

Abstract
Biocathode extracellular electron transfer (EET) may be exploited for biotechnology applications, including microbially mediated O2 reduction in microbial fuel cells and microbial electrosynthesis. However, biocathode mechanistic studies needed to improve or engineer functionality have been limited to a few select species that form sparse, homogeneous biofilms characterized by little or no growth. Attempts to cultivate isolates from biocathode environmental enrichments often fail due to a lack of some advantage provided by life in a consortium, highlighting the need to study and understand biocathode consortia in situ. Here, we present metagenomic and metaproteomic characterization of a previously described biocathode biofilm (+310 mV versus a standard hydrogen electrode [SHE]) enriched from seawater, reducing O2, and presumably fixing CO2 for biomass generation. Metagenomics identified 16 distinct cluster genomes, 15 of which could be assigned at the family or genus level and whose abundance was roughly divided between Alpha- and Gammaproteobacteria. A total of 644 proteins were identified from shotgun metaproteomics and have been deposited in the the ProteomeXchange with identifier PXD001045. Cluster genomes were used to assign the taxonomic identities of 599 proteins, with Marinobacter, Chromatiaceae, and Labrenzia the most represented. RubisCO and phosphoribulokinase, along with 9 other Calvin-Benson-Bassham cycle proteins, were identified from Chromatiaceae. In addition, proteins similar to those predicted for iron oxidation pathways of known iron-oxidizing bacteria were observed for Chromatiaceae. These findings represent the first description of putative EET and CO2 fixation mechanisms for a self-regenerating, self-sustaining multispecies biocathode, providing potential targets for functional engineering, as well as new insights into biocathode EET pathways using proteomics.
INTRODUCTION
Bioelectrochemical systems (BES) use microorganisms as catalysts to drive complex electrochemical reactions, such as electricity generation by microbial fuel cells (MFCs) (1), wastewater treatment (2), and microbial electrosynthesis (3–6), that would not be possible without living cells. The term “biocathode” refers to a biofilm, constituted of a single organism or microbial consortium, that has formed on the cathode of a BES and consumes electrons (e−). Cathodes hold great potential as a stable electron source to drive microbial metabolism (7); however, little is known about the underlying extracellular electron transfer (EET) pathways that could be exploited for biocathode functional engineering. Although biocathode EET has been demonstrated for a variety of microorganisms, including acetogens (5) and a methanogenic archaeon (6), studies aimed at identifying EET conduits from the electrode to cells have mostly been confined to the model organisms Geobacter (8) and Shewanella (9), due to the massive effort put forth to understand how these iron-reducing bacteria are able to catalyze EET at bioanodes (10–12). The ability of iron-reducing bacteria to reduce anodes led to the hypothesis that iron-oxidizing bacteria (FeOB) would be able to perform EET with cathodes, which has been demonstrated for at least two FeOB, Mariprofundus ferrooxydans and Rhodopseudomonas palustris (13, 14). While many bacteria have been shown to attach to electrodes and “consume” electrons, electrode-dependent growth has thus far been demonstrated only for M. ferrooxydans (13). Furthermore, most cathode EET processes studied to date rely on a fairly negative electrode potential (between 0 and −0.400 V standard hydrogen electrode [SHE]) to catalyze CO2 or O2 reduction. Biocathodes developed at higher potentials, such as those used in this study, need to rely on EET mediators with much higher potentials (>+0.300 V SHE). Identifying and understanding such mediators could provide flexibility to engineering functionality in BES applications where higher operating potentials are desired.
A challenge in developing the use of BES under environmentally relevant conditions (i.e., in seawater and under changing pH and changing temperature) is a lack of understanding of cathodic microbial communities. Little effort has been put into developing microbial consortia as biocathode catalysts, even though they have been shown to outperform homogeneous bacterial populations in terms of current density (15). Marshall et al. (3, 16) have demonstrated long-term biocommodity production using an acetogenic biocathode consortium but have not yet reported on the underlying EET pathways. Attempts to cultivate isolates from biocathode environmental enrichments typically result in loss of the electrochemical phenotype (4), as individual biofilm constituents may lack some essential cofactor provided by life in a consortium, such as vitamins or amino acids.
Advances in systems biology tools and large-scale, culture-independent, community level “omic” (e.g., metagenomic, metatranscriptomic, and metaproteomic) measurements now allow us to predict and potentially direct interactions in naturally occurring, stable microbial consortia (17). Metaomics data provide a perspective on the physiological state of organisms thriving from their associations with one another that may be different than when they are grown in homogeneous microbial populations (18). When applied to study biocathode microbial consortia, combined metagenomic and metaproteomic analyses may be particularly useful for generating information about the functions of biofilm constituents in relation to the biocathode lifestyle (19, 20). Biocathode metaproteomics can provide functional information from electrode-grown cells in order to predict biofilm EET pathways for targeted biofilm engineering, particularly when key EET biofilm constituents cannot be cultivated or when ex situ cultivation conditions are not relevant to electrode growth. In relation to BES, the use of metaproteomics to study bioanodes has been limited (21), and there are currently no published reports on the proteome of a biocathode.
We previously reported on the electrochemical features of an aerobically grown, nonphototrophic biocathode community enriched from seawater (22). This biocathode is a durable, multicell-layer-thick biofilm that is self-regenerating and self-sustaining. We hypothesized that the biocathode uses electrons supplied by a poised electrode (+310 mV versus SHE) to drive CO2 fixation and O2 reduction, since no other electron donor or carbon source is provided. Portions of the biocathode biofilm can be removed and used to inoculate subsequent biocathode reactors that achieve reproducible electrochemical characteristics of the parent biofilm. 16S rRNA gene clone libraries initially showed the biocathode biofilm to be a low-complexity consortium that consisted primarily of Marinobacter, a ubiquitous biofilm-forming member of the Gammaproteobacteria known to oxidize iron under aerobic and circumneutral conditions (23), as well as other bacteria most closely related to marine Alpha- and Gammaproteobacteria.
The primary objective of the current study was to obtain an initial understanding of in situ biocathode EET and carbon fixation pathways at maximum current. We expand upon our previous work using metagenomics, reverse transcription (RT)-PCR, and shotgun metaproteomics to (i) confirm the identities of the primary biofilm constituents, (ii) provide an initial survey of the biocathode metaproteome, (iii) identify proteins of putative EET pathways, and (iv) identify carbon fixation pathways that may confer autotrophy on the biocathode community using electrons from the electrode as an energy source. We show that an unknown member of the family Chromatiaceae expresses proteins for both CO2 fixation and EET. While Marinobacter may have some capacity for EET, no known EET pathways were identified. Roles for other abundant biofilm constituents, including Labrenzia and Kordiimonas, could not readily be predicted but are likely important for biofilm formation and carbon cycling.
MATERIALS AND METHODS
Biocathode biofilm cultivation.
The bioelectrochemical reactors were 2-liter dual-chambered microbial fuel cell reactors (Adams and Chittenden Scientific Glass) without membrane separation. The working electrodes were either graphite coupons (length, 3.0 cm; height, 10.0 cm; width, 0.2 cm; total geometric surface area, 65.2 cm2, or 0.00652 m2) or carbon cloth flags (length, 3.5 cm; height, 3.5 cm; total geometric surface area, 24.5 cm2, or 0.00245 m2). Following initial proteomics analysis from graphite coupons, carbon cloth electrodes were used to grow biocathodes for protein analysis to improve protein recovery, since their electrochemical features were identical. Further descriptions of reactor and electrochemical measurements can be found in the supplemental material. The reactors were filled with artificial seawater medium (ASW) (see the supplemental material for the composition), and scrapings from previously described enriched biocathode biofilms were used as a source of inoculum (22). The reactors were maintained at 30°C with stirring (VWR standard multiposition stirrer; setting 2 [150 to 200 rpm]) under atmospheric conditions. The working electrodes were maintained at approximately +0.310 V versus SHE (+0.100 V versus Ag/AgCl) using a multichannel potentiostat (Solartron 1470E) under software control (Multistat; Scribner). All the potentials reported here are versus SHE unless specifically noted otherwise. Once maximum current was reached, biocathode electrochemical features were characterized and confirmed to be consistent with those previously reported (see Fig. S1 and S2 in the supplemental material) (22).
Metagenomic DNA sequencing and assembly.
Metagenomic DNA was extracted from a graphite coupon electrode grown as described above using the MoBio PowerBiofilm DNA isolation kit. DNA integrity and concentration were verified using a Bioanalyzer 2100 (Agilent, Palo Alto, CA). Approximately 1.0 μg of high-quality DNA was processed using an Illumina TruSeq DNA sample preparation kit following the manufacturer's instructions (Illumina, San Diego, CA, USA). The library was validated and sequenced on an Illumina HiSeq 2000 sequencer using 100-bp paired-end reads. Approximately 31.3 million filtered raw read pairs were generated for this study. Raw sequence reads from the whole-genome shotgun (WGS) approach were first trimmed using SolexaQA (24), a Perl-based software package calculating quality statistics from FASTQ files generated by Illumina sequencers. The default setting (P = 0.05) was used. Additional quality control steps included the removal of low-quality reads based on error probabilities and trimming low-quality tails. After these steps, only paired-end reads of >90 bp were retained. These processed reads were then assembled using Velvet v1.2.10 (25) (kmer length = 55; insertion library length = 400). The expected coverage was set to auto.
ORF calling and annotation.
Metagene (26) was used to predict 79,765 open reading frames (ORFs) from the biocathode metagenome, of which 63,097, 86,537, and 77,136 ORFs were assigned COG, pfam, and KEGG identifiers (IDs), respectively (see Table S3 in the supplemental material). Annotations were assigned using RPS-BLAST and the NCBI Conserved Domain Database (27). The predicted ORFs were submitted to WebMGA (28) to generate KEGG identifiers (29). Contigs from clusters containing fewer than 500 contigs (see below) were submitted as individual groups to the RAST pipeline for annotation (http://rast.nmpdr.org). Amino acid sequences for known proteins of functional interest were used to identify homologous proteins from the translated metagenome using the BLASTp algorithm and the NCBInr protein database with default settings (http://blast.ncbi.nlm.nih.gov).
Taxonomic/phylogenetic analysis.
Predicted ORFs were processed using AMPHORA2 to identify and assign taxonomy to 31 different housekeeping genes (30). Single-copy housekeeping genes were used to infer the relative abundances of the biofilm constituents based on the Velvet assembly coverage of each unique gene. Since ORF calling on contigs could potentially identify paralogs or partial fragments, results returned from AMPHORA2 were further curated manually (see the supplemental material for details). During manual curation, ORFs identified by AMPHORA2 as containing a particular housekeeping gene from the same species, and from contigs with similar coverage, were grouped together. Relative abundance was estimated for each housekeeping gene by summing the Velvet assembly coverage of the contigs from which the genes were identified. These individual gene coverage results were averaged across a given taxonomic assignment and are reported in Fig. 1. Additionally, filtered raw reads were analyzed using MetaPhyler v1.25 to generate an abundance count at various taxonomic levels using taxonomic marker genes from complete genomes (31).
FIG 1.

Relative abundances (weighted by sequence coverage) and taxonomic distribution of the biocathode community based on the most likely phylogenetic assignment of 31 distinct housekeeping genes identified from assembled metagenomic contigs. Class, order, and family level identifications with at least 1% relative abundance are shown (except for Flavobacteriia, which is shown at 0.6% due to a high-confidence identification).
Clustering of assembled contigs.
Clusters were defined for groups of AMPHORA2 genes that resolved to the same taxonomic level and for which similar coverage was found for the contigs from which the ORFs were generated. Contigs longer than 200 bp were processed using the Metawatt binner (v1.7), which uses multivariate statistics of tetranucleotide frequencies combined with use of the interpolated Markov model (IMM) (GLIMMER 3.02) to cluster contigs (32). Initial clusters based only on tetranucleotide frequencies were built of at least 0.1 Mbp using the medium confidence level. The resulting clusters were merged after inspection if contigs contained therein were previously identified as being in the same cluster according to AMPHORA2 analysis. The IMM modeling step was then run to obtain a refined group of clusters. The average and standard deviation of coverage of contigs longer than 2 kb in each cluster were computed. In clusters where the standard deviation was more than 10% of the average, the large contigs (>5,000 bp) were compared to this average, and if the coverage deviated significantly from the average, it was subjected to further checks. ORFs from contigs identified as being suspect were searched against the NCBInr database using BLASTp. Consensus identification was attempted for each contig by counting the top genus hit of each ORF from that contig. If the closest match for more than 37% of predicted proteins on a contig were assigned to the same genus, the contig was designated as belonging to that genus. Contigs that had 25 to 37% proteins with their closest matches within a single genus were submitted for discontiguous megablast. If a large fraction of the contig was assigned to an organism in the same genus as those with >37% call identity, it was assigned that designation. For purposes of assigning functional proteins to a specific cluster genome, all unclassified clusters were treated as a single cluster genome.
Housekeeping genes.
The KEGG orthology (KO) functional annotation has been determined for 107 marker genes that are typically found to be in single copies in more than 95% of bacterial genomes (33, 34). The number of these marker genes in each Metawatt cluster was determined from the KO assignments made in the KEGG annotation of the metagenome contigs.
Multiheme c-type cytochrome prediction.
Multiheme c-type cytochromes (c-Cyts) were predicted using hmmsearch in the HMMER software package v3.1b1 (E value < 1E−5) (35). In silico-translated protein sequences derived from the metagenome were searched using a doubled CXXCH motif (pfam09699), which represents two copies of the heme-binding CXXCH motif. Predicted multiheme c-Cyts were screened for lipoprotein domains using the LipoP 1.0 server (36). The average molecular weight (MW) was predicted using the MoreFASTA utility of the DTASelect program (37).
Metaproteomics.
Proteins were extracted from graphite coupon and cloth biofilms using several extraction methods to help alleviate some of the issues surrounding protein extraction bias (38). A low protein yield per electrode prevented simultaneous sampling for both DNA and protein; therefore, separate samples were grown for proteomics and evaluated for electrochemical traits identical to those of reactors used for DNA extraction (see Table S1 in the supplemental material). The methods of protein extraction and analysis were adapted from previously developed methods (38, 39) and are briefly described below (a complete description can be found in the supplemental material). For graphite block biofilms, biofilms were scraped with razor blades from graphite blocks into 2% sodium dodecyl sulfate (SDS) in 50 mM ABC (ammonium bicarbonate) or B-Per Tris solution (Thermo Scientific, Rockford, IL). All samples were sonicated on ice, mixed with Tris-buffered phenol (1:1 ratio), incubated for 30 min at room temperature with mixing, and then subjected to centrifugation for phase separation. The phenol phase was collected from all samples and combined with ice-cold 100 mM ammonium acetate in 100% methanol (1:4 ratio) prior to incubation at −80°C for a minimum of 16 h. For graphite cloth biofilms, biofilm samples from graphite cloth were submerged in B-Per Tris solution (Life Technologies), sonicated on ice, and centrifuged (4°C; 5,000 × g; 10 min) to sediment small graphite particles. The cleared supernatants were transferred into new tubes and precipitated using 100 mM ammonium acetate in 100% methanol as described above. The extracted and precipitated proteins were collected by centrifugation, dissolved in SDS-PAGE running buffer, separated by SDS-PAGE, and stained using Coomassie blue. Distinct protein bands were cut, washed, and destained. Each band was then in-gel digested overnight using modified porcine trypsin, and the tryptic digests were collected into new tubes, dried via speed-vac, and stored at −20°C. The dried digests were reconstituted in 5% acetonitrile, 0.1% formic acid in water immediately prior to analysis by reverse-phase liquid chromatography-tandem mass spectrometry (LC–MS-MS) using a TempoMDLC system coupled to a QStar Elite mass analyzer. Tandem mass spectra were extracted using the AB Sciex MS Data Converter (version 2.0) and searched by Mascot (Matrix Science, London, United Kingdom; version 2.4.1) and X! Tandem (The Global Proteome Machine [http://thegpm.org]; version CYCLONE [2010.12.01.1]). Scaffold (version Scaffold_4.2.1; Proteome Software Inc., Portland, OR) was used to validate MS-MS-based peptide and protein identifications.
All identified proteins were assigned to the cluster genomes described above. In order to predict protein functions and metabolic pathways, amino acid sequences were annotated using the BLASTp algorithm and the NCBInr protein database with default settings (http://blast.ncbi.nlm.nih.gov). An annotation of the first highest-scoring protein was accepted if the E value was <5. For hypothetical proteins, conserved domains were used to predict function if they were present. Protein localization was predicted by PSORTb (v3.0.2 [http://www.psort.org/psortb/]). All annotated proteins were manually assigned to three functional categories: (i) enzymes (all annotated proteins for which an EC number could be obtained or a domain known to be in enzymes was present; cytochromes, peroxiredoxins, thioredoxin, molybdopterin, and ferredoxins were included in this category, and the enzyme database BRENDA [http://www.brenda-enzymes.org/index.php] was used to obtain EC numbers and KEGG pathway numbers for all identified enzymes); (ii) receptors, transporters, and membrane proteins (which were grouped according to their substrates/cargo); and (iii) structural proteins and proteins with unknown function. All information regarding protein annotation, predicted function, and predicted localization can be found in Data Set S2 in the supplemental material.
RT-PCR.
Biofilm RNA was extracted using a previously described method (40) with the following modification: 250 μl of Zirconia beads (Life Technologies) was used instead of 0.8 g of 0.5-mm glass beads. The extracted RNA samples were subjected to Turbo DNase (Life Technologies) treatment according to the manufacturer's recommended protocol and purified using the RNA Clean and Concentrator-5 kit (Zymo Research). The DNase treatment was repeated once for each sample to ensure the removal of potential contaminating metagenomic DNA. The Complete Whole Transcriptome Amplification kit (Sigma-Aldrich, St. Louis, MO) was used to amplify biofilm total RNA according to the manufacturer's recommended protocol. The amplified products were purified using the DNA Clean and Concentrator-5 kit (Zymo Research) and quantified using a NanoDrop 2000c UV-Vis Spectrophotometer (Thermo Scientific, Waltham, MA). The quantitative PCRs (qPCRs) were carried out in 25-μl reaction volumes containing 1× iQ SYBR Green Supermix (Bio-Rad Laboratories, Inc., Hercules, CA), 200 nM (each) forward and reverse primers, and 25 ng of amplified products using a MyiQ single-color real-time PCR detection system (Bio-Rad Laboratories, Inc.). Reactions were performed with an initial denaturation at 95°C for 3 min, followed by 35 cycles of 95°C for 10 s, 56°C for 20 s, and 72°C for 20 s. The sequences of the primers used in this study are listed in Table S2 in the supplemental material.
Accession numbers.
The mass spectrometry proteomics data (raw sequence reads) have been deposited in the ProteomeXchange Consortium (41) via the PRIDE partnership repository with the data set identifier PXD001045. The Sequence Read Archive (SRA) accession (SRX621521) of the raw Illumina reads can be found at http://www.ncbi.nlm.nih.gov/sra/SRX621521. Other proteins (see Tables 4 and 5) were also deposited in the PRIDE partnership repository under the same identifier.
TABLE 4.
Key components of the CBB cycle from the biocathode
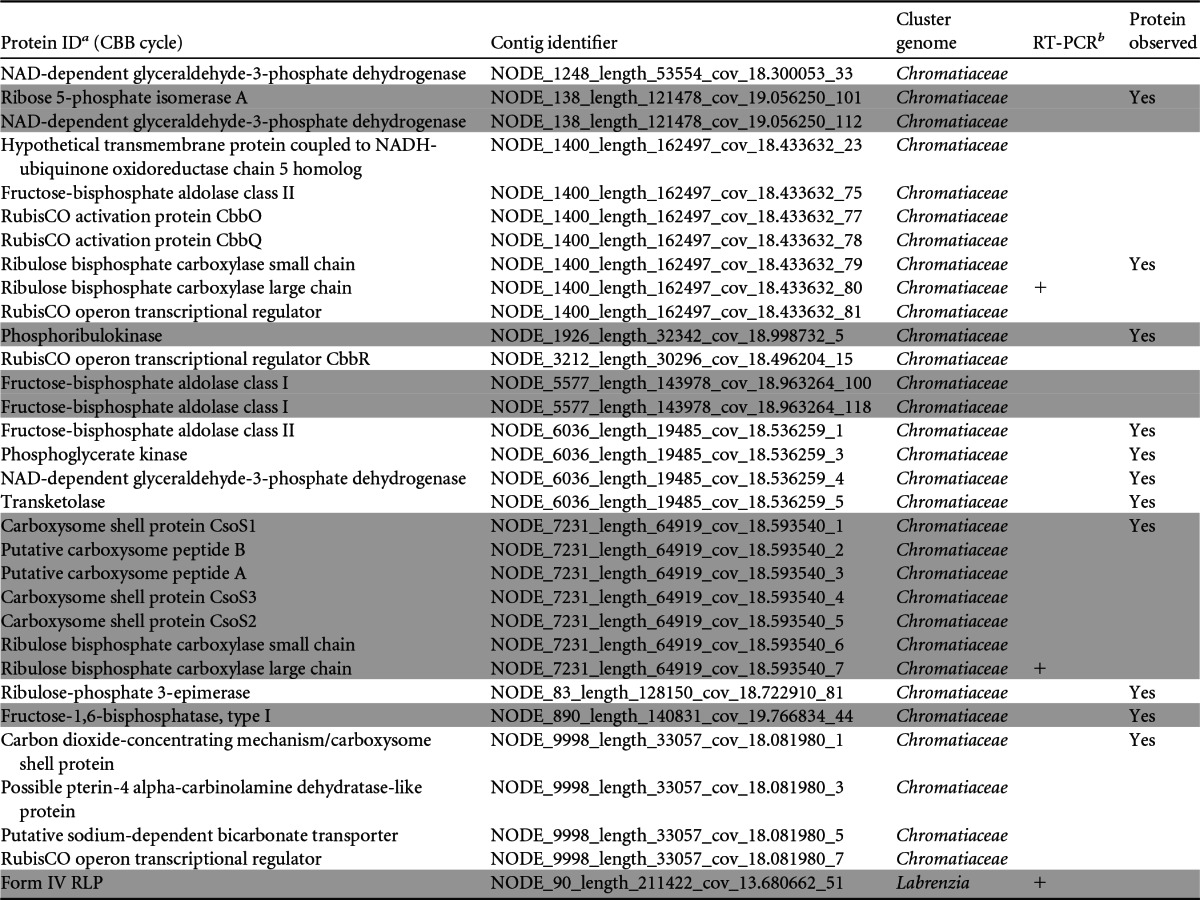
Shaded and unshaded proteins were identified as belonging to distinct contigs.
+, RT-PCR was positive for gene expression for the protein.
TABLE 5.
Distribution of the coxLMS operon among biocathode cluster genomes
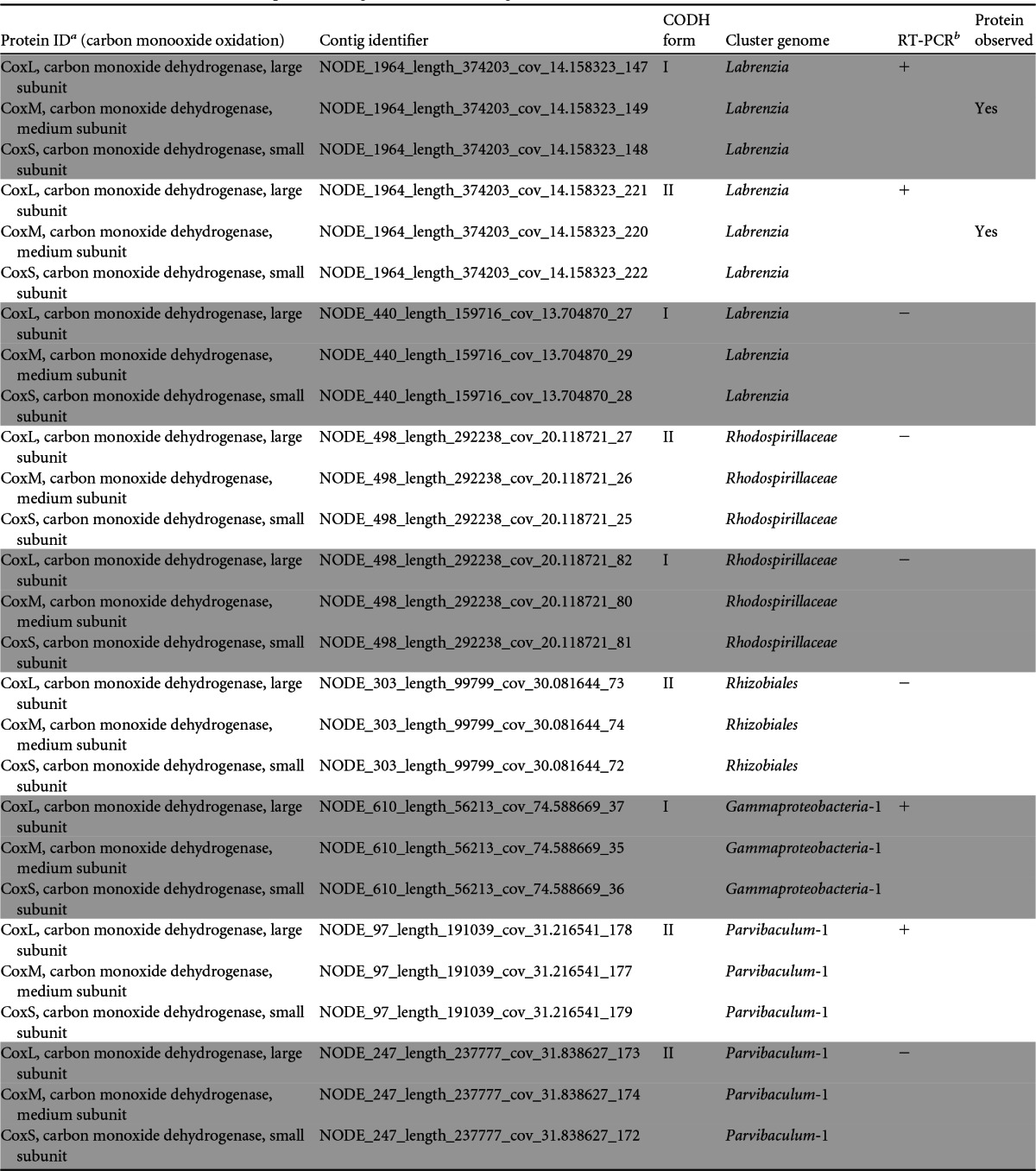
Shaded and unshaded proteins were identified as belonging to distinct contigs.
+, RT-PCR was positive for gene expression for the protein; −, no gene expression was detected.
RESULTS
Biocathode metagenome.
To further characterize the biological composition and functional potential of the community while simultaneously providing a matched-sample database for subsequent metaproteomic studies, a representative biocathode was subjected to whole-metagenome sequencing. Biocathode metagenome sequencing resulted in approximately 31.3 million filtered raw read pairs that were assembled into 32,870 contigs using the Velvet assembler (mean contig length = 2,016 bp; maximum contig length = 627,596 bp; N50 = 55,338 bp) (see Table S3 in the supplemental material). N50 is defined as the length (N) for which 50% of all bases are represented in fragments of length L (<N). Multiple data analysis techniques focusing on single- to low-copy-number housekeeping genes were used to determine the taxonomic composition of the biofilm and to estimate the relative abundances of constituents. A summary of the results from all the techniques used can be found in Table S4 in the supplemental material. First, AMPHORA2 was used to determine the average biofilm composition by examining the taxonomic assignment of 31 housekeeping genes from the assembled contigs (Fig. 1). Fifteen distinct cluster genomes could be identified by AMPHORA2 from 316 contigs that contained at least one of the housekeeping genes. Taxonomic identifications were found to be reliable down to the family level, with 99% of the housekeeping genes identified to the class level. A direct taxonomic analysis performed on the filtered but unassembled reads using MetaPhyler was consistent with those derived from the AMPHORA2 analysis (see Fig. S3 in the supplemental material). In both instances, sequencing fragments that could not be sorted into distinct cluster genomes were taxonomically classified as Betaproteobacteria or Flavobacteriia. These fragments may represent biocathode constituents present at very low abundance that could not be further resolved with the analysis techniques used here. Second, an additional clustering approach based on tetranucleotide frequency was performed on all contigs that were >200 bp in size using Metawatt (Fig. 2). Metawatt analysis resulted in 23 refined cluster genomes, 16 of which contained at least 1 Mb of sequence. All except the Unclassified-1 cluster were considered to be representative of a single organism. The RAST pipeline provided further confirmation of taxonomic assignments of refined cluster genomes with fewer than 500 contigs. Overall, the combined RAST, AMPHORA2, and Metawatt analyses agreed on all but one identification and even enabled genus level identification of some cluster genomes. The sole discrepancy was the RAST-identified Kordiimonas cluster genome, which the AMPHORA2/Metawatt analyses identified as a member of the Sphingomonadaceae. A draft genome sequence of Kordiimonas was recently deposited in GenBank (NZ_AQXF00000000.1) and was not part of the database version used in the AMPHORA2 analysis. As such, the RAST identification of Kordiimonas was used for this cluster genome designation. Finally, 107 essential single-copy genes with conserved KEGG identifiers in 95% of all sequenced bacteria were enumerated from the predicted ORFs (see Data Set S1 in the supplemental material) (33, 42). Out of the 16 cluster genomes predicted by AMPHORA2 and Metawatt, all but Parvibaculum-2 contained at least 73 of the 107 housekeeping genes. The total length of all cluster genomes makes up 89% of the total assembled metagenome length. Cluster genomes were subsequently used to predict the origins of proteins identified by biocathode metaproteomics.
FIG 2.

Contig sequence coverage is shown as points whose sizes are proportional to the contig length versus its GC content The color of each contig is based on the cluster to which it was assigned. (A) Sequence coverage in log scale depicting only higher-coverage organisms (>10). (B) Sequence coverage in linear scale depicting organisms with lesser coverage (<10).
Biocathode metaproteome.
A total of 644 proteins were identified from the biocathode biofilm (see Data Set S2 in the supplemental material), 599 (93%) of which could be mapped to cluster genomes (Table 1). The Marinobacter, Chromatiaceae, and Labrenzia cluster genomes were the most represented taxa in the metaproteome, with 177, 137, and 59 proteins identified, respectively. Annotation, classification, and predicted localization information for each of the identified proteins can be found in Data Set S2 in the supplemental material; it was used to search for proteins associated with biocathode EET and carbon fixation, as described below.
TABLE 1.
Summary of proteins identified from the biocathode metaproteome
| Identification | No. of proteins | No. of enzymesa | No. of receptors, transporters, and membrane proteins | No. of structural and hypothetical proteins |
|---|---|---|---|---|
| Marinobacter | 177 | 69 | 50 | 58 |
| Chromatiaceae | 137 | 63 | 33 | 41 |
| Labrenzia | 59 | 20 | 32 | 7 |
| Unclassified | 45 | 5 | 22 | 18 |
| Kordiimonas | 44 | 8 | 20 | 16 |
| Gammaproteobacteria-2 | 30 | 14 | 9 | 7 |
| Rhodospirillaceae | 29 | 12 | 10 | 7 |
| Phaeobacter | 26 | 6 | 15 | 5 |
| Alcanivoracaceae | 23 | 10 | 6 | 7 |
| Rhizobiales | 19 | 2 | 12 | 5 |
| Gammaproteobacteria-1 | 12 | 7 | 1 | 4 |
| Phyllobacteriaceae | 11 | 4 | 2 | 5 |
| Parvibaculum-1 | 10 | 3 | 4 | 3 |
| Muricauda | 8 | 2 | 6 | 0 |
| Hyphomonas | 7 | 3 | 3 | 1 |
| Parvibaculum-2 | 7 | 4 | 2 | 1 |
| Total | 644 | 232 | 227 | 185 |
Enzymes include all proteins for which an EC number could be obtained or for which an enzyme domain was found to be present from the NCBI annotation. Cytochromes, peroxiredoxins, thioredoxin, molybdopterin, and ferredoxins were included in the enzyme category.
Evidence for biocathode EET by an unknown member of the Chromatiaceae.
A suite of consensus genetic markers is not known for cathode EET. Based on the identification and relative abundance of Marinobacter bacteria (a known FeOB) from our previous work (22), as well as recent demonstrations of cathode EET by other FeOB (13, 14), we hypothesized that our biocathode consortium utilizes iron oxidation pathways for biocathode EET. We explored the biocathode metagenome for genetic evidence of known or predicted iron oxidation pathways from all characterized FeOB (43). Table 2 summarizes the results of this search, including instances in which either the protein product was identified by proteomics or gene expression was confirmed by RT-PCR. The most interesting finding was the discovery that the Chromatiaceae cluster genome contains homologs of genes for proteins from two different putative FeOB EET pathways. The family Chromatiaceae is most known for the purple sulfur bacteria, which use light energy to oxidize sulfide under anoxygenic conditions. The pufLM (photosynthetic unit forming) operon represents a conservative marker for photosynthesis within the Chromatiaceae (44); however, we were not able to find pufLM operon homologs within the Chromatiaceae cluster genome or within the entire metagenome, suggesting that the Chromatiaceae cluster genome probably represents a nonphotosynthetic member of the family. The entire genomic locus for a MopB-containing alternative complex III (ACIII), recently described as part of a putative iron oxidation pathway in Zetaproteobacteria (45, 46), was observed on a single contig within the Chromatiaceae cluster genome. The cluster includes genes for two multiheme c-Cyts, a 4Fe-4S ferredoxin, iron-sulfur binding protein, an integral transmembrane polysulfide reductase (NrfD), and two quinol-cytochrome c oxidoreductases and showed synteny with M. ferrooxydans and Sideroxydans lithotrophicus, as well as Geobacter uraniireducens (see Table S5 in the supplemental material). Proteins for four out of seven components of the putative ACIII cluster were observed by proteomics (Table 2). No specific iron oxidase has been assigned linking the ACIII to EET in Zetaproteobacteria, but the current model suggests that an outer membrane c-Cyt may be involved, as predicted for other FeOB. Two potential electrode oxidase proteins were identified within the Chromatiaceae cluster genome. Expression of a monoheme c-Cyt with sequence similarity to the monoheme Cyc2 from Acidithiobacillus ferrooxidans (also known as Cyt572) was identified by proteomics (Table 2). Cyc2 has been demonstrated to be important for iron oxidation (47, 48) and is suspected of directly oxidizing Fe(II) at low pH. In spite of significant divergence in the overall protein sequences (sequence alignments are shown in Fig. S4A and B in the supplemental material), the positions of the heme-binding site and some surrounding residues in the N termini are conserved among the three proteins. The predicted average molecular mass of the putative monoheme Cyc2-like protein from Chromatiaceae is 56 kDa, compared to 46 kDa for A. ferrooxidans Cyc2 and 61 kDa for Cyt572.
TABLE 2.
Distribution of proteins among cluster genomes reported to be important for iron oxidation in FeOB

Shaded and unshaded proteins were identified as belonging to distinct cluster genomes.
+, RT-PCR was positive for gene expression for the protein; −, no gene expression was detected.
Another c-Cyt of note identified from the Chromatiaceae cluster genome is a predicted undecaheme c-Cyt residing on the same contig (Node_83) as genes for the ACIII (Table 3). If such a protein were associated with the outer membrane, it could potentially participate in EET, as is proposed for membrane-associated multiheme c-Cyts of Shewanella and Geobacter (49, 50). Other predicted proteins of interest within the same contig are genes for the c-Cyt biogenesis system, an outer membrane porin (whose expression was confirmed by proteomics [see Data Set S2 in the supplemental material), and three periplasmic triheme c-Cyt family proteins (see Data Set S3 in the supplemental material).
TABLE 3.
Distribution of predicted multiheme c-type cytochromes among cluster genomes
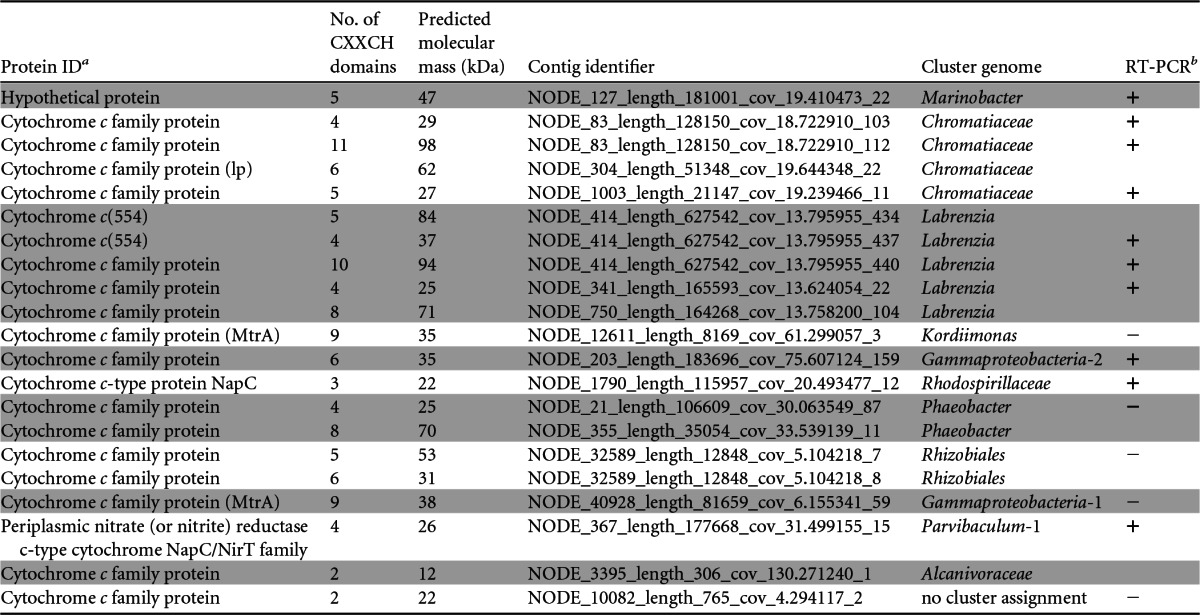
Shaded and unshaded proteins were identified as belonging to distinct cluster genomes. lp, predicted lipoprotein.
+, RT-PCR was positive for gene expression for the protein; −, no gene expression was detected.
We did not identify homologs of the well-known A. ferrooxidans rusticyanin, of Cyc1, or of the aa3-type oxidase pathway linking Cyc2 to oxygen reduction from the biocathode metagenome, leading us to investigate other oxygen reduction pathways associated with FeOB. With some known exceptions, cytochrome cbb3 oxidases are unique to Proteobacteria and are found in FeOB, such as M. ferrooxydans (45). Cytochrome cbb3 oxidases play a significant role in microaerobic respiration, with a high affinity for O2 (51). Genes encoding CcoN (conserved subunit I of cytochrome cbb3) were found in 12 different cluster genomes as part of the full ccoNOQP operon, and most were found by RT-PCR to be expressed (see Table S6 in the supplemental material). The only CcoN protein identified by proteomics was from the Chromatiaceae but was not part of the full ccoNOPQ operon (only ccoNO were present). A similar observation has been made for M. ferrooxydans, where only ccoNOP were identified in the genome (45). Partial reduction of O2 during aerobic iron oxidation can lead to production of potentially toxic reactive oxygen species (ROS), such as hydrogen peroxide. Bacterial cytochrome c peroxidases (CCP) are responsible for catalyzing the two-electron reduction of hydrogen peroxide to water and are critical for detoxification of ROS (52). CCP have also been suggested to be important for iron oxidation by Marinobacter aquaeolei (53, 54). Six full-length genes encoding CCP were identified from the biocathode metagenome (see Table S7 in the supplemental material). The expression of four of these genes, of which three belong to the Chromatiaceae cluster genome and one belongs to the Gammaproteobacteria-1 cluster genome, was confirmed by RT-PCR. Expression of one of the three Chromatiaceae CCP was also observed by proteomics. The role of CCP in biocathode EET is not clear, but given a potential role in iron oxidation in other organisms, it warrants further investigation.
Aside from the FeOB pathways described above, homologs for mtrAB genes from both the Kordiimonas and Gammaproteobacteria-1 cluster genomes were identified in the metagenome (Table 2). The decaheme c-Cyt MtrA and the outer membrane protein MtrB are involved in Shewanella oneidensis EET (43, 55), and the homologs PioA and PioB from R. palustris are known to be involved in iron oxidation (43). Additionally, the MtrA/MtrB homologs MtoA and MtoB from S. lithotrophicus have been demonstrated to oxidize iron in vitro (56). Interestingly, gene expression was not detected by either RT-PCR or proteomics.
Genes for known iron oxidation proteins were not identified in any other cluster genome, including Marinobacter. We therefore surveyed the metagenome for additional putative EET pathways based on the assumption that electron transfer at the biocathode may be mediated by a membrane-associated multiheme, c-Cyt, as is thought to be the case for anode-respiring bacteria. Approximately 187 genes encoding putative c-Cyts were identified in the biofilm metagenome by searching for the conserved CXXCH heme-binding motif. Twenty-one of them contained multiple heme-binding sites and are listed in Table 3, along with their predicted molecular masses. Gene expression for most predicted c-Cyts was observed by RT-PCR for Labrenzia, Marinobacter, Rhodospirillaceae, Chromatiaceae, Parvibaculum-1, and Gammaproteobacteria-2; however, no proteins were observed. This could, in part, be due to known difficulties associated with detecting c-Cyts by mass spectrometry (57). Although at this time it is not possible to assign a role in EET to the identified c-Cyts, it is interesting that they were present in a number of biofilm constituents.
Eighteen representative isolates from six different cluster genomes could be cultivated and were qualitatively evaluated for the ability to oxidize iron as an initial screening for EET (see Table S8 in the supplemental material). A representative member of the Chromatiaceae could not be cultivated using the methods reported here, and efforts are ongoing. Since Marinobacter and Labrenzia are predicted to be relatively abundant in the biofilm and showed some indication of iron oxidation, isolates were also evaluated for EET with a poised electrode (+0.310 V SHE) with and without supplementation with acetate (2 mM) (see Table S8 and Fig. S5A and B in the supplemental material). Supplementation with acetate resulted in Marinobacter EET, with a maximum current 2 orders of magnitude lower than that typically measured for the biocathode community. No electrode EET was observed for Labrenzia. When acetate was omitted, an initial spike in current was observed for Marinobacter, possibly associated with cells initially attaching to the electrode surface; however, no further increase in current was observed. This sharp spike in current by the Marinobacter-inoculated reactor contrasts with an increase in current over time for biocathode community reactors and suggests that the biocathode consortium is needed to develop and sustain Marinobacter when no organic carbon source is provided.
Potential for biocathode-linked carbon fixation by an unknown member of the Chromatiaceae.
The biocathodes described here are grown at equilibrium with atmospheric concentrations of O2 (i.e., no additional aeration is provided), while CO2 is the only carbon source provided and the electrode is assumed to be the primary electron donor. Purging the reactor of O2 was previously shown to eliminate the current (22); therefore, we hypothesized that the biocathode biofilm was fixing CO2 to support growth through an aerobic pathway. Table 4 summarizes 32 key Calvin-Benson-Bassham (CBB) cycle (58) genes and accessory genes that were identified in the biocathode metagenome almost exclusively from the Chromatiaceae cluster genome. Three genes encoding RubisCO were identified: two RubisCO form I (IAq and IAc) genes sharing 79% identity at the amino acid level in the Chromatiaceae cluster and one gene encoding a form IV RubisCO-like protein (RLP) in the Labrenzia cluster. The IAq locus included the RubisCO structural genes rbcL, rbcS, and cbbQO, encoding proteins important in RubisCO assembly, and the cbbR gene, encoding a Lys-type regulator. The IAc operon includes rbcL and rbcS, followed by genes encoding carboxysome shell peptides that are assumed to enhance the effectiveness of CO2 capture (see Fig. S6 in the supplemental material). Of the 32 genes identified in the metagenome, 11 were identified in the metaproteome analysis, including IAq RbcS, phosphoribulokinase, and two carboxysome shell proteins. RT-PCR analysis confirmed that both form I rbcL genes were expressed.
The gene for form IV RLP was identified in the Labrenzia cluster genome. RLPs are structural homologs of RubisCO that are unable to catalyze CO2 fixation (59). They form six deeply branching subclades, only two of which have defined biochemical functions, participating in methionine salvage pathways (60, 61). Another clade (form IV-Photo) has an unknown role during growth using thiosulfate as an electron donor (62). The RLP present in the Labrenzia bin is related to the form IV-NonPhoto clade, which has no known function. The gene encoding this protein was expressed; however, the protein was not detected (Table 4). Marker genes associated with alternative CO2 fixation pathways were not observed, including genes for ATP citrate lyase, bifunctional carbon monoxide dehydrogenases (CODH)/acetyl-coenzyme A (CoA) synthase, malonyl-CoA reductase, propionyl-CoA synthase, and 4-hydroxybutyryl-CoA dehydratase.
Taken together, genomic, RT-PCR, and proteomic evidence of CBB cycle proteins and complex inner membrane respiratory proteins (NADH/quinone oxidoreductase, cytochrome bc1 complex, cytochrome cbb3 oxidase, and ACIII), as well as putative EET proteins, identified from the Chromatiaceae cluster genome could account for electrode-driven autotrophy under microaerobic conditions. Due to the positive potential of the electrode, CO2 fixation through the CBB cycle in Chromatiaceae must to be linked to O2 reduction in order to generate proton motive force to power reverse electron transport from the cytochrome bc1 complex through the NADH-quinone oxidoreductase complex, as in other chemolithoautotrophs (reviewed in references 43 and 63). The ACIII could take the place of the cytochrome bc1 complex, accepting electrons from a transperiplasm redox module and reducing the quinone pool. Energy conservation may occur via electron bifurcation or via proton motive force (64). The estimated ΔG′ (′ denotes standard conditions) (−nFΔE°, where n = 1 mol, F = 96,485 J, and ΔE° is the formal potential ) for reduction of oxygen (+0.8 V SHE) with an electrode poised at +0.310 V SHE is −47 kJ/mol e−, which is within the range for other organisms powering cell biosynthesis by reverse electron transport using iron as an electron donor (65). A schematic summarizing the predicted EET pathway identified from the Chromatiaceae cluster genome, and supported by proteomics and RT-PCR, is presented in Fig. 3. This proposed scheme does not explain how fixed carbon from Chromatiaceae may be distributed to other members of the biocathode consortia for biomass formation or how much total biomass could be generated. The total extractable cell biomass measured from surrogate reactors increased by an order of magnitude or more before reaching maximum current and ranged from 1.41E6 to 6.03E6 cells (see Fig. S7 and Table S9 in the supplemental material). The accumulated biomass was independent of the number of cells in the inoculum (2.0E4 versus 2.0E5 cells), indicating that the initial number of cells available to attach to the electrode surface does limit current. If all attached cells are assumed to contribute to the current, either by direct electron transfer or by driving EET through syntrophy, the rate of cell-normalized EET ranges from 0.29 to 2.62 pmol electrons h−1 cell−1. This estimate is ca. 4 to 40 times greater than the 0.075 pmol electrons h−1 cell−1 reported for M. ferrooxydans cathodes (13). This could be due to a real increase in the rate of EET for our biocathode community or to an underestimation of the number of electrode-associated cells by an order of magnitude or more due to challenges in disaggregating the biofilm for flow cytometry.
FIG 3.
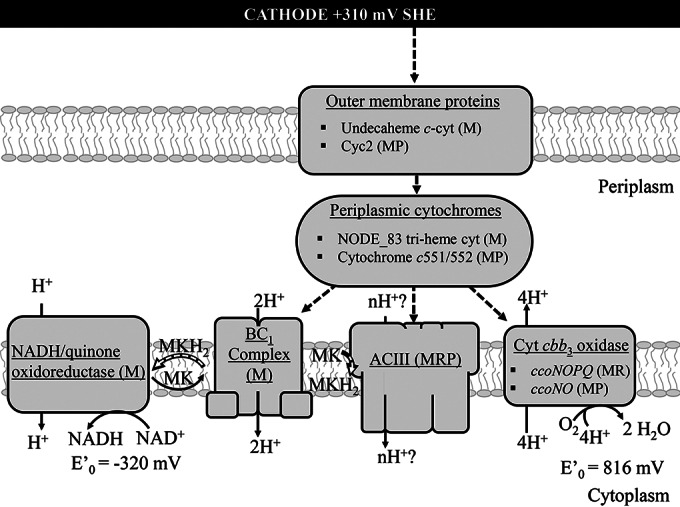
Schematic of the hypothetical electron transfer pathway between the electrode and Chromatiaceae respiratory proteins. The oxidative branch of the pathway generates energy and proton motive force through cytochrome cbb3 oxidase. The proton motive force is used to generate ATP and to power the reductive branch, which produces NADH to provide reducing equivalents for CO2 fixation. Both the cytochrome bc1 complex and the putative ACIII are capable of reducing quinones, such as menaquinone (MK), accepting electrons from cytochromes at potentials within the range expected here. Support for the presence of these pathways in the uncultivated Chromatiaceae is indicated by the following: evidence from the metagenome only (M), an RT-PCR product found (for at least one gene in a complex) (R), and/or a protein identified (for at least one gene in a complex) (P). E′0 indicates the standard reduction potential.
An alternative source of biofilm carbon and energy: CO oxidation.
CODHs have recently been found to be abundant in diverse marine bacteria (66, 67), suggesting that aerobic CO oxidation is a widespread metabolic strategy in the marine environment. CO is present in the atmosphere at 0.35 to 0.5 ppm (68) and could conceivably be available to biofilm organisms as an inorganic carbon source or a supplemental energy source. Labrenzia spp. are among the bacteria known to oxidize CO via carbon monoxide dehydrogenase to CO2 (69) and are estimated to make up ca. 5% of the biofilm composition, leading us to examine the genomic potential of the biocathode biofilm for CO oxidation. CODH genes were classified into two known forms based on sequence divergence of the large subunit CoxL: form I, known as the OMP group (from Oligotropha, Mycobacterium, and Pseudomonas), is well characterized in the classic carboxydotrophs and is able to oxidize CO; form II, known as the BMS group (from Bradyrhizobium, Mesorhizobium, and Sinorhizobium), is a putative CODH inferred from sequence homology to form I. The function of form II CoxL remains unknown, but it may preferentially oxidize alternate substrates (66). A total of nine CODH operons (coxLMS) were found in the biofilm metagenome and were distributed among the Labrenzia, Rhodospirillaceae, Rhizobiales, Parvibaculum-1, and Gammaproteobacteria-1 cluster genomes (Table 5). Of those nine, one of two form I and the form II CoxM protein from the Labrenzia cluster genome were observed by proteomics. RT-PCR of the large-subunit CoxL protein from the same operons in which CoxM was observed showed that it was also expressed. Expression of the second coxL form I operon of the Labrenzia cluster genome was not observed by either proteomics or RT-PCR. RT-PCR of genes encoding CoxL from the remaining six operons detected expression of a form II from the Parvibaculum-1 cluster genome and a form I from the Gammaproteobacteria-1 cluster genome. It is interesting that the Labrenzia, Rhodospirillaceae, and Parvibaculum-1 cluster genomes contain multiple copies or both forms of the cox operons. The presence of both forms may provide an ecological advantage under a range of CO conditions and substrates, as has been noted previously for a diversity of Labrenzia spp. (69). As noted above, genetic evidence for CO2 fixation could not be found for CODH-containing cluster genomes, and a possible role for this metabolism is discussed further below.
DISCUSSION
In this study, we used metagenomics and metaproteomics to confirm the identities of major biofilm constituents, to identify the major CO2 fixation pathway, and to provide an initial characterization of putative EET pathways of a previously described self-regenerating and self-sustaining biocathode community. The community is of low diversity, with Gammaproteobacteria estimated to make up ca. 60% of the constituents, while the remaining 35 to 40% belong mainly to the Alphaproteobacteria. This distribution is consistent with previous clone library analyses, where the majority of clones were Gammaproteobacteria, including Marinobacter and Chromatiaceae (22). Alphaproteobacteria, Planctomycetes, and Actinobacteria were also previously identified by clone library analysis; however, the last two phyla were not detected here by metagenomics, suggesting they were not essential to biocathode performance and may have been lost during subsequent transfers.
In general, the distribution of biocathode proteins among cluster genomes correlated with the predicted taxonomic distribution of the metagenome, with more abundant taxa representing a higher proportion of identified proteins. With the exception of Parvibaculum-1, cluster genomes with higher sequence coverage also had a greater number of proteins identified. For example, Labrenzia, Phaeobacter, and Kordiimonas had the highest sequence coverage among Alphaproteobacteria identified to the genus level and together represent 18% of all identified proteins. Likewise, Marinobacter and Chromatiaceae are two of the most abundant Gammaproteobacteria and made up 27% and 21% of all identified proteins, respectively. The family Alcanivoraceae was predicted to make up 33% of Gammaproteobacteria but was not previously identified by clone library analysis and accounted for only 4% of all identified proteins. This discrepancy may be due to short contig lengths, which can result in artificially high abundance counts and/or may result in undercounting Alcanivoraceae proteins, as short peptide sequences could be insufficient for MS identification.
Cluster genomes generated using metagenomics were used to address two fundamental questions currently facing biocathode technologies: can microorganisms gain energy for growth through electrode-driven autotrophy and, if so, what are the biocathode EET and CO2 fixation pathways? The proteomic and genomic observations presented here confirm that an uncharacterized member of the family Chromatiaceae expresses proteins for autotrophic CO2 fixation and could potentially use the electrode as an electron donor. Genes encoding proteins of proposed iron oxidation pathways, including Cyc2 (Cyt572) and the Zetaproteobacteria ACIII, were identified from the Chromatiaceae cluster genome, and expression was observed by proteomics or RT-PCR. Experimental evidence is still needed to confirm the involvement of ACIII in EET during iron oxidation and has thus far been inferred only from genomic and proteomic observations. The fact that a known metal reducer, G. uraniireducens, also contains a homologous ACIII but is not known to oxidize iron further confounds its role.
Key enzymes from the CBB cycle were identified only in the Chromatiaceae cluster genome. The presence of form I RubisCO suggests that these enzymes are directly involved in carbon metabolism associated with the CBB cycle (59, 70, 71). At this time, we have no direct evidence for electrode-driven growth of Chromatiaceae, as we have been unable to cultivate a representative isolate. Electrode-associated cell biomass increases over time as the magnitude of the current increases while no other electron donor is provided other than the poised electrode and no other source of carbon is intentionally provided other than CO2. The rates of cell-normalized EET are at least as high as that previously noted for M. ferrooxydans growing on a cathode (13), suggesting that the number of electrons from the electrode is sufficient for autotrophic growth. Future studies using stable-isotope labeling need to address what proportion of accumulated biomass results from CO2 fixation by Chromatiaceae and just how this fixed carbon is distributed to other biofilm constituents. Temporal analysis of biofilm development using fluorescence in situ hybridization (FISH) may also help to determine the dependence of the growth of one organism on another and may shed light on what limits biocathode current. Biocathodes are limited by oxygen diffusion to the electrode, as well as the buildup of ROS from oxygen reduction, both of which could limit biofilm growth. Due to the fact that the biocathode community is enriched from the environment and many constituents have not been characterized, we cannot rule out the possibility that degradation of reactor components (nylon screws, wire insulation, or chelating agents in mineral solution) could contribute to the biocathode carbon supply. Although degradation has not been specifically observed, at least two biocathode constituents, Kordiimonas and Alcanivoraceae, are related to known hydrocarbon-degrading bacteria with appetites for unconventional sources of carbon.
A significant role in CO2 fixation or biocathode EET could not be predicted for biocathode constituents other than Chromatiaceae using the search parameters in this study. The Marinobacter, Labrenzia, Gammaproteobacteria-2, Rhodospirillaceae, and Parvibaculum-1 cluster genomes all contained genes for multiheme c-Cyt that may participate in EET, and expression was observed by RT-PCR. Several biocathode isolates exhibited the capacity to oxidize iron in gradient tubes when small amounts of organic carbon were supplied, and Marinobacter had some capacity for electrode EET, indicating possible mixotrophy or, as others have noted, “opportunitrophy” (54). Given its estimated abundance in the biofilm and observed interaction with the electrode, it was surprising that specific EET proteins could not be prescribed for Marinobacter. Other potential Marinobacter EET pathways need to be explored, aside from those currently known for FeOB, including those containing proteins with redox cofactors other than iron, such as molybdenum and copper.
This study focused on EET and CO2 fixation; however, proteins that may be important for life at the biocathode were also identified. In general, enzymes from more abundant biofilm constituents were those involved in amino acid and nucleotide biosynthesis, carbohydrate metabolism, fatty acid metabolism, and mitigation of oxygen stress. With the exception of the Chromatiaceae, most cluster genomes contained a number of ABC or TRAP transporters and TonB receptors (see Data Set S2 in the supplemental material). This supports the idea that Chromatiaceae are the primary biofilm producers, while other constituents are most likely involved in carbon cycling. Proteins prevalent among all the cluster genomes included structural proteins for motility and biofilm formation, such as flagella, type IV pili, fimbria, and chemotaxis. Members of the order Rhodobacterales, which includes Phaeobacter, Labrenzia, and Hyphomonas spp., have been reported to be ubiquitous and dominant primary surface colonizers of biocorroding communities in temperate coastal waters (72) and deep-sea environments (23) and to be significantly abundant in acetogenic multispecies biocathodes (16). The Kordiimonas genome cluster was highly represented among the Alphaproteobacteria; however, the role of the organism within the biocathode community is also not clear. Many of the identified proteins from Kordiimonas were hypothetical due to the genome sequence only recently being made available. Few proteins were identified from cluster genomes with very low sequence coverage (i.e., Muricauda, Phyllobacteriaceae, and Hyphomonas), which would indicate a specific role in the biocathode biofilm.
An unexpected finding was that CO oxidation appeared to be active in the biocathode community and may represent a dynamic metabolic strategy for energy and carbon acquisition. We identified expression of the coxL gene from the Labrenzia, Parvibaculum-1, and Gammaproteobacteria-1 cluster genomes. While not much is known about CO oxidation by Parvibaculum, L. aggregata is categorized as a carboxydovore, where only low concentrations of CO are oxidized during mixotrophic metabolism in the presence of other organic substrates (69). While it is not immediately apparent how CO oxidation is incorporated into the carbon cycle of the biofilm, we can imagine a scenario where CO is oxidized by members of Labrenzia, Parvibaculum-1, or Gammaproteobacteria-1 to CO2 for fixation by Chromatiaceae. Such a relationship may explain why these particular biocathode constituents remain part of the consortium and may also indicate a potential target for functional engineering to modify biocathode carbon acquisition.
Conclusions.
In order to improve biocathode performance through functional engineering for MFCs, microbial electrosynthesis, or other biotechnology applications, there is a need to understand EET and energy conservation in such systems. Naturally enriched biocathode consortia operating at more positive electrode potentials and under aerobic conditions may have an advantage over homogeneous cell populations in terms of their robustness and stability under changing conditions, such as pH, temperature, and salinity, which are relevant to operating in the marine environment. In this study, metagenomics combined with metaproteomics provided a more comprehensive understanding, beyond previous 16S rRNA gene clone libraries, of the primary constituents of a self-regenerating and self-sustaining biocathode biofilm. We confirmed expression of a major autotrophic CO2 fixation pathway (the CBB cycle) from a nonphotosynthetic, uncharacterized member of the family Chromatiaceae. We have also presented proteins for putative EET pathways from the electrode to this organism that could potentially drive CO2 fixation. Thus far, efforts to cultivate Chromatiaceae off the electrode have been unsuccessful, highlighting the importance of using an omics approach to study microbial communities. Although the greatest number of proteins were identified from Marinobacter, which was also shown to engage in EET with iron and the electrode, no specific EET pathways could be identified. The metagenomic and metaproteomic analyses reported here will serve as the basis for future studies to determine the roles of other biocathode constituents and whether their relationships can be exploited to manipulate biocathode performance and metabolism.
Supplementary Material
ACKNOWLEDGMENTS
We thank the DoD High Performance Computing Modernization Program's (HPCMP) PETTT staff at the Naval Research Laboratory for assistance with software configuration. We thank Martin Wu, University of Virginia, for assistance and guidance, particularly with the AMPHORA2 analysis. We also thank Daniel R. Bond, University of Minnesota, for helpful discussions regarding biocathode electron transfer.
This work was funded by the Office of Naval Research via U.S. NRL core funds, as well as under the following award numbers (to S.M.S.-G.): N0001413WX20995, N0001414WX20485, and N0001414WX20518.
The opinions and assertions contained here are ours and are not to be construed as those of the U.S. Navy, the military service at large, or the U.S. government.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02947-14.
REFERENCES
- 1.Bond DR, Holmes DE, Tender LM, Lovley DR. 2002. Electrode-reducing microorganisms that harvest energy from marine sediments. Science 295:483–485. doi: 10.1126/science.1066771. [DOI] [PubMed] [Google Scholar]
- 2.Virdis B, Rabaey K, Rozendal RA, Yuan Z, Keller J. 2010. Simultaneous nitrification, denitrification and carbon removal in microbial fuel cells. Water Res 44:2970–2980. doi: 10.1016/j.watres.2010.02.022. [DOI] [PubMed] [Google Scholar]
- 3.Marshall CW, Ross DE, Fichot EB, Norman RS, May HD. 2012. Electrosynthesis of commodity chemicals by an autotrophic microbial community. Appl Environ Microbiol 78:8412–8420. doi: 10.1128/AEM.02401-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rabaey K, Rozendal RA. 2010. Microbial electrosynthesis—revisiting the electrical route for microbial production. Nat Rev Microbiol 8:706–716. doi: 10.1038/nrmicro2422. [DOI] [PubMed] [Google Scholar]
- 5.Lovley DR, Nevin KP. 2013. Electrobiocommodities: powering microbial production of fuels and commodity chemicals from carbon dioxide with electricity. Curr Opin Biotechnol 24:385–390. doi: 10.1016/j.copbio.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 6.Lohner ST, Deutzmann JS, Logan BE, Leigh J, Spormann AM. 2014. Hydrogenase-independent uptake and metabolism of electrons by the archaeon Methanococcus maripaludis. ISME J 8:1673–1681. doi: 10.1038/ismej.2014.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenbaum M, Aulenta F, Villano M, Angenent LT. 2011. Cathodes as electron donors for microbial metabolism: which extracellular electron transfer mechanisms are involved? Bioresour Technol 102:324–333. doi: 10.1016/j.biortech.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 8.Strycharz SM, Glaven RH, Coppi MV, Gannon SM, Perpetua LA, Liu A, Nevin KP, Lovley DR. 2011. Gene expression and deletion analysis of mechanisms for electron transfer from electrodes to Geobacter sulfurreducens. Bioelectrochemistry 80:142–150. doi: 10.1016/j.bioelechem.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 9.Ross DE, Flynn JM, Baron DB, Gralnick JA, Bond DR. 2011. Towards electrosynthesis in shewanella: energetics of reversing the mtr pathway for reductive metabolism. PLoS One 6:e16649. doi: 10.1371/journal.pone.0016649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strycharz-Glaven SM, Snider RM, Guiseppi-Elie A, Tender LM. 2011. On the electrical conductivity of microbial nanowires and biofilms. Energy Environ Sci 4:4366–4379. doi: 10.1039/c1ee01753e. [DOI] [Google Scholar]
- 11.Snider RM, Strycharz-Glaven SM, Tsoi SD, Erickson JS, Tender LM. 2012. Long-range electron transport in Geobacter sulfurreducens biofilms is redox gradient-driven. Proc Natl Acad Sci U S A 109:15467–15472. doi: 10.1073/pnas.1209829109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El-Naggar MY, Wanger G, Leung KM, Yuzvinsky TD, Southam G, Yang J, Lau WM, Nealson KH, Gorby YA. 2010. Electrical transport along bacterial nanowires from Shewanella oneidensis MR-1. Proc Natl Acad Sci U S A 107:18127–18131. doi: 10.1073/pnas.1004880107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Summers ZM, Gralnick JA, Bond DR. 2013. Cultivation of an obligate Fe(II)-oxidizing lithoautotrophic bacterium using electrodes. mBio 4:e00420-12. doi: 10.1128/mBio.00420-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bose A, Gardel EJ, Vidoudez C, Parra EA, Girguis PR. 2014. Electron uptake by iron-oxidizing phototrophic bacteria. Nat Commun 5:3391. doi: 10.1038/ncomms4391. [DOI] [PubMed] [Google Scholar]
- 15.Huang L, Regan JM, Quan X. 2011. Electron transfer mechanisms, new applications, and performance of biocathode microbial fuel cells. Bioresour Technol 102:316–323. doi: 10.1016/j.biortech.2010.06.096. [DOI] [PubMed] [Google Scholar]
- 16.Marshall CW, Ross DE, Fichot EB, Norman RS, May HD. 2013. Long-term operation of microbial electrosynthesis systems improves acetate production by autotrophic microbiomes. Environ Sci Technol 47:6023–6029. doi: 10.1021/es400341b. [DOI] [PubMed] [Google Scholar]
- 17.Orphan VJ. 2009. Methods for unveiling cryptic microbial partnerships in nature. Curr Opin Microbiol 12:231–237. doi: 10.1016/j.mib.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 18.Wintermute EH, Silver PA. 2010. Dynamics in the mixed microbial concourse. Genes Dev 24:2603–2614. doi: 10.1101/gad.1985210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams TJ, Cavicchioli R. 2014. Marine metaproteomics: deciphering the microbial metabolic food web. Trends Microbiol 22:248–260. doi: 10.1016/j.tim.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 20.Hettich RL, Pan CL, Chourey K, Giannone RJ. 2013. Metaproteomics: harnessing the power of high performance mass spectrometry to identify the suite of proteins that control metabolic activities in microbial communities. Anal Chem 85:4203–4214. doi: 10.1021/ac303053e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pereira-Medrano AG, Knighton M, Fowler GJ, Ler ZY, Pham TK, Ow SY, Free A, Ward B, Wright PC. 2013. Quantitative proteomic analysis of the exoelectrogenic bacterium Arcobacter butzleri ED-1 reveals increased abundance of a flagellin protein under anaerobic growth on an insoluble electrode. J Proteomics 78:197–210. doi: 10.1016/j.jprot.2012.09.039. [DOI] [PubMed] [Google Scholar]
- 22.Strycharz-Glaven SM, Glaven RH, Wang Z, Zhou J, Vora GJ, Tender LM. 2013. Electrochemical investigation of a microbial solar cell reveals a nonphotosynthetic biocathode catalyst. Appl Environ Microbiol 79:3933–3942. doi: 10.1128/AEM.00431-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edwards KJ, Rogers DR, Wirsen CO, McCollom TM. 2003. Isolation and characterization of novel psychrophilic, neutrophilic, Fe-oxidizing, chemolithoautotrophic alpha- and, gamma-Proteobacteria from the deep sea. Appl Environ Microbiol 69:2906–2913. doi: 10.1128/AEM.69.5.2906-2913.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cox MP, Peterson DA, Biggs PJ. 2010. SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics 11:485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noguchi H, Park J, Takagi T. 2006. MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res 34:5623–5630. doi: 10.1093/nar/gkl723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marchler-Bauer A, Lu SN, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke ZX, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang DC, Zhang NG, Zheng CJ, Bryant SH. 2011. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res 39:D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu ST, Zhu ZW, Fu LM, Niu BF, Li WZ. 2011. WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genomics 12:444. doi: 10.1186/1471-2164-12-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanehisa M, Goto S, Kawashima S, Nakaya A. 2002. The KEGG databases at GenomeNet. Nucleic Acids Res 30:42–46. doi: 10.1093/nar/30.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu M, Scott AJ. 2012. Phylogenomic analysis of bacterial and archaeal sequences with AMPHORA2. Bioinformatics 28:1033–1034. doi: 10.1093/bioinformatics/bts079. [DOI] [PubMed] [Google Scholar]
- 31.Liu B, Gibbons T, Ghodsi M, Treangen T, Pop M. 2011. Accurate and fast estimation of taxonomic profiles from metagenomic shotgun sequences. BMC Genomics 12(Suppl 2):S4. doi: 10.1186/1471-2164-12-S2-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strous M, Kraft B, Bisdorf R, Tegetmeyer HE. 2012. The binning of metagenomic contigs for microbial physiology of mixed cultures. Front Microbiol 3:410. doi: 10.3389/fmicb.2012.00410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dupont CL, Rusch DB, Yooseph S, Lombardo MJ, Richter RA, Valas R, Novotny M, Yee-Greenbaum J, Selengut JD, Haft DH, Halpern AL, Lasken RS, Nealson K, Friedman R, Venter JC. 2012. Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J 6:1186–1199. doi: 10.1038/ismej.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishii S, Suzuki S, Norden-Krichmar TM, Tenney A, Chain PS, Scholz MB, Nealson KH, Bretschger O. 2013. A novel metatranscriptomic approach to identify gene expression dynamics during extracellular electron transfer. Nat Commun 4:1601. doi: 10.1038/ncomms2615. [DOI] [PubMed] [Google Scholar]
- 35.Finn RD, Clements J, Eddy SR. 2011. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Juncker AS, Willenbrock H, Von Heijne G, Brunak S, Nielsen H, Krogh A. 2003. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci 12:1652–1662. doi: 10.1110/ps.0303703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tabb DL, McDonald WH, Yates JR. 2002. DTASelect and contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res 1:21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leary DH, Hervey WJ IV, Deschamps JR, Kusterbeck AW, Vora GJ. 2013. Which metaproteome? The impact of protein extraction bias on metaproteomic analyses. Mol Cell Probes 27:193–199. doi: 10.1016/j.mcp.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 39.Leary DH, Hervey WJ IV, Li RW, Deschamps JR, Kusterbeck AW, Vora GJ. 2012. Method development for metaproteomic analyses of marine biofilms. Anal Chem 84:4006–4013. doi: 10.1021/ac203315n. [DOI] [PubMed] [Google Scholar]
- 40.Cury JA, Koo H. 2007. Extraction and purification of total RNA from Streptococcus mutans biofilms. Anal Biochem 365:208–214. doi: 10.1016/j.ab.2007.03.021. [DOI] [PubMed] [Google Scholar]
- 41.Vizcaino JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, Dianes JA, Sun Z, Farrah T, Bandeira N, Binz PA, Xenarios I, Eisenacher M, Mayer G, Gatto L, Campos A, Chalkley RJ, Kraus HJ, Albar JP, Martinez-Bartolome S, Apweiler R, Omenn GS, Martens L, Jones AR, Hermjakob H. 2014. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol 32:223–226. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH. 2013. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31:533–538. doi: 10.1038/nbt.2579. [DOI] [PubMed] [Google Scholar]
- 43.Bird LJ, Bonnefoy V, Newman DK. 2011. Bioenergetic challenges of microbial iron metabolisms. Trends Microbiol 19:330–340. doi: 10.1016/j.tim.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 44.Tank M, Thiel V, Imhoff JF. 2009. Phylogenetic relationship of phototrophic purple sulfur bacteria according to pufL and pufM genes. Int Microbiol 12:175–185. doi: 10.2436/20.1501.01.96. [DOI] [PubMed] [Google Scholar]
- 45.Singer E, Emerson D, Webb EA, Barco RA, Kuenen JG, Nelson WC, Chan CS, Comolli LR, Ferriera S, Johnson J, Heidelberg JF, Edwards KJ. 2011. Mariprofundus ferrooxydans PV-1 the first genome of a marine Fe(II) oxidizing Zetaproteobacterium. PLoS One 6:e25386. doi: 10.1371/journal.pone.0025386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singer E, Heidelberg JF, Dhillon A, Edwards KJ. 2013. Metagenomic insights into the dominant Fe(II) oxidizing Zetaproteobacteria from an iron mat at Lo'ihi, Hawai'i. Front Microbiol 4:52. doi: 10.3389/fmicb.2013.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jeans C, Singer SW, Chan CS, Verberkmoes NC, Shah M, Hettich RL, Banfield JF, Thelen MP. 2008. Cytochrome 572 is a conspicuous membrane protein with iron oxidation activity purified directly from a natural acidophilic microbial community. ISME J 2:542–550. doi: 10.1038/ismej.2008.17. [DOI] [PubMed] [Google Scholar]
- 48.Valdes J, Pedroso I, Quatrini R, Dodson RJ, Tettelin H, Blake R II, Eisen JA, Holmes DS. 2008. Acidithiobacillus ferrooxidans metabolism: from genome sequence to industrial applications. BMC Genomics 9:597. doi: 10.1186/1471-2164-9-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Richardson DJ, Butt JN, Fredrickson JK, Zachara JM, Shi L, Edwards MJ, White G, Baiden N, Gates AJ, Marritt SJ, Clarke TA. 2012. The porin-cytochrome model for microbe-to-mineral electron transfer. Mol Microbiol 85:201–212. doi: 10.1111/j.1365-2958.2012.08088.x. [DOI] [PubMed] [Google Scholar]
- 50.Liu Y, Wang Z, Liu J, Levar C, Edwards MJ, Babauta JT, Kennedy DW, Shi Z, Beyenal H, Bond DR, Clarke TA, Butt JN, Richardson DJ, Rosso KM, Zachara JM, Fredrickson JK, Shi L. 19 August 2014. A trans-outer membrane porin-cytochrome protein complex for extracellular electron transfer by Geobactersulfurreducens PCA. Environ Microbiol Rep. doi: 10.1111/1758-2229.12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pitcher RS, Watmough NJ. 2004. The bacterial cytochrome cbb3 oxidases. Biochim Biophys Acta 1655:388–399. doi: 10.1016/j.bbabio.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 52.Becker CF, Watmough NJ, Elliott SJ. 2009. Electrochemical evidence for multiple peroxidatic heme states of the diheme cytochrome c peroxidase of Pseudomonas aeruginosa. Biochemistry 48:87–95. doi: 10.1021/bi801699m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Waite J. 2012. Characterization of cytochrome c peroxidase of Marinobacter aquaeolei. University of Southern California, Los Angeles, CA. [Google Scholar]
- 54.Singer E, Webb EA, Nelson WC, Heidelberg JF, Ivanova N, Pati A, Edwards KJ. 2011. Genomic potential of Marinobacter aquaeolei, a biogeochemical “opportunitroph”. Appl Environ Microbiol 77:2763–2771. doi: 10.1128/AEM.01866-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coursolle D, Gralnick JA. 2012. Reconstruction of extracellular respiratory pathways for iron(III) reduction in Shewanella oneidensis strain MR-1. Front Microbiol 3:56. doi: 10.3389/fmicb.2012.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu J, Wang Z, Belchik SM, Edwards MJ, Liu C, Kennedy DW, Merkley ED, Lipton MS, Butt JN, Richardson DJ, Zachara JM, Fredrickson JK, Rosso KM, Shi L. 2012. Identification and characterization of MtoA: a decaheme c-type cytochrome of the neutrophilic Fe(II)-oxidizing bacterium Sideroxydans lithotrophicus ES-1. Front Microbiol 3:37. doi: 10.3389/fmicb.2012.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang F, Bogdanov B, Strittmatter EF, Vilkov AN, Gritsenko M, Shi L, Elias DA, Ni SS, Romine M, Pasa-Tolic L, Lipton MS, Smith RD. 2005. Characterization of purified c-type heme-containing peptides and identification of c-type heme-attachment sites in Shewanella oneidenis cytochromes using mass spectrometry. J Proteome Res 4:846–854. doi: 10.1021/pr0497475. [DOI] [PubMed] [Google Scholar]
- 58.Berg IA, Kockelkorn D, Ramos-Vera WH, Say RF, Zarzycki J, Fuchs G. 2010. Autotrophic carbon fixation in biology: pathways, rules, and speculations, p 33–53. In Aresta M. (ed), Carbon dioxide as chemical feedstock. Wiley-VCH, Weinheim, Germany. [Google Scholar]
- 59.Tabita FR, Hanson TE, Li HY, Satagopan S, Singh J, Chan S. 2007. Function, structure, and evolution of the RubisCO-like proteins and their RubisCO homologs. Microbiol Mol Biol Rev 71:576. doi: 10.1128/MMBR.00015-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Singh J, Tabita FR. 2010. Roles of RubisCO and the RubisCO-like protein in 5-methylthioadenosine metabolism in the nonsulfur purple bacterium Rhodospirillum rubrum. J Bacteriol 192:1324–1331. doi: 10.1128/JB.01442-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ashida H, Saito Y, Kojima C, Kobayashi K, Ogasawara N, Yokota A. 2003. A functional link between RuBisCO-like protein of Bacillus and photosynthetic RuBisCO. Science 302:286–290. doi: 10.1126/science.1086997. [DOI] [PubMed] [Google Scholar]
- 62.Hanson TE, Tabita FR. 2001. A ribulose-1,5-bisphosphate carboxylase/oxygenase (RubisCO)-like protein from Chlorobium tepidum that is involved with sulfur metabolism and the response to oxidative stress. Proc Natl Acad Sci U S A 98:4397–4402. doi: 10.1073/pnas.081610398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Simon J, Klotz MG. 2013. Diversity and evolution of bioenergetic systems involved in microbial nitrogen compound transformations. Biochim Biophys Acta 1827:114–135. doi: 10.1016/j.bbabio.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 64.Refojo PN, Teixeira M, Pereira MM. 2012. The alternative complex III: properties and possible mechanisms for electron transfer and energy conservation. Biochim Biophys Acta 1817:1852–1859. doi: 10.1016/j.bbabio.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 65.Ferguson SJ, Ingledew WJ. 2008. Energetic problems faced by micro-organisms growing or surviving on parsimonious energy sources and at acidic pH. I. Acidithiobacillus ferrooxidans as a paradigm. Biochim Biophys Acta 1777:1471–1479. doi: 10.1016/j.bbabio.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 66.King GM, Weber CF. 2007. Distribution, diversity and ecology of aerobic CO-oxidizing bacteria. Nat Rev Microbiol 5:107–118. doi: 10.1038/nrmicro1595. [DOI] [PubMed] [Google Scholar]
- 67.Cunliffe M. 2011. Correlating carbon monoxide oxidation with cox genes in the abundant Marine Roseobacter Clade. ISME J 5:685–691. doi: 10.1038/ismej.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Crutzen PJ, Gidel LT. 1983. A two-dimensional photochemical model of the atmosphere. 2. The tropospheric budgets of the anthropogenic chlorocarbons Co, Ch4, Ch3cl and the effect of various nox sources on tropospheric ozone. J Geophys Res Ocean Atmos 88:6641–6661. [Google Scholar]
- 69.Weber CF, King GM. 2007. Physiological, ecological, and phylogenetic characterization of Stappia, a marine CO-oxidizing bacterial genus. Appl Environ Microbiol 73:1266–1276. doi: 10.1128/AEM.01724-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Badger MR, Bek EJ. 2008. Multiple Rubisco forms in proteobacteria: their functional significance in relation to CO(2) acquisition by the CBB cycle. J Exp Bot 59:1525–1541. doi: 10.1093/jxb/erm297. [DOI] [PubMed] [Google Scholar]
- 71.Tabita FR, Satagopan S, Hanson TE, Kreel NE, Scott SS. 2008. Distinct form I, II, III, and IV Rubisco proteins from the three kingdoms of life provide clues about Rubisco evolution and structure/function relationships. J Exp Bot 59:1515–1524. doi: 10.1093/jxb/erm361. [DOI] [PubMed] [Google Scholar]
- 72.Dang H, Li T, Chen M, Huang G. 2008. Cross-ocean distribution of Rhodobacterales bacteria as primary surface colonizers in temperate coastal marine waters. Appl Environ Microbiol 74:52–60. doi: 10.1128/AEM.01400-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.