2 is both necessary and sufficient for regulation of Nrg1, we reasoned that Hand2 replacement in Nfatc1Cre EfnB2CKOs would restore Nrg1 expression and improve ventricular trabeculation. To test this, we generated the Creactivatable Hand2 transgene CAG-CAT-Hand2 (CC-H2; Fig. S4A). Ectopic expression of Hand2 in limb mesenchyme results in preaxial polydactyly (McFadden et al., 2002). As predicted, Prx1-Cre mediated activation of CC-H2 within the limb results in polydactyly, indicating that the conditional transgene can be efficiently and specifically activated (Fig. S4B). Similarly, Nfatc1Cre efficiently activates CC-H2 in the endocardium (Fig. S4C). E13.5 Nfatc1Cre;CC-H2(+) embryos do not display any obvious cardiac phenotypes (data not shown). Subsequently, CC-H2(+); EfnB2fx/+ females were then crossed with Nfatc1Cre; EfnB2fx/+ males to generate Nfatc1Cre; CC-H2(+); EfnB2 CKOs. CC-H2(−); EfnB2 CKOs die by E11.5, with severe pericardial edema, hemorrhaging, and defects in cardiac looping and chamber development (Fig. 5C). These phenotypes closely resemble the defects observed in Tie2-Cre EfnB2 CKOs (Gerety and Anderson, 2002), indicating that while EfnB2 function within extracardiac vasculature is likely important, loss of endocardial EfnB2 is sufficient to cause mid-gestation lethality. Hand2 ISH at E10.5 confirms robust endocardial Hand2 expression in controls (Fig. 5E, F), and loss of Hand2 in EfnB2 CKOs (Fig. 5G arrow). The presence of the CC-H2 allele restores Hand2 expression within the EfnB2 CKOs (Fig. 5H arrow; n = 3). Nrg1 ISH reveals robust expression in controls (Fig. 5I, J), and loss of Nrg1 expression in EfnB2 CKOs (Fig. 5K arrow). CC-H2(+); EfnB2 CKOs display a significant increase in Nrg1 expression (Fig. 5L, arrow), while assessment of trabeculation by Bmp10 ISH reveals a marked improvement in Bmp10 expressing trabecular myocardium (Fig. 5P arrow) when compared to EfnB2 CKOs lacking the CC-H2 allele (Fig. 5O arrow). Expression analysis of isolated E10.5 ventricles by qPCR demonstrates that while still well below control levels, Nrg1 expression in EfnB2 mutants that carry the CC-H2 allele (n = 4) is twice as high as in EfnB2 mutants lacking the CC-H2 allele (upregulated 112%). Bmp10 expression is increased by 79% with obvious improvement in trabeculation (Fig. 5Q).
Figure 5. Conditional CAG-CAT-Hand2 transgene expression in Nfatc1Cre EfnB2 CKOs partially improves trabeculation and Nrg1 expression.

Wholemount images of E10.5 control, CC-H2(+) control, EfnB2 CKO, and CC-H2(+); EfnB2 CKO embryos (A–D). Hand2 section ISH (E–H). Arrow in G indicates loss of Hand2; arrow in H indicates restoration of endocardial Hand2 via CC-H2. Nrg1 section ISH (I–L). Arrow in K indicates loss of Nrg1 in EfnB2 CKOs; arrow in L indicates restored Nrg1 expression in EfnB2 CKOs that are also CC-H2(+).Bmp10 section ISH (M–P). Arrow in O indicates Bmp10 expressing myocardium and lack of trabeculation; arrow in P indicates restoration of Bmp10 expressing trabeculations. qPCR analysis of isolated E10.5 ventricles (Q). Asterisks denote significant difference (p≤0.05). CC-H2, CAG-CAT-Hand2. Scale bars in E – P represent 100μm.
Notch-dependent Hand2 function also regulates coronary angiogenesis
The above analyses demonstrate that endocardial Hand2 plays a crucial role in myocardial morphogenesis. Myocardial trabeculation and compaction are intimately linked to endothelial cell behavior during development of the coronary vasculature by a complex signaling network that includes Vegf, Fgf, Tgf-β and Notch components (Smart et al., 2009). Furthermore, early embryonic lethality in genetic models of dysfunctional Notch signaling has precluded an in vivo assessment of the role of Notch signaling in formation of the coronary arteries. As we have established Hand2 as an integral component of endocardial Notch signaling, and as coronary endothelium is derived at least in part from the endocardium (Wu et al., 2012), the survival of H2CKOs to E14.5 allows us a unique opportunity to assess Notch function in early coronary development. Analysis of hearts at E13.5, one day after initiation of intra-myocardial coronary formation (Tian et al., 2013), shows that Nfatc1Cre H2CKO hearts (Fig. 6B, D arrows) exhibit an increased density of primitive coronary vessels when compared to control ventricles (Fig. 6A, C). Subsequent analyses revealed a comparable degree of hyper-vascularization in Tie2-Cre H2CKOs (data not shown, Table S2). To ascertain the mechanism underlying this hyper-vascularization, we analyzed genes associated with vascular development in E10.5 and E13.5 Nfatc1Cre H2CKO isolated ventricles by qPCR (n = 4, Fig. 6E and F respectively). As expected, expression levels of Hand2 and Nrg1 are down at both time points. Analysis of vascular markers at E10.5 reveals dysregulation of select components of Vegf signaling (Fig. 6E). VegfR2 is downregulated by 30%, and its co-receptor Nrp1 is downregulated by 14%. VegfD, which encodes a VegfR3 specific ligand, is downregulated by 25%. By E13.5, expression levels of VegfR2 and VegfD have recovered (Fig. 6F), whereas Nrp1 remains downregulated. Surprisingly, expression of VegfA, which encodes the primary Vegf ligand that regulates vascular development (Ferrara et al., 1996), is upregulated by over 200%. Expression of VegfR3 is upregulated by 120%, and expression of Dll4, which encodes a notch ligand regulated by VegfRs (Wythe et al., 2013), is upregulated by approximately 85%. Differential regulation of several additional key vascular factors was also observed at E13.5. The venous markers CouptfII and EphB4 are downregulated by 21% and 24% respectively, while the arterial marker EfnB2 is downregulated by 33%. Surprisingly, Sox18 and Lyve1, which encode factors typically associated with endothelium of the lymphatic system, were upregulated by 45% and 73% respectively. To determine what cardiac cell populations were upregulating Lyve-1, we conducted Lyve-1 immunostaining at E10.5 (Fig. 6G, H) and E13.5 (Fig. 6I–N) in control and H2CKO hearts. Sections from E13.5 hearts were co-stained with VegfR2 to mark cardiac endothelium (see Table S3 for glossary of terminology pertaining to endothelial cell populations). E10.5 immunostaining reveals that Lyve-1 is expressed robustly within endocardium of both control and H2CKO hearts. In contrast, by E13.5, Lyve-1 immunostaining in control hearts is restricted to a population of peripheral cardiac macrophages (Fig. 6I; Pinto et al., 2012), whereas robust Lyve-1 immunostaining persists in the ventricular endocardium of H2CKOs (Fig. 6J, L). However, while Lyve-1 still robustly marks H2CKO ventricular endocardium (Fig. 6N white arrow), Lyve-1 expression does not mark the expanded coronary endothelium of H2CKOs (Fig. 6N yellow arrow).
Figure 6. Hand2 controls coronary development and endocardial maturation via regulation of Vegf signaling.

R26RlacZ stained E13.5 hearts (A, B). LV outer curvature (C, D). Nfatc1Cre H2CKOs display hyper-vascularization (arrows in D). qPCR analysis of E10.5 and E13.5 gene expression (n = 4) in Nfatc1Cre H2CKOs (E, F; Asterisks denote significant difference (p≤0.05). Lyve-1 immunostaining in E10.5 Control and Tie2-CreH2CKO hearts (G, H). Lyve-1 immunostaining in E13.5 Control and Nfatc1Cre H2CKO hearts (I–N). In E13.5 control hearts Lyve-1 expression is restricted to endothelial lymphatic precursors (K), while H2CKOs continue to express Lvye-1 within ventricular endocardium (L white arrow). Persistent Lyve-1 expression marks ventricular endocardium (N white arrow) but not coronary endothelium (N yellow arrow). Dotted lines denote the border between compact and trabecular myocardium. Scale bars in A and B represent 250μm, 100μm for C and D, 200μm for G and H, 250μm for I and J, and 50μm for K–N.
As previously mentioned, qPCR demonstrates differential expression of several Vegf signaling components. We therefore investigated, using ChIP, ChIP-seq, EMSA, and luciferase reporter assays, the possibility that Hand2 directly regulates at least a subset of these components (Fig. 7A). Given that our results establish Hand2 as a downstream effector of Notch signaling, and Notch also regulates expression of VegfRs within endothelial cells (Herbert and Stainier, 2011), we first addressed the possibility that Hand2 directly regulates VegfR3 expression. Using the previously described HeLa ChIP samples, we show that immunoprecipitation of Hand2-Myc/E12 transfected cell lysates results in selective enrichment of a region within the VegfR3 promoter that has been previously reported to have regulatory activity (Fig. 7B; Shawber et al., 2007). Furthermore, by EMSA we show that Hand2 E12 heterodimers specifically bind an oligo corresponding to one of the two E-boxes within this human promoter region (Fig. 7C), which consists of the 500bp upstream of the VegfR3 translation start site. Finally, a luciferase reporter containing the homologous 500bp mouse promoter, was significantly repressed by Hand2 E12 heterodimers (~ 50% repression; p≤0.05; Fig. 7D). Collectively these data indicate that Hand2 may directly repress VegfR3 expression within cardiac endothelium.
Figure 7. Hand2 regulates Vegf signaling.

Differentially expressed genes that were analyzed for direct regulation by Hand2 (A). ChIP of a region within the 500bp VegfR3 promoter (B). EMSA demonstrates Hand2-E12 heterodimers specifically bind an oligo corresponding to an E-box within the ChIP amplicon of the VegfR3 promoter (C). Black arrow indicates E12 homodimer binding, red arrow indicates Hand2-E12 heterodimer binding, and asterisk indicates nonspecific binding. Hand2-E12 heterodimers repress significantly (p≤0.05) a luciferase reporter containing the 500bp VegfR3 promoter (D). Hand2-3xFLAG ChIP-seq demonstrates a prominent region of enrichment approximately 356kb upstream of the Nrp1 coding region (E). A luciferase reporter containing this potential enhancer region is significantly (p≤0.05) trans-activated 5-fold by H2-E12 heterodimers (F). ISH demonstrates strong myocardial expression of VegfA within the compact zone of control E13.5 hearts (G). Red dots denote the border between compact and trabecular myocardium. In H2CKOs, the distinction between VegfA expression levels in compact versus trabecular myocardium is more poorly defined (H). Scale bars in G and H represent 100μm.
To detect additional Hand2 targets we utilized a Hand2 ChIP-seq data set that was generated for an alternate study (Osterwalder et al., Dev Cell, in press). This ChIP-seq data set employed a Hand2-3xFLAG knock-in allele. E10.5 Hand2 expressing tissues, including the heart, were collected and used for an immunoprecipitation of Hand2-bound regions of genomic DNA. This data set was cross referenced with our H2CKO gene expression data, where in addition to increased VegfR3, we observed decreased expression of the VegfR2 co-factor Nrp1. Analysis of Hand2-3xFLAG ChIP-seq data revealed a region of high enrichment 356kb upstream of the Nrp1 coding region (Fig. 7E and Table S4). An approximately 3kb region containing this potential enhancer was cloned upstream of a luciferase reporter driven by the thymidine kinase (TK) minimal promoter. When co-transfected into HeLa cells with Hand2 and E12, a significant 5.2 fold trans-activation was observed (Fig. 7F). Analysis of VegfR3 and Nrp1 expression by ISH reveals expression within cardiac endothelium (Fig. S5A, B) although differential expression between controls and H2CKOs is not detectable by this non-quantitative assay.
Given that VegfA is upregulated by over 200% in E13.5 H2CKOs, we analyzed the Hand2-3xFLAG ChIP-seq data-set for evidence of direct regulation. No prominent peaks were detected within the VegfA locus. Indeed, ISH demonstrates that VegfA expression is not detectable within endocardium. However, VegfA is strongly expressed within the myocardial compact zone, with low levels of expression observed within trabeculae (Fig. 7G). In H2CKOs, the difference in expression levels between compact and trabecular myocardium is visibly less distinct, with high VegfA expressing regions appearing to extend beyond the compact zone (Fig. 7H).
Discussion
Loss of Hand2 within the SHF cardiac progenitors that give rise to both endocardial and myocardial lineages has been associated with TA. In this study, we show that loss of Hand2 specifically in the endocardium, a cell population in which robust Hand2 expression has previously gone uninvestigated, can cell autonomously generate TA. These results underscore the importance of effective endocardial-myocardial signaling during early cardiac morphogenesis. Mechanistically, our data show that endocardial Hand2 sits downstream of Notch1 and EfnB2, and is itself directly upstream of Nrg1, being necessary and sufficient for Nrg1 expression in vivo. Notch signaling is a well-established component of endocardium-to-myocardium communication. Hand2 endocardial roles include regulation of trabeculation, positioning of the IVS, and IVS morphogenesis. Indeed, endocardial H2CKOs present multiple septums that are marked by Irx2 and compact zone/septal marker Hey2, while excluding trabecular marker Anf (Fig. S2). These ectopic right-sided septa do not express Hand1 or Tbx5 at their left side base (Fig. S2I–L), suggesting that these septa form via a non-canonical mechanism from RV cardiomyocytes.
These data demonstrate that endocardial Hand2 regulates the specification of trabecular and septal myocardium from primitive myocardium. Given the striking similarities between the cardiac defects of EfnB2 knockouts (Fig. 3F, G), which do not express Nrg1, and the reported phenotypes of Nrg1 knockouts (Kramer et al., 1996), the most logical conclusion is that loss of Nrg1 expression is the root cause of the hypotrabeculation, septal, and TA phenotypes observed in H2CKOs.
We show that trabeculation and Nrg1 expression within EfnB2 CKO mice is improved by Hand2 replacement via the CC-H2 transgene (Fig. 5). Hand2 is necessary for normal expression of Nrg1 (Fig. 2), and we demonstrate that Hand2 is sufficient for partial restoration of Nrg1 expression levels in EfnB2 CKOs. While trabeculation is markedly improved in CC-H2(+) EfnB2 CKOs, these embryos still display hallmarks of cardiovascular failure such as pericardial edema and hemorrhage. The incomplete restoration of normal cardiac phenotype is not surprising given that restoration of Nrg1 expression is incomplete, and EfnB2 signaling likely has a much broader range of endocardial downstream targets then just Hand2. Our observation that approximately 37% of Nrg1 expression remains in Tie2-Cre H2CKOs (Fig. 2K) while less than 10% remains in Tie2-Cre EfnB2 CKOs (Fig. 3G) indicates that EfnB2 also influences Nrg1 expression via Hand2-independent inputs.
While it is clear that endocardial loss of Hand2 impairs the Notch-dependent processes of trabeculation and septal development, it is less clear how this impairment results in TA. It is possible that multiple morphogenic inputs contribute to the TA observed in H2CKOs. Previous studies have concluded that TA occurs when the atrial connection to the AV canal expands rightward, but the ventricular inner curvature fails to remodel (Kim et al., 2001). Given the dramatic myocardial defects observed in H2CKOs, Hand2-dependent remodeling of AV canal myocardium could be involved; however, histological analysis of H2CKOs suggests a simpler model wherein the septum shifts rightward in H2CKOs and interferes with development of the primitive right AV canal, thus resulting in TA. Indeed, we show that Tie2-Cre H2CKOs have a smaller RV, which is not due to increased cell death within SHF progenitors, increased cell death within the RV, or decreased proliferation within the RV (Fig. S6A–D). Furthermore, analysis of ventricle areas reveals that Tie2-Cre H2CKOs have a smaller RV, larger LV, but no change in total area (Fig. S6E), thus supporting histological observations of a rightward shifted septum. In extreme rightward septal shifts, AV cushion maturation is not hindered but the tricuspid valve forms above the LV, thus resulting in DILV. Finally, Hand2 ablation within the developing OFT and AV valve mesenchyme does not alter septation or valve morphogenesis, and does not result in TA (VanDusen et al., 2014). In total, these data demonstrate a novel cell-autonomous Hand2-dependent role of the endocardium in the etiology of TA/DILV.
In addition to the dramatic defects in myocardial morphogenesis observed in endocardial H2CKOs, histological analyses at E13.5 also reveal significant vascular phenotypes. H2CKOs display precocious and disorganized development of the coronary vascular plexus. Analysis of vascular related gene expression in E10.5 and E13.5 H2CKOs surprisingly reveals that major components of cardiac Vegf signaling are dysregulated. VegfR2 and VegfD are downregulated at E10.5, while VegfR3 and VegfA are significantly upregulated by E13.5. Additionally, VegfR2 co-receptor Nrp1 is downregulated at both E10.5 and E13.5, while expression of the notch ligand Dll4 is increased at E13.5 – an indication of enhanced Vegf signaling (Fig. 6E, F). Furthermore, qPCR and immunostaining reveals elevated expression of the receptor Lyve-1 within endocardium of E13.5 H2CKOs. Analysis at E10.5 demonstrates that Lyve-1 is initially expressed throughout the early endocardium, and is subsequently down-regulated as the endocardium matures, such that by E13.5 expression is no longer detectable within ventricular endocardium, but is observed only within cardiac macrophages. Given the early endocardial expression of Lyve-1, this persistence within H2CKOs most likely represents a defect in endocardial maturation, rather than ectopic activation of the lymphatic gene program.
Of the genes we observed to be differentially expressed in E13.5 H2CKOs, both the Dll4 and Nrp1 loci contain prominent Hand2 Chip-Seq peaks (Table S4). Nrp1 encodes an isoform-specific VegfA receptor that acts in concert with VegfR2. Interestingly, an RNA subtractive hybridization screen previously identified Nrp1 as being downregulated in Hand2 systemic null embryos (Yamagishi et al., 2000). Our data indicate that Nrp1 is an endocardial target of Hand2, although regulation may also occur in additional tissues where these factors are co-expressed (Fig. S5A). Co-transfection of Hand2 and E12 with a luciferase reporter containing the potential upstream enhancer region yielded over 5 fold trans-activation, further supporting direct regulation of Nrp1 by Hand2. In contrast, luciferase reporter assays failed to demonstrate Hand2 mediated repression via the enriched Dll4 upstream region, indicating that the increased Dll4 expression in H2CKOs may be secondary to changes in expression of Vegf receptors, which are known to regulate Dll4 (Wythe et al., 2013). However, our data do not rule out Hand2 direct regulation of Dll4 by alternate undetected enhancers. Expression of VegfR3 is upregulated in E13.5 H2CKOs by over 100%. VegfR3 is capable of functioning as both a homo-dimer and VegfR3–VegfR2 hetero-dimer, and distinct functions have been associated with different interactions (Dixelius et al., 2003). VegfR3 plays crucial roles in angiogenic sprouting and development of the lymphatic system (Benedito et al., 2012), and we show that VegfR3 is expressed within at least a subset of the endocardium (Fig. S5B arrow). While no high-ranking ChIP-Seq peaks were observed within the VegfR3 locus, cardiac endothelium represents only a small portion of the total Hand2 expressing tissue that was utilized, and so sensitivity may be a limiting factor of this assay. Our HeLa ChIP results indicate that Hand2 interacts with the VegfR3 promoter, while transactivation assays correlate with H2CKO expression data, indicating that Hand2 may repress VegfR3 transcription. In addition to VegfR3, we observe a 45% increase in Sox18 expression at E13.5. As Sox18 specifically marks endothelium of the coronary vasculature at this time-point (Fig. S5C), this increase most likely reflects the hyper-vascularization phenotype.
Similar to several extracardiac angiogenic models of Notch signaling obstruction (Benedito et al., 2012; Tammela et al., 2008), we show that endocardial Hand2 ablation results in a hyper-vascularization phenotype featuring the formation of an excessive number of new vessels. Furthermore, we show that this phenotype is accompanied by broad dysregulation of Vegf signaling. Homeostasis of Vegf signaling is crucial during embryonic development, as mice heterozygous for a VegfA null mutation die at E9.5, while an increase in VegfA expression (~3 fold) results in lethality at approximately E13.5 (Miquerol et al., 2000). In the present study we demonstrate that expression of VegfA is expanded in H2CKOs. This could reflect aberrant specification of trabecular myocardium, or secondary pathological effects of compromised cardiac function. VegfA is the most differentially regulated gene that we observe, while VegfR2, VegfR3, and Nrp1 are also dysregulated within H2CKOs. Given the complex interactions that take place between these molecules, disrupted receptor stoichiometry and upregulated VegfA, a growth factor that is well known for its pro-angiogenic qualities, most likely accounts for the observed coronary phenotype. These data not only provide insight into a second novel function of Hand2 within the endocardium, but also reveal a wider role of Notch signaling during coronary vessel development. Coronary heart disease is a major cause of mortality in developed nations, being responsible for approximately one of every six deaths in the United States (Go et al., 2013). Consequently, further assessment of Hand2's regulatory role in Vegf signaling during coronary vascularization, as well as potential adult homeostatic roles of Hand2-dependent Notch signaling, are interesting avenues of future investigations.
Experimental Procedures
Mice and Genotyping
Tie2-Cre(+) and Nfatc1-Cre(+) mice were crossed with Hand2+/− mice to generate Tie2-Cre(+);Hand2+/− and Nfatc1-Cre(+);Hand2+/− males. These males were then crossed with Hand2fx/fx;ROSA26R reporter mice (lacZ or eYFP) to generate conditional null Hand2 embryos. Tie2-Cre(+) females were crossed with EfnB2fx/fx males to generate Tie2-Cre(+);EfnB2fx/+ males. These males were then crossed with EfnB2fx/fx females to generate conditional null EfnB2 embryos. The cre-activatable transgene CAG-CAT-Hand2 was constructed by replacing the Myc-Twist1 cDNA of CAG-CAT-Twist (Connerney et al., 2006) with the murine Myc-Hand2 cDNA. This construct was used for microinjection to establish a transgenic line. For genotyping information see Supplemental Procedures. Mouse maintenance and experimentation was performed according to protocols approved by the IUSOM IACUC.
Hand2-3xFLAG ChIP-Seq
The Hand2 ChIP-Seq dataset was generated as part of a previous study using homozygous Hand2-3xFLAG embryos, which express a 3xFLAG epitope-tagged Hand2 protein (Osterwalder et al., Dev Cell, submitted). Briefly, Hand2 expressing tissues (heart, limbs and pharyngeal arches) from 150 Hand2-3xFLAG E10.5 embryos were dissected and pooled. Samples were cross-linked in 1% formaldehyde for 5 minutes and ChIP was performed using FLAG antibodies (Sigma F1804). ChIP-Seq reads were mapped to the mouse genome (NCBI37/mm9) and peaks were detected using MACS (version 1.3.7.1) for the HAND2-3xFLAG and input control samples. Statistically validated peaks were sorted according to fold-enrichment and number of reads.
Section RNA In Situ Hybridization
Antisense digoxygenin labeled riboprobes were transcribed with T7, SP6, or T3 (Roche). Section In Situ hybridization was performed as previously described (Vincentz et al., 2008). The cDNA for a Fog2 ribo-probe was kindly supplied by Eric Svensson, Tbx20 by Simon Conway, Anf and Irx2 by Vincent Christoffels, TgfβR2 from Henry Sucov, and EfnB2 from Hai Wang. All data reflect assessment in n≥3 embryos for ISH, and immunostaining, and n≥4 for qRT-PCR analyses.
Quantitative RT-PCR
Total RNA was isolated from E10.5 or E13.5 ventricles using the High Pure RNA Isolation Kit (Roche). This RNA served as a template to generate cDNA using the Transcriptor First Strand cDNA Synthesis Kit (Roche). For qPCR, cDNA was amplified using Taqman Probe-Based Gene Expression Assays (Applied Biosystems). Relative gene expression was determined after normalization to GAPDH. The Student's t-test was used to detect significant differences between sample groups; asterisks denotes p-values ≤ 0.05. Error bars represent standard error (SE).
EMSAs, luciferase assays and ChIP
Hand2 and E12 were in vitro translated using the Promega Reticulocyte Lysate System. 5ul of translated protein was incubated with radio-labeled oligos corresponding to E-boxes and D-boxes within the Nrg1 and VegfR3 loci, in binding buffer for 30 minutes at 25°C (Firulli et al., 2007). Transcription factor/oligo complexes were run out on a non-denaturing 6% polyacrylamide gel and assessed on a phospho-imager. For luciferase assays, HeLa cells were transfected using X-tremeGENE HP (Roche) at a ratio of 3:1 with Nrg1-500bp+pGL4.10 (luciferase), VegfR3-500bp+pGL4.10 (luciferase), pGL4.73 (SV40-renilla) or pGL4.74 (TK-renilla), Pcs2+Myc-Hand2, Pcs2+Myc-E12, pcDNA+Myc-ΔBasic-Hand2 (kindly provided by Eric Olson), or pcDNA3.1, and cultured for 48 hours. After harvest and processing, luciferase and renilla reporter activity was assessed in equal amounts of cell lysate using Luminoskan Ascent software and a ThermoLabsystems luminometer. Luciferase results were normalized to protein concentration or renilla. Asterisks denote significant difference from pcDNA control (pvalue≤0.05); error bars represent SE. For construction of Nrp1(−358/−355kb) and Dll4(−23/−20kb) luciferase reporters, genomic regions containing a Hand2-3xFLAG ChIP-Seq peak were PCR amplified and cloned into TK-pGL4.10. For chromatin immunoprecipitation (ChIP) of the Nrg1 promoter, HeLA cells were again transfected using X-tremeGENE HP at a ratio of 3:1. After culturing for 48 hours, cells were processed as previously described (Barnes et al., 2011). Briefly, equal amounts of sheered chromatin were immunoprecipitated overnight at 4°C with 50ul of αMyc-conjugated agarose beads (Sigma), or beads without antibody for a negative control. After reversing crosslinks, eluted immunoprecipitated DNA was phenol chloroform extracted, resuspended in ddH2O, and used for subsequent PCR reactions. After 37 cycles of PCR amplification, product was analyzed on an agarose gel. ChIP transfection constructs included Pcs2+Myc-Hand2, Pcs2+E12, and Pcs2+Myc. See supplemental procedures for oligo information.
Supplementary Material
Acknowledgements
We thank Danny Carney and Hannah Lohr for technical assistance and support. We also thank the Riley Heart Research Center Group for discussion and helpful feedback. Furthermore, we thank Thomas Coate for kindly providing EfnB2fx/fx mice. Infrastructural support at the Herman B Wells Center is partially supported by the Riley Children's Foundation and the Carleton Buehl McCulloch Chair. Grant support for this work was provided by NIH 1R01HL120920-01, 1R01HL122123-01, and 1R0AR061392-03 (ABF) and American Heart Association predoctoral fellowship 12PRE11700006 (NJV).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions NJV wrote manuscript, conceived of experiments, and carried out experiments. JC carried out experiments and critically read manuscript. JWV and BAF interpreted data, provided reagents, and critically read manuscript. RZ, MO, and JLR conceived of and carried out Hand2-3xFLAG Chip-seq. BZ aided in experimental planning and provided Nfatc1Cre mice. JGB and JLP assayed Hand2 expression in RBPJk−/− mice. WS interpreted data and evaluated manuscript. ABF wrote and edited manuscript and conceived of experiments.
References
- Anderson R, Becker A, Macartney F, Shinebourne E, Wilkinson J, Tynan M. Is “tricuspid atresia” a univentricular heart? Pediatric Cardiology. 1979;1:51–56. [Google Scholar]
- Barnes RM, Firulli BA, VanDusen NJ, Morikawa Y, Conway SJ, Cserjesi P, Vincentz JW, Firulli AB. Hand2 Loss-of-Function in Hand1-Expressing Cells Reveals Distinct Roles in Epicardial and Coronary Vessel Development. Circulation Research. 2011;108:940–949. doi: 10.1161/CIRCRESAHA.110.233171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedito R, Rocha SF, Woeste M, Zamykal M, Radtke F, Casanovas O, Duarte A, Pytowski B, Adams RH. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature. 2012;484:110–114. doi: 10.1038/nature10908. [DOI] [PubMed] [Google Scholar]
- Chen H, Shi S, Acosta L, Li W, Lu J, Bao S, Chen Z, Yang Z, Schneider MD, Chien KR, et al. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development. 2004;131:2219–2231. doi: 10.1242/dev.01094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connerney J, Andreeva V, Leshem Y, Muentener C, Mercado MA, Spicer DB. Twist1 dimer selection regulates cranial suture patterning and fusion. Dev Dyn. 2006;235:1345–1357. doi: 10.1002/dvdy.20717. [DOI] [PubMed] [Google Scholar]
- Dixelius J, Mäkinen T, Wirzenius M, Karkkainen MJ, Wernstedt C, Alitalo K, Claesson-Welsh L. Ligand-induced vascular endothelial growth factor receptor-3 (VEGFR-3) heterodimerization with VEGFR-2 in primary lymphatic endothelial cells regulates tyrosine phosphorylation sites. Journal of Biological Chemistry. 2003;278:40973–40979. doi: 10.1074/jbc.M304499200. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Carver Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. 1996. [DOI] [PubMed] [Google Scholar]
- Firulli BA, Redick BA, Conway SJ, Firulli AB. Mutations within helix I of Twist1 result in distinct limb defects and variation of DNA binding affinities. J Biol Chem. 2007;282:27536–27546. doi: 10.1074/jbc.M702613200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frensing T, Kaltschmidt C, Schmitt-John T. Characterization of a neuregulin-1 gene promoter: positive regulation of type I isoforms by NF-kappaB. Biochim Biophys Acta. 2008;1779:139–144. doi: 10.1016/j.bbagrm.2007.11.007. [DOI] [PubMed] [Google Scholar]
- Gerety SS, Anderson DJ. Cardiovascular ephrinB2 function is essential for embryonic angiogenesis. Development. 2002;129:1397–1410. doi: 10.1242/dev.129.6.1397. [DOI] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, et al. Heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grego-Bessa J, Luna-Zurita L, del Monte G, Bolos V, Melgar P, Arandilla A, Garratt AN, Zang H, Mukouyama YS, Chen H, et al. Notch signaling is essential for ventricular chamber development. Dev Cell. 2007;12:415–429. doi: 10.1016/j.devcel.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert SP, Stainier DY. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat Rev Mol Cell Biol. 2011;12:551–564. doi: 10.1038/nrm3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman JI. Incidence of congenital heart disease: I. Postnatal incidence. Pediatr Cardiol. 1995;16:103–113. doi: 10.1007/BF00801907. [DOI] [PubMed] [Google Scholar]
- Jiao K, Langworthy M, Batts L, Brown CB, Moses HL, Baldwin HS. Tgfbeta signaling is required for atrioventricular cushion mesenchyme remodeling during in vivo cardiac development. Development. 2006;133:4585–4593. doi: 10.1242/dev.02597. [DOI] [PubMed] [Google Scholar]
- Kim JS, Viragh S, Moorman AF, Anderson RH, Lamers WH. Development of the myocardium of the atrioventricular canal and the vestibular spine in the human heart. Circ Res. 2001;88:395–402. doi: 10.1161/01.res.88.4.395. [DOI] [PubMed] [Google Scholar]
- Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- Kramer R, Bucay N, Kane DJ, Martin LE, Tarpley JE, Theill LE. Neuregulins with an Ig-like domain are essential for mouse myocardial and neuronal development. Proceedings of the National Academy of Sciences. 1996;93:4833–4838. doi: 10.1073/pnas.93.10.4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden DG, McAnally J, Richardson JA, Charité J, Olson EN. Misexpression of dHAND induces ectopic digits in the developing limb bud in the absence of direct DNA binding. Development. 2002;129:3077–3088. doi: 10.1242/dev.129.13.3077. [DOI] [PubMed] [Google Scholar]
- Miquerol L, Langille BL, Nagy A. Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development. 2000;127:3941–3946. doi: 10.1242/dev.127.18.3941. [DOI] [PubMed] [Google Scholar]
- Pinto AR, Paolicelli R, Salimova E, Gospocic J, Slonimsky E, Bilbao-Cortes D, Godwin JW, Rosenthal NA. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PloS one. 2012;7:e36814. doi: 10.1371/journal.pone.0036814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Red-Horse K, Ueno H, Weissman IL, Krasnow MA. Coronary arteries form by developmental reprogramming of venous cells. Nature. 2010;464:549–553. doi: 10.1038/nature08873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shawber CJ, Funahashi Y, Francisco E, Vorontchikhina M, Kitamura Y, Stowell SA, Borisenko V, Feirt N, Podgrabinska S, Shiraishi K, et al. Notch alters VEGF responsiveness in human and murine endothelial cells by direct regulation of VEGFR-3 expression. J Clin Invest. 2007;117:3369–3382. doi: 10.1172/JCI24311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart N, Dubé KN, Riley PR. Coronary vessel development and insight towards neovascular therapy. International journal of experimental pathology. 2009;90:262–283. doi: 10.1111/j.1365-2613.2009.00646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson EC, Huggins GS, Lin H, Clendenin C, Jiang F, Tufts R, Dardik FB, Leiden JM. A syndrome of tricuspid atresia in mice with a targeted mutation of the gene encoding Fog-2. Nat Genet. 2000;25:353–356. doi: 10.1038/77146. [DOI] [PubMed] [Google Scholar]
- Takeuchi JK, Ohgi M, Koshiba-Takeuchi K, Shiratori H, Sakaki I, Ogura K, Saijoh Y, Ogura T. Tbx5 specifies the left/right ventricles and ventricular septum position during cardiogenesis. Development. 2003;130:5953–5964. doi: 10.1242/dev.00797. [DOI] [PubMed] [Google Scholar]
- Tammela T, Zarkada G, Wallgard E, Murtomaki A, Suchting S, Wirzenius M, Waltari M, Hellstrom M, Schomber T, Peltonen R, et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature. 2008;454:656–660. doi: 10.1038/nature07083. [DOI] [PubMed] [Google Scholar]
- Tian X, Hu T, Zhang H, He L, Huang X, Liu Q, Yu W, Yang Z, Zhang Z, Zhong TP, et al. Subepicardial endothelial cells invade the embryonic ventricle wall to form coronary arteries. Cell Res. 2013;23:1075–1090. doi: 10.1038/cr.2013.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchihashi T, Maeda J, Shin CH, Ivey KN, Black BL, Olson EN, Yamagishi H, Srivastava D. Hand2 function in second heart field progenitors is essential for cardiogenesis. Dev Biol. 2011;351:62–69. doi: 10.1016/j.ydbio.2010.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDusen NJ, Vincentz JW, Firulli BA, Howard MJ, Rubart M, Firulli AB. Loss of Hand2 in a population of Periostin lineage cells results in pronounced bradycardia and neonatal death. Developmental biology. 2014;388:149–158. doi: 10.1016/j.ydbio.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzi MP, McCulley DJ, De Val S, Dodou E, Black BL. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Developmental Biology. 2005;287:134–145. doi: 10.1016/j.ydbio.2005.08.041. [DOI] [PubMed] [Google Scholar]
- Vincentz JW, Barnes RM, Rodgers R, Firulli BA, Conway SJ, Firulli AB. An absence of Twist1 results in aberrant cardiac neural crest morphogenesis. Dev Biol. 2008;320:131–139. doi: 10.1016/j.ydbio.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Zhang Z, Lui W, Chen X, Wang Y, Chamberlain AA, Moreno-Rodriguez RA, Markwald RR, O'Rourke BP, Sharp DJ, et al. Endocardial cells form the coronary arteries by angiogenesis through myocardial-endocardial VEGF signaling. Cell. 2012;151:1083–1096. doi: 10.1016/j.cell.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wythe JD, Dang LT, Devine WP, Boudreau E, Artap ST, He D, Schachterle W, Stainier DY, Oettgen P, Black BL, et al. ETS Factors Regulate Vegf-Dependent Arterial Specification. Dev Cell. 2013;26:45–58. doi: 10.1016/j.devcel.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi H, Olson EN, Srivastava D. The basic helix-loop-helix transcription factor, dHAND, is required for vascular development. J Clin Invest. 2000;105:261–270. doi: 10.1172/JCI8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.