

Abstract
Nanoparticulate (NP) drug carrier systems are attractive vehicles for selective drug delivery to solid tumors. Ideally, NPs should evade clearance by the reticuloendothelial system while maintaining the ability to interact with tumor cells and facilitate cellular uptake. Great effort has been made to fulfill these design criteria, yielding various types of functionalized NPs. Another important consideration in NP design is the physical and functional stability during circulation, which, if ignored, can significantly undermine the promise of intelligently designed NP drug carriers. This commentary reviews several NP examples with stability issues and their consequences, ending in a discussion of experimental methods for reliable prediction of NP stability.
Keywords: Nanomedicine, Nanoparticles, Drug delivery, Stability, Sensitivity
Introduction
Nanoparticulate (NP) systems represented by liposomes, polymeric micelles, and polymeric nanospheres (NS) have been widely pursued as a carrier of chemotherapeutic drugs. While low molecular-weight compounds diffuse into both normal and tumor tissues, NPs cannot pass the capillaries in normal tissues but enter tumors via relatively leaky vasculature of tumor tissues (Kratz 2008). Moreover, due to the relatively defective lymphatic drainage system of tumors, nanomedicines reaching tumors cannot be easily removed (Kratz 2008). These unique features of vascular and lymphatic structures of many solid tumors, called the enhanced permeability and retention (EPR) effect (Matsumura and Maeda 1986), give an opportunity for NPs to access tumors selectively. In addition, NPs have large surface area per mass, which may be functionalized to enhance interactions with tumors. These features make NPs an attractive carrier to increase drug delivery to solid tumors and reduce non-specific toxicity to healthy tissues.
For effective drug delivery to tumors, NPs should evade the surveillance of the reticuloendothelial system (RES) during circulation. Once NPs reach the tumors, they should be able to interact with tumor cells for efficient entry into the cells. Often these requirements are difficult to meet simultaneously. For example, an approach to prolong NP circulation may interfere with NP-cell interactions (Hatakeyama et al. 2011), whereas functional ligands attached to NP surface to enhance NP-cell interactions may negatively affect long-term circulation of NPs (Gu et al. 2008). To meet these conflicting requirements, NPs are designed in an increasingly complex manner so that they present both properties simultaneously or one after the other on demand. On the other hand, one of the often ignored problems in developing new NP systems is that such complexity can compromise its stability in size and/or drug encapsulation, another critical feature of a NP as a drug delivery system.
Physical (size) and functional (drug retention) stability are the most fundamental attributes of NPs, responsible for their selective accumulation in tumors and alteration of pharmacokinetics of the drug payload. Despite the significance, NP stability has largely been underestimated and/or improperly evaluated during development. The ever increasing gap between the complexity of NP technology and modest increase of therapeutic effects is at least partly attributable to the oversight of NP stability. Without an active effort to accommodate this critical requirement in design of new NP systems, it will be difficult to develop a product with clinical impact. In an attempt to increase the recognition of its significance, we provide an overview of stability issues observed in selected NP systems and discuss the needs and potential ways to diagnose similar issues in the early stage of NP development.
Liposomes
Liposomes are spherical vesicles consisting of one or more concentric lipid bilayers separated by aqueous phases. Liposomal formulations containing doxorubicin (Doxil®, Caelyx®) were approved by the FDA for treatment of metastatic breast cancer, ovarian cancer, and Kaposi sarcoma. Liposomes are generally known to be stable in blood (Gregoriadis and Davis 1979), but an effort to make them stimulisensitive may reduce stability of the system. For example, de Smet et al. studied in vivo drug release from temperaturesensitive liposomes (TSL) based on dipalmitoylphosphatidylcholine, which contained doxorubicin as a model drug and a magnetic resonance imaging agent for an imaging-guided drug delivery (de Smet et al. 2011). For differential determination of the blood kinetics of drug and carrier, TSLs were labeled with indium-111 (111In), and doxorubicin level and radioactivity of the blood were separately monitored. Interestingly, both the 111In-TSLs and doxorubicin levels in blood showed a biexponential decrease yet with distinct profiles, where doxorubicin was cleared faster than TSLs (Fig. 1). This indicates that doxorubicin leached out of TSLs upon injection, probably due to the thermal sensitivity leading to instability, and was cleared as free doxorubicin.
Fig. 1.
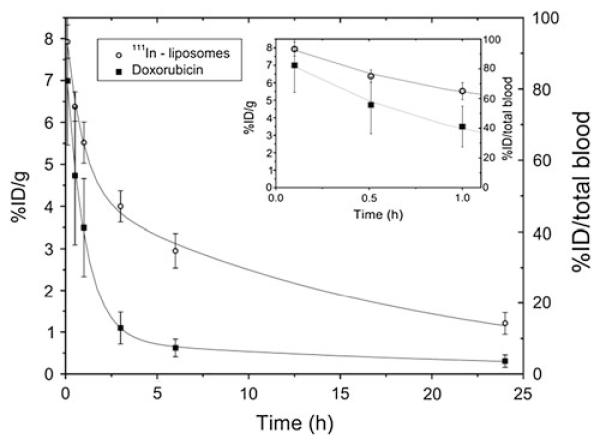
Blood kinetics of 111In labeled TSLs containing doxorubicin. The percentage of the injected dose (%ID) is plotted per gram blood (left axis) and for the total blood (right axis). Reprinted with permission from (de Smet et al. 2011). Copyright © 2011 Elsevier
The balance between stability and sensitivity may be achieved with judicious choice of liposomal components. A TSL was prepared with poly[2-(2-ethoxy)ethoxyethyl vinyl ether]-b-octadecyl vinyl ether (poly(EOEOVE)-OD4) as a thermosensitive component (Kono et al. 2010). The polymer amphiphile underwent a transition from a hydrophilic coil to a hydrophobic globule at temperatures above 40 °C, leading to destabilization of liposomal membrane followed by drug release. In achieving sharp transition in liposomal integrity, it was critical to include PEG-lipid and prevent premature interaction of partially dehydrated poly(EOEOVE) chains with liposomal membrane. Consequently, doxorubicin-loaded in the poly(EOEOVE)-based TSL showed a significant contrast in tumor growth between heated and non-heated animals (Kono et al. 2010).
Polymeric nanospheres (NS)
Polymeric NS refer to a spherical matrix of hydrophobic polymers, in which a drug is molecularly dispersed or embedded as groups of molecules. Poly(lactic-co-glycolic acid) (PLGA) is widely used due to its biocompatibility and biodegradability. Its utility in sustained drug delivery is well established with various dosage forms; however, drug retention in NS has been challenging. Xu et al. used PLGA NS with physically entrapped Nile red, a hydrophobic fluorescent dye, to study cellular uptake of the NS via fluorescent microscopy (Xu et al. 2009). While alternative techniques suggested the lack of cellular uptake of PLGA NS, Nile red fluorescence appeared in the cytoplasm in less than 30 min. The rapid increase in intracellular fluorescence was attributed to extracellular dye release followed by uptake of free dyes and/or direct dye transfer to contacting cells. Due to the premature drug release, NS-encapsulated paclitaxel (PTX) showed similar cytotoxicity as free PTX, even when NS was separated from cells via Transwells. Of note, such a rapid drug release was not seen when phosphate buffered saline (PBS) was used as release medium but evident in the presence of amphiphilic cellular components such as lipids or proteins.
Poor drug retention was also observed in other types of PLGA NS. Amoozgar et al. developed a pH-sensitive NS with a covalent conjugate of PLGA and a low molecular weight chitosan (LMWC), where PLGA formed a core and LWMC a surface layer (Amoozgar et al. 2012). The pH-sensitivity of LMWC layer provided the NS with the ability to deliver a drug in a tumor-specific manner by serving as a stealth layer in neutral pH and facilitating cellular uptake in weakly acidic pH of tumor stroma. Due to the increased cellular uptake at acidic pH, it was expected that PTX delivered by PLGA-LMWC NS (PTX/PLGA-LMWC NS) would be more cytotoxic than that by pH-insensitive PLGA NS (PTX/PLGA NS) at pH 6.2. The expected difference was observed when cells were allowed to contact the NS for 3 h and then grow for additional 3 days under the influence of NS that established interaction with the cells in the first 3 h (Fig. 2). However, when cells were exposed to the drug sources for 72 h, cytotoxic effects of PTX/PLGA-LMWC NP and PTX/PLGA NP were not different from each other and that of free PTX. This result suggests that drug release from NS was substantial in 72 h and over-whelmed the effect of preferential uptake of the carrier.
Fig. 2.

Viability of SKOV-3 cells exposed to PTX or PTX/NS at different pHs for 3 or 72 h. Reprinted with permission from (Amoozgar et al. 2012). Copyright © 2012 American Chemical Society
A similar phenomenon was observed with PLGA NS modified with a cell-penetrating peptide, TAT (Gullotti and Yeo 2012). TAT was added to enhance cellular uptake of the NS, thereby facilitating drug delivery to multidrug resistant cells. As expected, PLGA-TAT NS were readily taken up by SKOV-3 and NCI/ADR-RES ovarian cancer cells in 3 h, in contrast to unmodified PLGA NS barely taken up by the cells. Due to the increased cellular uptake of PLGA-TAT NS, it was expected that PTX delivered by the PLGA-TAT NS would show greater cytotoxicity in drug resistant NCI/ADR-RES cells by bypassing drug efflux mechanisms. However, no difference in cytotoxicity was observed between PTX/PLGA-TAT NS and PTX/PLGA NS. This result was attributed to premature extracellular PTX release from PLGA-TAT NS, estimated by the drug release kinetics study performed in PBS with a surfactant. This result demonstrates that enhancing cellular uptake of NPs does not necessarily translate to a greater therapeutic effect unless NPs retain the payload until they reach target locations.
Polymeric micelles
Polymeric micelles are self-assemblies of amphiphilic block-copolymers, consisting of a hydrophilic polymer block (typically polyethylene glycol, PEG) and a hydrophobic polymer block (e.g., polycaprolactone, polypeptide, or polylactide), where the hydrophobic part forms a core away from aqueous environment encapsulating hydrophobic payloads. Excessive dilution or hydrophobic environment is a common cause of micelle destabilization. Savic et al. studied the stability of poly(caprolactone)-b-poly-ethylene glycol (PCL21-b-PEG45) micelles in media and in cells using fluorogenic dye fluorescein-5-carbonyl azide diacetate covalently attached to the PCL block (Savić et al. 2006). For 2 days at 37 °C, the micelles remained stable in distilled water and showed 18 % loss of micelle integrity in PBS. On the other hand, when micelles were incubated in 100 % serum, complete dissociation of micelles occurred in 1 h, estimated by an increase in fluorescent intensity of the released dye. This result highlights the role of serum proteins in destabilization of polymeric micelles, which are likely overlooked in studies performed with buffers or distilled water as a dispersion medium.
A relevant observation was made with poly(benzyl aspartic acid)-PEG micelles, which encapsulate camptothecin (CPT) in the core. CPT loaded in the micelles and free CPT were compared with respect to drug stability in media containing serum and serum albumin of various species (Opanasopit et al. 2005). Although CPT in micelles was more stable than free CPT, those incubated in serum or human serum albumin underwent much faster hydrolysis than in PBS, attributable to preferential binding of serum proteins to carboxylate form of CPT, which shifts the lactone (active form)-carboxylate (inactive form) equilibrium to an unfavorable direction. This result indicates that serum proteins had direct influence on CPT encapsulated in polymeric micelles; i.e., the micelles did not completely isolate the drug from the serum-containing medium. Interestingly, such instability of micellar CPT was not observed with mouse albumin. This suggests that in vivo studies using a mouse model may overestimate the effectiveness of CPT loaded in the polymeric micelles and have little values in predicting its therapeutic effects in human.
The instability of polymeric micelles is reflected by differential biodistribution of drug and micellar polymers and rapid elimination of polymers, which indicate premature drug release and micelle dissociation, respectively. Burt et al. (1999) used a block co-polymer of d, l-lactic acid and methoxypolyethylene glycol (PDLLA-MePEG) to form polymeric micelles encapsulating PTX. PTX and the polymer were labeled with 3H and 14C, respectively, and their biodistributions were evaluated in rats. 1 h after intravenous injection, the kidneys and bladder showed greater 14C radioactivity than in blood plasma, indicating dissociation of micellar structure at this point (Burt et al. 1999). In contrast, 3H radioactivity was more evenly distributed in different organs than with 14C (Burt et al. 1999), mirroring the biodistribution of released and then protein-bound PTX. Consequently, PTX (3H) and PDLLA-MePEG (14C) showed distinct elimination kinetics: While >95 % of the polymer portion of the micelles was eliminated in 15 h, PTX levels in tissues slowly decreased over 12 h after injection. These results indicate rapid dissociation of PTX from micelles in blood.
The instability of a similar micelle system (PDLLA-PEG) was demonstrated using Forster resonance energy transfer (FRET) imaging (Chen et al. 2008a, b). DiIC18 and DiOC18, a hydrophobic FRET pair, were employed to mimic the entrapment of a hydrophobic drug in the micelle. The FRET signal was observed when the micelles were intact, and DiIC18 and DiOC18 were within proximity to each other, whereas the loss of FRET signal indicated dissociation of micellar structure leading to dye release (Fig. 3). When PDLLA-PEG micelles containing DilC18 and DiOC18 were injected in the bloodstream of a live animal, a decrease of FRET signal was observed within 15 min of intravenous injection indicating instability of micelles in the bloodstream (Chen et al. 2008a). An in vitro FRET analysis of the different components of the blood revealed that α- and β-globulins were two major factors in the fast destabilization of micelles (Chen et al. 2008a).
Fig. 3.

Fluorescence spectra of PEG-PDLLA FRET micelles diluted by ten times with a water and b acetone. Insets show diagram of a FRET micelle prepared with 0.75 %DiO and 0.75 %DiI at 2 mg/mL polymer concentration. c Fluorescence spectra of FRET micelles in blood vessels during 3 h post IV injection. Reprinted with permission from (Chen et al. 2008b). Copyright © 2008 American Chemical Society
pH-sensitive polymeric micelles
Environment-sensitive NPs is a reasonable strategy to satisfy conflicting requirements for ideal NPs, but it can be challenging to find a balance between stability and sensitivity. Bae et al. developed a pH-sensitive micelle system for tumor-specific release of an anticancer drug (Lee et al. 2003b). The micelle was prepared with poly(l-histidine)-poly(ethylene glycol) (polyHis-b-PEG) (PolyHis at 3 or 5 kDa, and PEG at 2 kDa). The imidazole group of His ionizes at a pH less than 7 (Lee et al. 2003b). This allows for polyHis to possess hydrophobic properties at pH greater than 7 and hydrophilic properties at pH less than 7. Therefore, at pH above 7, polyHis-b-PEG diblock copolymer forms a micelle that encapsulates a hydrophobic drug. At pH below 7, this micelle is destroyed and releases the drug due to ionization of polyHis. This pH sensitivity helps achieve tumor-specific drug release in slightly acidic tumor microenvironments (pH 5.8–7.3) (Gerweck and Seetharaman 1996). Micellar stability was evaluated by measuring turbidity change at different pHs. PolyHis5K-b-PEG2K was found stable in pH 8 over 2 days, while polyHis3K-b-PEG2K showed instability over this period of time. The critical micelle concentrations (CMC) of polyHis5K-b-PEG2K and polyHis3K-b-PEG2K at pH 8 were determined to be 2.3 and 62 μg/mL, respectively, reflecting greater stability of the former. The authors suggest that the lower stability of polyHis3K-b-PEG2K is likely due to the shorter and, thus, less hydrophobic polyHis block length. At pH below 7.2, CMC of polyHis5K-b-PEG2K increased, due to the protonation of the imidazole group, i.e., reduction in hydrophobicity of polyHis, reflected by an increase in transmittance of the micelle solution. The acid-labile micelles based on pH-sensitive block-co-polymers have become a popular approach for drug delivery to the tumor extracellular matrix (ECM). However, the authors report that this formulation was relatively unstable at pH7.4, evidenced by an increase in light transmittance through the micelle solution, decrease in fluorescence intensity of the encapsulated pyrene, and particle size reduction (Lee et al. 2003b).
To address the instability issue in the polyHis-b-PEG micelle system, pH-insensitive PLLA-b-PEG-folate was blended with polyHis5K-b-PEG2K to form “mixed” micelles. Blending PLLA-b-PEG-folate formed micelles with greater stability due to the consistent hydrophobicity of PLLA block in the micelle core (Lee et al. 2003a). The pH for micelle dissociation was lowered by increasing the ratio of PLLA-b-PEG to polyHis-b-PEG in polymer blends (Lee et al. 2003a). One disadvantage of this approach was that >40 % PLLA-b-PEG removed pH-sensitivity of the micelles, limiting further reduction of triggering pH, which would have been desirable for endosomal drug release (Lee et al. 2003a). This problem was addressed by replacing polyHis-b-PEG with a copolymer of l-phenylalanine (Phe) and l-His N-carboxyl-anhydride (poly(His-co-Phe)-b-PEG) (Kim et al. 2008, 2009). The inclusion of Phe lowered the pKa of the co-polymer, enabling flexible control of the destabilization pH while maintaining the stability at pH 7.4.
Drug-polymer conjugates
Zhou et al. utilized cationic polymer poly(l-lysine) (PLL) as a carrier of anti-cancer drugs for its ability to localize in the nucleus (Zhou et al. 2009). To overcome the toxicity of PLL due to the cationic nature, its primary amines were amidized as acid labile β-carboxylic amines (PLL/amide), which shielded the primary amines during circulation but underwent hydrolysis at lysosomal pH to restore amines for nuclear translocation. Therefore, the PLL/amide showed low toxicity, minimal cellular interactions, and low hemolytic activity at neutral pH, while maintaining the ability to enter the nucleus once in the cells. Anticancer drug CPT was incorporated to PLL via disulfide bond, cleavable in the reducing intracellular environment. In addition, folate was conjugated for cellular uptake of the conjugate. Due to the folate and intracellularly released CPT, the drug-PLL/amide conjugate showed IC50 of 3.5 and 0.41 μg/mL for SKOV-3 and MCF-7 cells, respectively, while free CPT showed limited cytotoxicity. The enhanced toxicity of CPT released from FA-PLL/DCA-CPT is attributable to the enhanced nuclear drug delivery via the drug carrier. On the other hand, it was noted that some of CPT might have been prematurely released in the cytosol during transit to the nucleus due to the short halflife of 3-thiol-propionate-based disulfide bond, which remains to be improved for more efficient nuclear delivery of CPT (Zhou et al. 2009).
Stability of commercial NP products
Clinical pharmacokinetics studies of commercial NP products suggest that their stability in blood remains to be improved. For example, Abraxane is a surfactant-free NP formulation of PTX, where PTX is bound to human serum albumin via hydrophobic interactions. Abraxane is present as stable NPs with a diameter of 130 nm in saline but quickly dissolves into albumin-PTX complexes in blood (Desai 2008). Due to the lack of toxic surfactants, Abraxane can deliver a higher dose of taxane than surfactant-based formulations such as Taxol without toxicity related to vehicles (Nyman et al. 2005; Miele et al. 2009; Harries et al. 2005). However, since PTX circulates as albumin-PTX complexes with a size identical to endogenous albumin molecules (Desai 2008), there is no substantial improvement in pharmacokinetic parameters such as increase in circulation half-life (t1/2) or area under the curve (AUC) (t1/2: 14.6 h; AUC: 17.6 μg h/mL) over Taxol (t1/2: 13.3 h; AUC: 23.2 μg h/mL) (Hamaguchi et al. 2007; Ernsting et al. 2012). Similarly, Genexol, a polymeric micelle formulation of PTX, shows no improvement in t1/2 (11.4 h) or AUC (11.6 μg h/mL) compared to Taxol (Hamaguchi et al. 2007), suggesting that PTX quickly partitions out of micelles in blood and binds to serum proteins (Ernsting et al. 2012). On the other hand, newer products evaluated in early clinical (NK105, polymeric micelles) or preclinical studies (Cellax, PTX-polymer conjugate NPs) show greater t1/2 and/or AUC (Hamaguchi et al. 2007; Ernsting et al. 2012), providing better effects on primary tumors or metastasis in animal models (Ernsting et al. 2012; Murakami et al. 2013), as compared to Abraxane. These examples indicate that circulation stability of NP products is critical to taking full advantage of the EPR effect, thereby improving pharmacokinetics profiles of the carried drug.
Current evaluation methods of NP stability
It is often observed in the literature that NPs are determined to be stable in one way or another, yet the outcomes in vivo or in vivo-mimicking conditions deviate from the expectation. For example, the previously mentioned PDLLA-MePEG micelles were expected to be stable, given the relatively low CMC and good colloidal stability lasting 24 h; however, they were dissociated in less than 1 h in blood (Burt et al. 1999). We expected that PLGA NS would retain PTX well based on in vitro release kinetics study (Xu et al. 2009) but found that the drug was released prematurely in the presence of amphiphilic components, cancelling potential advantages afforded by the carrier (Gullotti and Yeo 2012). The gaping difference between the estimated stability of NPs and their performance in biological systems is partly attributable to the lack of reliable methods to predict NP stability.
NPs are typically characterized with respect to the size, surface charge, colloidal stability and drug release kinetics in a physiologically relevant medium–in most cases, buffered saline. However, many studies, including those discussed in this article, report that NPs can behave differently in blood than in saline due to the abundant serum proteins, lipids, and cellular components, changing the surface properties from their “factory settings” and losing the stability and ability to carry drug. In order to overcome this disconnect between the measured NP properties and their in vivo performance, it is necessary to develop a medium system in which NPs may be suspended and evaluated, yielding useful results for predicting their biological effects. In accommodating the complexity of composition and rheological properties, there would be no replacement for blood. However, in cases where NP characterization requires spectroscopic techniques, such as absorbance or light scattering, blood is not a feasible medium due to the interference of particulate blood components and color of heme. Ideally, the in vivo-like medium should reflect amphiphilicity and viscosity of blood, such as proteins and lipids, which collectively contribute to the instability of NP during circulation. Moreover, the medium should not interfere with measurements of particle properties; therefore, it is preferable that it has low optical density and minimal particulate contents. In measuring drug retention in NPs, it is important to consider the extent by which NP is diluted upon IV injection and check if the NPs will withstand dilution and avoid dose dumping driven by the concentration gradient. Human body has an average of 5 L of blood (Sherwood 2011); thus, if NP is delivered in 50 mL IV bolus injection, it is diluted at least 100 times, assuming the NPs become evenly dispersed in the blood with minimal tissue distribution. The opposite situation is also possible, when the NPs are administered to body parts with a limited amount of fluid such as the lungs or peritoneal cavity. In this case, NPs delivering a drug with low transepithelial permeability and water solubility may face a period where the released drug is saturated and becomes unavailable for absorption due to precipitation (Bajaj et al. 2012).
Considering the complexity of physiological fluid and the volume to which the NPs will be subjected, one may employ several complementary methods to predict physical and functional stability of NP systems. Critical association or micelle concentration (CAC/CMC), the concentration at which a self-assembled particle associates and dissociates, can be measured in an in vivo-like medium to predict the stability of self-assembled NP systems, such as polymeric micelles. CAC/CMC can be measured using a variety of different detection methods such as conductivity, chromatography, surface tension, fluorescent probes, and light scattering. For temperature-sensitive polymeric micelle systems, the low critical solution temperature (LCST) can be utilized for studying stability. The LCST is the temperature at which phase transition of a thermosensitive polymer occurs and, thus, can trigger drug release. Changes in NP size and/or turbidity can also be an indicator of NP degradation, particularly in systems containing multiple components that are non-covalently associated with each other. Gel permeation chromatography can also be used to measure the physical stability of self-assembled NP systems. This method separates destabilized NP components from intact NPs since the molecular weights of the components are less than that of the NPs as a whole. Lastly, FRET is gaining popularity in studies testing the stability of NPs both in vivo and at the molecular level (Fig. 3), as previously described. Readers interested in more detail are referred to our recent review article (Cho et al. 2013).
Future perspectives
For NP drug delivery systems to substantially modify pharmacokinetics of the carried drug thereby improving its safety and efficacy profiles as compared to traditional formulations, at least two pairs of critical requirements must be met simultaneously: (i) be stealthy during circulation but sticky at the target tissues; and (ii) retain drug during circulation but release drug at the target locations be they ECM or intracellular organelles (Sun et al. 2012). While significant technical advances have been made in several aspects of these requirements, NP stability has largely been overlooked. Several studies reviewed in this article show that the potential advantages of smartly designed carriers may be undermined if drug is prematurely released during circulation or prior to arriving at intracellular targets. Such an oversight results partly from improper methods of stability assessment, where NPs are evaluated without consideration of the complexity of the in vivo environments that accommodate NPs. Development of a methodology to interrogate the NP behavior in conditions simulating physiological fluid with high fidelity is an essential precursor to the clinical success of nanomedicine.
Acknowledgments
This work was supported by NSF DMR-1056997, a Grant from the Lilly Endowment, Inc. to College of Pharmacy, Purdue University, and Intramural Research Program (Global RNAi Carrier Initiative) of Korea Institute of Science and Technology. This study was also partly supported by the NIH/NCRR-Indiana Clinical and Translational Sciences Institute Pre-doctoral Fellowship (TL1 RR025759, PI: A. Shekhar) to KCL.
Contributor Information
Karen C. Liu, Weldon School of Biomedical Engineering, Purdue University, 206 South Martin Jischke Drive, West Lafayette, IN 47907, USA
Yoon Yeo, Weldon School of Biomedical Engineering, Purdue University, 206 South Martin Jischke Drive, West Lafayette, IN 47907, USA; Department of Industrial and Physical Pharmacy, Purdue University, 575 Stadium Mall Drive, West Lafayette, IN 47907, USA; Biomedical Research Institute, Korea Institute of Science and Technology, Hwarangno 14-gil 5, Seongbuk-gu, Seoul 136-791, Republic of Korea.
References
- Amoozgar Z, Park J, Lin Q, Yeo Y. Low molecularweight chitosan as a pH-sensitive stealth coating for tumorspecific drug delivery. Molecular Pharmaceutics. 2012;9:1262–1270. doi: 10.1021/mp2005615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajaj G, Kim MR, Mohammed SI, Yeo Y. Hyaluronic acid-based hydrogel for regional delivery of paclitaxel to intraperitoneal tumors. Journal of Controlled Release. 2012;158:386–392. doi: 10.1016/j.jconrel.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt HM, Zhang X, Toleikis P, Embree L, Hunter WL. Development of copolymers of poly(d, l-lactide) and methoxypolyethylene glycol as micellar carriers of paclitaxel. Colloids and Surfaces B. 1999;16:161–171. [Google Scholar]
- Chen H, Kim S, He W, Wang H, Low PS, Park K, Cheng J-X. Fast release of lipophilic agents from circulating PEG-PDLLA micelles revealed by in vivo Förster resonance energy transfer imaging. Langmuir. 2008a;24:5213–5217. doi: 10.1021/la703570m. [DOI] [PubMed] [Google Scholar]
- Chen H, Kim S, Li L, Wang S, Park K, Cheng J-X. Release of hydrophobic molecules from polymer micelles into cell membranes revealed by Forster resonance energy transfer imaging. Proceedings of the National Academy of Sciences. 2008b;105:6596–6601. doi: 10.1073/pnas.0707046105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho EJ, Holback H, Liu KC, Abouelmagd SA, Park J, Yeo Y. Nanoparticle characterization: State of the art, challenges, and emerging technologies. Molecular Pharmaceutics. 2013;10:2093–2110. doi: 10.1021/mp300697h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Smet M, Heijman E, Langereis S, Hijnen NM, Grüll H. Magnetic resonance imaging of high intensity focused ultrasound mediated drug delivery from temperature-sensitive liposomes: An in vivo proof-of-concept study. Journal of Controlled Release. 2011;150:102–110. doi: 10.1016/j.jconrel.2010.10.036. [DOI] [PubMed] [Google Scholar]
- Desai N. Nab technology: A drug delivery platform utilising endothelial gp60 receptor-based transport and tumour-derived SPARC for targeting drug delivery report. 2008 winter;:37–41. 2007/2008. [Google Scholar]
- Ernsting MJ, Murakami M, Undzys E, Aman A, Press B, Li SD. A docetaxel-carboxymethylcellulose nanoparticle outperforms the approved taxane nanoformulation, abraxane, in mouse tumor models with significant control of metastases. Journal of Controlled Release. 2012;162:575–581. doi: 10.1016/j.jconrel.2012.07.043. [DOI] [PubMed] [Google Scholar]
- Gerweck LE, Seetharaman K. Cellular pH gradient in tumor versus normal tissue: Potential exploitation for the treatment of cancer. Cancer Research. 1996;56:1194–1198. [PubMed] [Google Scholar]
- Gregoriadis G, Davis C. Stability of liposomes invivo and invitro is promoted by their cholesterol content and the presence of blood cells. Biochemical and Biophysical Research Communications. 1979;89:1287–1293. doi: 10.1016/0006-291x(79)92148-x. [DOI] [PubMed] [Google Scholar]
- Gu F, Zhang L, Teply BA, Mann N, Wang A, Radovic-Moreno AF, Langer R, Farokhzad OC. From the cover: Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers. Proceedings of the National Academy of Sciences. 2008;105:2586–2591. doi: 10.1073/pnas.0711714105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullotti E, Yeo Y. Beyond the imaging: Limitations of cellular uptake study in the evaluation of nanoparticles. Journal of Controlled Release. 2012;164:170–176. doi: 10.1016/j.jconrel.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaguchi T, Kato K, Yasui H, Morizane C, Ikeda M, Ueno H, Muro K, Yamada Y, Okusaka T, Shirao K, Shimada Y, Nakahama H, Matsumura Y. A phase I and pharmacokinetic study of NK105, a paclitaxel-incorporating micellar nanoparticle formulation. British Journal of Cancer. 2007;97:170–176. doi: 10.1038/sj.bjc.6603855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harries M, Ellis P, Harper P. Nanoparticle albumin-bound paclitaxel for metastatic breast cancer. Journal of Clinical Oncology. 2005;23:7768–7771. doi: 10.1200/JCO.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Hatakeyama H, Akita H, Harashima H. A multifunctional envelope type nano device (MEND) for gene delivery to tumours based on the EPR effect: A strategy for overcoming the PEG dilemma. Advanced Drug Delivery Reviews. 2011;63:152–160. doi: 10.1016/j.addr.2010.09.001. [DOI] [PubMed] [Google Scholar]
- Kim D, Gao ZG, Lee ES, Bae YH. In vivo evaluation of doxorubicin-loaded polymeric micelles targeting folate receptors and early endosomal pH in drug-resistant ovarian cancer. Molecular Pharmaceutics. 2009;6:1353–1362. doi: 10.1021/mp900021q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Lee ES, Oh KT, Gao ZG, Bae YH. Doxorubicin-loaded polymeric micelle overcomes multidrug resistance of cancer by double-targeting folate receptor and early endosomal pH. Small. 2008;4:2043–2050. doi: 10.1002/smll.200701275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono K, Ozawa T, Yoshida T, Ozaki F, Ishizaka Y, Maruyama K, Kojima C, Harada A, Aoshima S. Highly temperature-sensitive liposomes based on a thermosensitive block copolymer for tumor-specific chemotherapy. Biomaterials. 2010;31:7096–7105. doi: 10.1016/j.biomaterials.2010.05.045. [DOI] [PubMed] [Google Scholar]
- Kratz F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. Journal of Controlled Release. 2008;132:171–183. doi: 10.1016/j.jconrel.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Lee ES, Na K, Bae YH. Polymeric micelle for tumor pH and folate-mediated targeting. Journal of Controlled Release. 2003a;91:103–113. doi: 10.1016/s0168-3659(03)00239-6. [DOI] [PubMed] [Google Scholar]
- Lee ES, Shin HJ, Na K, Bae YH. Poly(l-histidine)–PEG block copolymer micelles and pH-induced destabilization. Journal of Controlled Release. 2003b;90:363–374. doi: 10.1016/s0168-3659(03)00205-0. [DOI] [PubMed] [Google Scholar]
- Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Research. 1986;46:6387–6392. [PubMed] [Google Scholar]
- Miele E, Spinelli GP, Miele E, Tomao F, Tomao S. Albumin-bound formulation of paclitaxel (Abraxane® ABI-007) in the treatment of breast cancer. International Journal of Nanomedicine. 2009;4:99–105. doi: 10.2147/ijn.s3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Ernsting MJ, Undzys E, Holwell N, Foltz WD, Li SD. Docetaxel conjugate nanoparticles that target alpha-smooth muscle actin-expressing stromal cells suppress breast cancer metastasis. Cancer Research. 2013;73:4862–4871. doi: 10.1158/0008-5472.CAN-13-0062. [DOI] [PubMed] [Google Scholar]
- Nyman DW, Campbell KJ, Hersh E, Long K, Richardson K, Trieu V, Desai N, Hawkins MJ, Von Hoff DD. Phase I and pharmacokinetics trial of ABI-007, a novel nanoparticle formulation of paclitaxel in patients with advanced nonhematologic malignancies. Journal of Clinical Oncology. 2005;23:7785–7793. doi: 10.1200/JCO.2004.00.6148. [DOI] [PubMed] [Google Scholar]
- Opanasopit P, Yokoyama M, Watanabe M, Kawano K, Maitani Y, Okano T. Influence of serum and albumins from different species on stability of camptothecin-loaded micelles. Journal of Controlled Release. 2005;104:313–321. doi: 10.1016/j.jconrel.2005.02.014. [DOI] [PubMed] [Google Scholar]
- Savić R, Azzam T, Eisenberg A, Maysinger D. Assessment of the integrity of poly(caprolactone)-b-poly(ethylene oxide) micelles under biological conditions: A fluorogenic-based approach. Langmuir. 2006;22:3570–3578. doi: 10.1021/la0531998. [DOI] [PubMed] [Google Scholar]
- Sherwood L. Human physiology: From cells to systems. Cengage Learning; Belmont, CA: 2011. [Google Scholar]
- Sun Q, Radosz M, Shen Y. Challenges in design of translational nanocarriers. Journal of Controlled Release. 2012;164:156–169. doi: 10.1016/j.jconrel.2012.05.042. [DOI] [PubMed] [Google Scholar]
- Xu P, Gullotti E, Tong L, Highley CB, Errabelli DR, Hasan T, Cheng J-X, Kohane DS, Yeo Y. Intracellular drug delivery by poly(lactic-co-glycolic acid) nanoparticles, revisited. Molecular Pharmaceutics. 2009;6:190–201. doi: 10.1021/mp800137z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Shen Y, Tang J, Fan M, Van Kirk EA, Murdoch WJ, Radosz M. Charge-reversal drug conjugate for targeted cancer cell nuclear drug delivery. Advanced Functional Materials. 2009;19:3580–3589. [Google Scholar]