

Abstract
Objectives:
For most adults with initial clinical presentation of multiple sclerosis (MS), biological disease was likely initiated many years prior. Pediatric-onset MS provides an opportunity to study early disease processes.
Methods:
Using antigen microarrays, including CNS-related proteins, lipids, and other autoantigens, we studied early immunologic events involved in clinical onset of pediatric MS. Serum samples were collected at the time of incident acquired CNS demyelinating syndromes (ADS) in children who, in subsequent prospective follow-up, were ascertained to have either pediatric MS (ADS-MS) or a monophasic illness (ADS-mono). Samples were obtained both at the time of ADS presentation and 3 months into follow-up. We used an initial training set of samples to implicate antibody signatures associated with each group, and then a test set. An additional set of follow-up samples (stability set) was used as a form of internal validation.
Results:
Children with ADS-MS tended to have distinguishable serum antibody patterns both at the time of ADS presentation and 3 months into follow-up. At the time of ADS, serum samples from patients with ADS-MS or ADS-mono reacted against similar numbers of CNS antigens, although CNS antigens implicated in adult MS were more often targeted in children with ADS-MS. The follow-up ADS-MS samples reacted against a broader panel of CNS antigens, while corresponding ADS-mono samples exhibited a contraction of the initial antibody response.
Conclusions:
Our findings in this prospective cohort of pediatric-onset CNS demyelinating diseases point to an active process of epitope spreading during early stages of MS, not seen in monophasic CNS inflammatory conditions.
While most patients developing multiple sclerosis (MS) experience initial clinical symptom onset in adulthood, the biological mechanisms involved in disease initiation likely manifested many years prior. This has limited the ability to study early MS disease pathogenesis and has also posed a challenge to understanding whether, and to what extent, initiating disease mechanisms identified in animal models such as experimental autoimmune encephalomyelitis translate into the human disease. In comparison to adult-onset MS, pediatric-onset MS, which accounts for 2% to 5% of all MS cases,1,2 provides an opportunity to study immunologic mechanisms that may contribute to early disease pathogenesis. Only a portion of all children presenting with initial symptoms of acquired CNS demyelinating syndrome (ADS) will develop further disease activity establishing the diagnosis of MS.1–3 The remaining children with ADS represent a population with monophasic CNS-directed inflammation, who will not develop further disease activity. Comparing immune responses of children with ADS who are prospectively ascertained as having either MS (ADS-MS) or monophasic disease (ADS-mono) may therefore provide important insights into early MS disease mechanisms.
A practical challenge in studying children relates to the limited availability of biological samples, and assays that can be applied to small volumes of serum or plasma may provide the greatest utility. Antigen arrays are high-throughput tools for the characterization of antibody response profiles.4,5 We and others have used antigen arrays to examine humoral immune responses in patients with MS6–10 and in animals with experimental autoimmune encephalomyelitis.8,11,12 Here, we applied antigen arrays to examine the profile and evolution of serum antibodies in a large, prospectively followed cohort of children with ADS, to gain insights into early immunologic mechanisms that may contribute to MS development.
METHODS
Standard protocol approvals and patient consents.
Details of the Prospective Canadian Pediatric Demyelinating Disease Study protocol, inclusion and exclusion criteria, and clinical characteristics of all participants have been recently described.13 Protocols and informed consents, obtained for all participants, were approved by the institutional ethics-review boards.
Samples.
Serum samples were collected and stored (−80°C) using a standardized protocol as part of the Canadian Study,13 in which participants are followed from time of ADS onset with comprehensive clinical and imaging assessments to ascertain whether they develop new MS-defining disease activity. Serum samples, collected at the time of acute ADS presentation (attack) and 3 months later (follow-up), were randomly allocated to training or test sets. Longer-term (6- to 12-month) follow-up samples from a subset of children were used (stability set) for additional internal validation to assess stability of antibody signatures over time. Only samples from children with ≥2.5 years of prospective follow-up were included. MS diagnosis was conferred in follow-up based on either a second (investigator-confirmed) clinical demyelinating attack or by MRI confirmation of dissemination in time according to established criteria.14 Clinical characteristics of the overall ADS cohort are shown in table 1, characteristics of children contributing to analysis of samples collected at the time of acute attack (divided into training and test sets) are shown in table e-1 on the Neurology® Web site at Neurology.org, and characteristics of children contributing to analysis of samples collected at 3-month follow-up (divided into training and test sets) and in later follow-up (stability set) to assess stability of the signatures over time are shown in table e-2.
Table 1.
Clinical demographics of participating children with ADS

Antigens.
Peptide antigens were synthesized at the Biopolymers Facility, Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School. Recombinant proteins and lipids were purchased from Sigma (St. Louis, MO), Abnova (Taipei City, Taiwan), Matreya LLC (Pleasant Gap, PA), Avanti Polar Lipids (Alabaster, AL), Calbiochem (San Diego, CA), Chemicon (Temecula, CA), GeneTex (San Antonio, TX), Novus Biologicals (Littleton, CO), Assay Designs (Ann Arbor, MI), ProSci Inc. (Poway, CA), EMD Biosciences (San Diego, CA), Cayman Chemical (Ann Arbor, MI), HyTest (Turku, Finland), Meridian Life Science (Memphis, TN), and Biodesign International (Saco, ME). Antigens used in the construction of antigen microarrays are listed in table e-3. Among them were CNS-related antigens, including putative targets previously implicated in adult MS,9,10 as well as lipids and other autoantigens.
Antigen microarray production, development, and data analysis.
Antigens were spotted in replicates of 6 on SuperEpoxy 2 slides (TeleChem, Sunnyvale, CA) as previously described.4 The microarrays were blocked with 1% bovine serum albumin, and incubated (2 hours, 37°C) with the test serum in blocking buffer. The arrays were then washed and incubated (45 minutes) with 1:500 dilution of goat anti-human immunoglobulin G (IgG) Cy3-conjugated detection antibodies (Jackson ImmunoResearch Labs, West Grove, PA). Arrays were scanned with a ScanArray 4000X scanner (GSI Lumonics, Billerica, MA). Normalized raw data were analyzed using the GeneSpring software (Silicon Genetics, Redwood City, CA). Antigen reactivity was defined by the mean intensity of binding to the replicates of that antigen on the microarray, and expressed as relative fluorescence units.
RESULTS
Serum autoantibody reactivities in ADS-MS compared with ADS-mono during the incident acute demyelinating event.
Using serum samples obtained at the time of incident presentation with ADS, we initially analyzed antibody response profiles in a training set of 40 children with ADS, including 20 children subsequently confirmed to have MS (ADS-MS) and 20 children who remained monophasic (ADS-mono). Differences between the 2 groups were observed for antibodies directed against 48 antigens (figure 1A and table e-4). Reactivities to 24 of these antigens were higher in the ADS-MS group while reactivities to the 24 other antigens were higher in the ADS-mono group. Thirteen of the 24 antibodies (54%) exhibiting higher reactivity in the ADS-MS sera were reactive to CNS antigens (figure 1B); 12 of these 13 antigens (86%) were among those previously implicated in adult MS (figure 1C). Fourteen of the 24 antibodies (58%) that exhibited higher reactivity in the ADS-mono samples were reactive to CNS antigens (figure 1B), while only 2 of these 14 (15%) were implicated as targets in adult MS (p < 0.0004, figure 1C). There was no effect of sex or age at time of sampling, nor did differences between polysymptomatic vs mono-symptomatic presentations skew the results (data not shown). These findings indicate that, although CNS-reactive antibodies can be detected in both ADS-mono and ADS-MS samples, the antibodies of children with ADS-MS more frequently react with CNS antigens that were previously linked to adult MS pathogenesis.
Figure 1. IgG antibody reactivities in ADS-mono and ADS-MS serum samples.

(A) Heatmap in which each column represents the mean IgG antibody reactivities in the ADS-mono or the ADS-MS group, and each row shows the antibody reactivity to an antigen according to the colorimetric scale depicted on the left. Represented are samples taken at the time of an acute demyelinating attack (attack). The antibody reactivities included in this heatmap are listed in table e-4. (B) The total number of antigens to which antibodies are detected in ADS-mono and ADS-MS serum samples. These antigens are designated as CNS antigens (CNS), and non-CNS antigens (other). (C) The total number of CNS antigens to which antibodies are detected in ADS-mono and ADS-MS serum samples. These CNS antigens are designated into antigens previously linked to MS in adult cohorts (MS), as well as CNS antigens not previously linked to MS (other CNS). (D) Heatmap of the mean IgG antibody reactivities in the ADS-mono or the ADS-MS group, 3 months after the attack (follow-up). The antibody reactivities included in this heatmap are listed in table e-6. (E) Heatmap depicting the mean IgG antibody reactivities against MBP, PLP, and CNPase in ADS-MS serum samples. ADS = acquired CNS demyelinating syndrome; IgG = immunoglobulin G; MBP = myelin basic protein; MOG = myelin oligodendrocyte glycoprotein; mono = monophasic; MS = multiple sclerosis; PLP = proteolipid protein.
Discriminating power of serum autoantibody profiles in ADS-MS and ADS-mono during the incident acute demyelinating event.
To initially assess the potential discriminating power of the IgG antibody reactivity profiles shown in table e-4, we performed a leave-one-out cross-validation (LOOCV) analysis within the same training set. The number of true (correct) and false (incorrect) classifications was computed to estimate success rate, positive predictive value (PPV; the fraction of patients with ADS-MS correctly identified as ADS-MS by their antigen microarray reactivity), and negative predictive value (NPV; the fraction of patients with ADS-mono correctly identified as ADS-mono by their antigen microarray reactivity) in the training set. The LOOCV revealed a PPV of 0.900 and an NPV of 0.950. We also generated receiver operating characteristic curves by plotting the sensitivity against 1 − specificity and calculating the area under the curve (AUC) for each population. The AUC is a measure of the ability of the model to discriminate between ADS-mono and ADS-MS.15 Receiver operating characteristic analysis of the classifier built using the IgG reactivities in the training set produced an AUC of 0.952 (figure 2A and table e-5). Because the LOOCV was performed on the same sample cohort (training set) used to generate the model, performance of the classifier may reflect overfitting of the model. Thus, we next evaluated the performance of the same classifier on an independent test set composed of additional samples (14 ADS-mono and 14 ADS-MS samples), also collected at the time of ADS presentation. We found that the pattern of IgG antibody reactivity identified in the training set could classify the test set of samples with a PPV of 0.785, an NPV of 0.857, and an AUC of 0.872 (figure 2B and table e-5).
Figure 2. Performance of IgG antigen array reactivity classifiers in discriminating ADS-mono from ADS-MS at the time of an acute demyelinating attack.
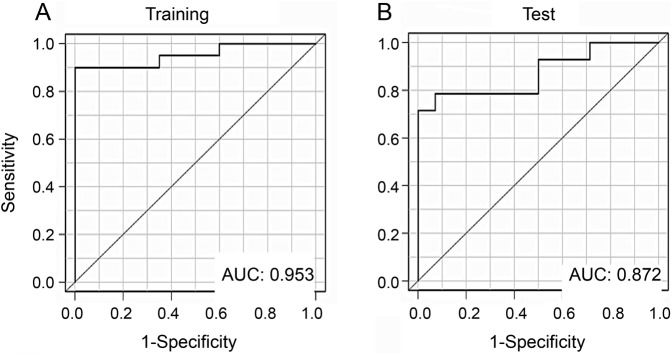
Receiver operating characteristic curves were obtained with classifiers constructed based on IgG reactivities and evaluated on (A) training set samples, and (B) test set samples (all obtained at the time of an acute demyelinating attack). ADS = acquired CNS demyelinating syndrome; AUC = area under the curve; IgG = immunoglobulin G; mono = monophasic; MS = multiple sclerosis.
Serum autoantibody reactivities and their discriminating power in ADS-MS and ADS-mono samples obtained in follow-up (3 months after the incident acute demyelinating event).
Serum antibodies recognizing myelin oligodendrocyte glycoprotein have been previously reported at the time of ADS in both ADS-mono and ADS-MS children, however these antibodies tended to persist only in the children with ADS-MS.16 We therefore wished to examine whether the evolution of serum antibody reactivities measured in our antigen array would differ between our ADS-MS and ADS-mono groups over time. We applied the identical antigen arrays in parallel to a training set of serum samples obtained 3 months after the acute ADS presentations of 19 patients with ADS-MS and 20 with ADS-mono. We identified a pattern of 84 IgG antibody reactivities that appeared to discriminate ADS-MS from ADS-mono in these follow-up samples (figure 1D and table e-6). Sixty-eight of the 84 IgG reactivities were higher in ADS-MS samples; 55 of these 68 antibody reactivities (81%) targeted CNS antigens (figure 1B). In comparison, only 7 of the 16 antibody reactivities (44%) associated with ADS-mono were directed against CNS antigens (p < 0.0001, Fisher exact test) (figure 1B). Moreover, 54 of the 55 CNS antigens (98%) targeted in ADS-MS correspond to putative disease targets implicated in adult MS, as opposed to 2 of the 7 antigens (29%) targeted in ADS-mono samples (p < 0.0001, Fisher exact test) (figure 1C).
To analyze the discriminating power of the serum IgG antibody reactivities detected during follow-up of the demyelinating event, we initially performed an LOOCV in a training set of 20 ADS-mono and 19 ADS-MS serum samples taken 3 months after an acute demyelinating attack. The LOOCV produced a PPV of 0.842 and an NPV of 0.900; the AUC was 0.866 (figure 3A and table e-7). Analysis of the performance of this IgG-based classifier on a test set of additional 15 ADS-mono and 8 ADS-MS samples also taken 3 months after an acute demyelinating attack revealed a PPV of 1.000 and an NPV of 0.625, and an AUC of 0.833 for the classifier (figure 3B and table e-7). Finally, we evaluated the stability of the classifiers constructed based on the IgG antibody reactivities measured at 3 months’ follow-up using samples obtained in longer-term follow-up (up to 12 months after ADS) from a subset of children. Testing the antibody reactivity classifier in this stability cohort (comprised of 10 ADS-mono and 11 ADS-MS samples) revealed an AUC of 0.727, a PPV of 0.700, and an NPV of 0.545 (figure 3C and table e-7).
Figure 3. Performance of IgG antigen array reactivity classifiers in discriminating ADS-mono from ADS-MS 3 months after an acute demyelinating attack, and stability in later follow-up.
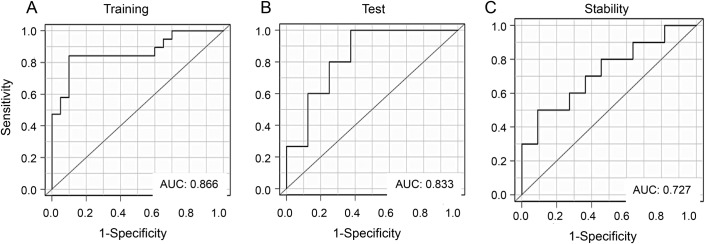
Receiver operating characteristic curves were obtained with classifiers constructed based on IgG reactivities and evaluated on (A) training set samples, (B) test set samples (all obtained 3 months after an acute demyelinating attack), as well as (C) stability set samples, obtained in later follow-up (12 months after ADS presentations). ADS = acquired CNS demyelinating syndrome; AUC = area under the curve; IgG = immunoglobulin G; mono = monophasic; MS = multiple sclerosis.
Early detection of intra- and intermolecular antigen spreading in patients with ADS-MS.
Taken together, our data suggest an expansion of the initial antibody response to CNS antigens in ADS-MS samples, compatible with the development of intra- and intermolecular epitope spreading. To address whether this apparent expansion in CNS antibody reactivity in children with ADS-MS relative to those with ADS-mono results from increased CNS damage in children with MS, we took advantage of the prospective coregistered brain MRIs acquired on all participants in the Canadian Study, and calculated the volume of T2 hyperintense brain lesions as an overall measure of inflammatory disease burden. The T2 lesion volumes at initial presentation did not differ significantly between the ADS-MS and ADS-mono cohorts (p = 0.23) and, moreover, the change in T2 volumes assessed between the time of presentation and the follow-up time point (follow-up T2 volume minus presentation T2 volume) was also not different between groups (figure 4; p = 0.89) indicating that the observed expansion of the antibody response in the ADS-MS group relative to the ADS-mono group is not the result of an increased volume of injured CNS tissue. In further support that epitope spreading explains the expansion in CNS-reactive antibodies seen in the children with MS is the detailed analysis of antibody responses to particular myelin antigens and their epitopes (figure 1E). While only a few epitopes of proteolipid protein and myelin basic protein were targeted in ADS-MS samples taken at the time of the attack, a significant spreading of the antibody response was detected during follow-up, consistent with “intramolecular” epitope spreading. This intramolecular epitope spreading was not detected in the ADS-mono cohort. Moreover, while no antibodies to CNPase were detectable at the time of ADS in all children, we detected antibodies to multiple CNPase epitopes in follow-up samples of the children with MS consistent with “intermolecular” epitope spreading—again, a phenomenon not observed in the ADS-mono follow-up samples.
Figure 4. Changes in MRI T2 lesion volumes in children with ADS-mono or ADS-MS.
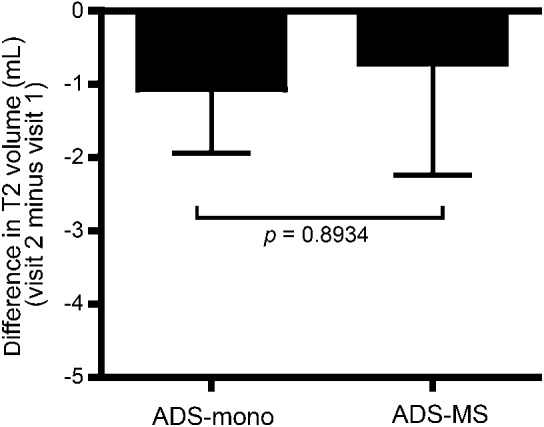
The change in average brain T2 lesion volumes (mL) in the ADS-mono and ADS-MS cohorts, calculated as the difference in volumes between the time of follow-up and the time of ADS presentation. ADS = acquired CNS demyelinating syndrome; mono = monophasic; MS = multiple sclerosis.
DISCUSSION
In the present study, we measured serum antibody profiles in samples obtained from children with incident ADS at the time of initial attack and 3 months later, and compared these response profiles between children subsequently ascertained as having MS (ADS-MS) vs those children who remained with a monophasic illness (ADS-mono). Although both groups of children harbored serum antibodies reactive with CNS antigens, the breadth and specificity of the antibody responses were different. At the time of incident attack, ADS-MS and ADS-mono sera reacted against similar numbers of CNS antigens; however, ADS-MS samples displayed increased antibody reactivities to antigens linked to adult MS. Three months after the incident attack, sera from patients with ADS-MS reacted against a broader panel of CNS antigens, while patients with ADS-mono exhibited a contraction in their CNS antibody responses over the same period. The observation that MRI T2 lesion loads did not differ between the ADS-MS and ADS-mono cohorts and, in particular, that the changes in T2 lesion burden diminished to the same extent between the ADS presentation and follow-up in both patient groups indicates that the expansion of the antibody response seen in the ADS-MS group was not simply attributable to an increase in the volume of involved tissue.
One notes that antibodies detected with antigen microarrays may be of low affinity, and their pathologic significance is unknown. At the same time, given their relative sensitivity,5,10 autoantibody patterns detected by the antigen microarrays used here might not be captured by more standard assays such as ELISA. We confirmed acceptable reproducibility of our antigen microarray technique, with repeat measurements exhibiting a coefficient of variation of 13.2 ± 1.2, and further supported by our finding that the stability set of follow-up samples also had a high classifier AUC value (0.727), which is similar to a previously reported MS disease classifier that was generated based on combined genetic and environmental factors in adult patients with MS15 (AUC approximately 0.7). While findings from our stability set provide a measure of internal validation of our results, external validation in an independent cohort will clearly be needed to more fully assess the generalizability of our findings.
Our observations are compatible with a process of molecular epitope spreading in children with ADS-MS, in contrast to the apparent control of CNS inflammation in patients with ADS-mono. Diversification of antigenic specificities from the initial epitope-specific immune response can be to additional epitopes of either the same (intramolecular) or different (intermolecular) antigens.17 T-cell epitope spread, previously described in experimental models of MS (mice and marmosets)18 and considered in patients with MS,19 is thought to be driven by antigen-presenting cells exposed to an expanding repertoire of CNS antigens.20,21 Epitope spread can also occur for autoantibody responses (humoral epitope spread). Robinson, Steinman, and coworkers described a broad diversification of antibody responses in experimental models of MS and rheumatoid arthritis, detectable as early as 1 month after disease induction,12,22 and autoantibody epitope spread has also been implicated in patients with type 1 diabetes23 and systemic lupus erythematosus.24 Our observation that pediatric ADS-MS serum samples obtained 3 months after incident ADS presentation exhibit expanded antibody reactivities (against additional epitopes of the same CNS antigens as well as additional CNS antigens that did not appear to be targeted at the time of the acute demyelinating attack) provides what we believe is the first evidence of autoantibody epitope spread in the serum of patients with MS, and indicates that both intra- and intermolecular humoral epitope spreading can occur as early features of the human disease.
Of interest is whether the contracted CNS reactivities observed over time in the ADS-mono group, in contrast to the epitope spreading noted in patients with ADS-MS, reflects a relative failure in these young patients with MS to regulate aberrant CNS-directed autoantibody responses. Such a defect in immune regulation would be consistent with abnormalities in T-cell immune-regulatory mechanisms described in adult- and pediatric-onset patients with relapsing-remitting MS.22–25 If true, our data may reflect deficits in immunoregulation of target-directed B-cell responses in early MS, allowing the development of humoral epitope spreading as part of the ongoing CNS inflammation observed in these children.3 We cannot comment on whether or not the autoantibody epitope spread we detect in children with ADS-MS actually contributes to further CNS injury, or whether it emerges as a secondary phenomenon due to ongoing tissue injury. Similarly, we are unable to determine which antigen or antigens initiate the CNS-specific immune response in these patients. It should also be noted that our current work is based on antibody analyses in serum samples as opposed to CSF,9 which would be expected to provide more direct insights into early disease targets and epitope spreading. Indeed, recent proteomic analysis of CSF obtained from pediatric ADS-MS and ADS-mono patients has implicated novel antigens of the axoglial apparatus (not present on the current antigen array) as potential early targets in MS.26 While epitope spreading may be more challenging to ascertain in serum, our study benefited from repeat sampling in a relatively large cohort of children with MS and controls; a similar approach using CSF samples would be very challenging. Future work incorporating newly implicated antigens into antigen arrays and access to both serum and CSF from well-characterized cohorts will be of considerable interest, as would development of other approaches assessing antigen-specific responses in early-onset MS. Such work may help to guide strategies aimed at boosting immunoregulatory mechanisms and/or controlling effector T cells at the antigen-specific level, such as DNA vaccines6,7 or the induction of regulatory T cells by stimulation of immunoregulatory molecular pathways with small molecules11,27 or nanoparticles,28 as used to control epitope spreading and CNS inflammation in experimental models of MS.
Supplementary Material
ACKNOWLEDGMENT
The authors thank all pediatric donors, their families, and caregivers, as well as members of the Montreal Neurological Institute Experimental Therapeutics Program for judicious care of high sample quality.
GLOSSARY
- ADS
acquired CNS demyelinating syndrome
- AUC
area under the curve
- IgG
immunoglobulin G
- LOOCV
leave-one-out cross-validation
- mono
monophasic
- MS
multiple sclerosis
- NPV
negative predictive value
- PPV
positive predictive value
Footnotes
Editorial, page 2200
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Dr. Amit Bar-Or (McGill University): development of the study concept and design, study supervision, acquisition of data, analysis and interpretation, writing, reviewing, and revising of manuscript. Dr. Francisco J. Quintana (Brigham and Women's Hospital): development of the study concept and design, study supervision, acquisition of data, analysis and interpretation, writing, reviewing, and revising of manuscript. Dr. Brenda Banwell (CHOP): development of the study concept and design, study supervision, acquisition of data, analysis and interpretation, writing, reviewing, and revising of manuscript. Dr. Howard L. Weiner (Brigham and Women's Hospital): data interpretation. Dr. Mukanthu Nyirenda (McGill University): critical review of the manuscript, data analysis. Sandra Magalhaes (McGill University): implementation of the study and data analysis. Melissa McGowan (Toronto Hospital for Sick Children): implementation of the study and data analysis. Dumitru Fetco (McGill University): data analysis and interpretation. Bonny Patel (Brigham and Women's Hospital): implementation of the study and data analysis. Ada Yeste (Brigham and Women's Hospital): implementation of the study and data analysis. Jessica Kenison (Brigham and Women's Hospital): implementation of the study and data analysis. Roya Rahbari (Brigham and Women's Hospital): implementation of the study and data analysis. Mohammad Hussain (Brigham and Women's Hospital): implementation of the study and data analysis. Julia O'Mahony (Toronto Hospital for Sick Children): implementation of the study, data analysis and interpretation. Sridar Narayanan (McGill University): data analysis and interpretation. Doug Arnold (McGill University): data analysis and interpretation. Trina Johnson (McGill University): implementation of the study and data analysis. Sathy Rajasekharan (McGill University): implementation of the study and data analysis.
STUDY FUNDING
The Canadian Pediatric Demyelinating Disease Study Group is supported by a Multiple Sclerosis Society of Canada Research Foundation grant to A.B.-O. and B.B. This work was also supported in part by grants to F.Q. and H.L.W. from EMD Serono and the National Multiple Sclerosis Society.
DISCLOSURE
F. Quintana has participated as a speaker at meetings sponsored by, received consulting fees, and/or received grant support from: Biogen Idec, Novartis, EMD Serono, Genzyme-Sanofi, GlaxoSmithKline, and Teva Neuroscience. He is also an associate editor of Immunology, and serves on the editorial boards of Systems Biomedicine, Seminars in Immunopathology, American Journal of Clinical and Experimental Immunology, and Immunologia. B. Patel, A. Yeste, M. Nyirenda, J. Kenison, R. Rahbari, D. Fetco, M. Hussain, J. O'Mahony, S. Magalhaes, M. McGowan, T. Johnson, S. Rajasekharan, S. Narayanan, and D. Arnold report no disclosures. H. Weiner has participated as a speaker at meetings sponsored by, received consulting fees, and/or received grant support from: Biogen Idec, EMD Serono, Genentech, Genzyme-Sanofi, GlaxoSmithKline, Guthy-Jackson Greater Good Foundation, NasVax, Novartis, and Teva Neuroscience. B. Banwell serves as an advisor to Biogen Idec, Novartis, and Sanofi-Aventis. She does not receive any financial remuneration for her advisory role. Dr. Banwell also serves as an advisor and as a site investigator for Eli Lilly. Dr. Banwell serves as a chief editor for Multiple Sclerosis and Related Disorders, and is on the editorial board for Neurology®. A. Bar-Or has participated as a speaker at meetings sponsored by, received consulting fees, and/or received grant support from: Amplimmune, Aventis, Bayhill Therapeutics, Berlex/Bayer, Biogen Idec, BioMS, DioGenix, Eli Lilly, EMD Serono, Genentech, Genzyme-Sanofi, GlaxoSmithKline, Guthy-Jackson Greater Good Foundation, MedImmune, Mitsubishi Pharma, Novartis, Ono Pharmacia, Receptos, Roche, Teva Neuroscience, and Wyeth. He also serves on the editorial boards of Clinical and Experimental Neurology and Neurology. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Banwell B, Ghezzi A, Bar-Or A, Mikaeloff Y, Tardieu M. Multiple sclerosis in children: clinical diagnosis, therapeutic strategies, and future directions. Lancet Neurol 2007;6:887–902. [DOI] [PubMed] [Google Scholar]
- 2.Yeh EA, Chitnis T, Krupp L, et al. Pediatric multiple sclerosis. Nat Rev Neurol 2009;5:621–631. [DOI] [PubMed] [Google Scholar]
- 3.Verhey LH, Branson HM, Shroff MM, et al. MRI parameters for prediction of multiple sclerosis diagnosis in children with acute CNS demyelination: a prospective national cohort study. Lancet Neurol 2011;10:1065–1073. [DOI] [PubMed] [Google Scholar]
- 4.Quintana FJ, Hagedorn PH, Elizur G, Merbl Y, Domany E, Cohen IR. Functional immunomics: microarray analysis of IgG autoantibody repertoires predicts the future response of mice to induced diabetes. Proc Natl Acad Sci USA 2004;101(suppl 2):14615–14621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robinson WH, Digennaro C, Hueber W, et al. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat Med 2002;8:295–301. [DOI] [PubMed] [Google Scholar]
- 6.Bar-Or A, Vollmer T, Antel J, et al. Induction of antigen-specific tolerance in multiple sclerosis after immunization with DNA encoding myelin basic protein in a randomized, placebo-controlled phase 1/2 trial. Arch Neurol 2007;64:1407–1415. [DOI] [PubMed] [Google Scholar]
- 7.Garren H, Robinson WH, Krasulová E, et al. Phase 2 trial of a DNA vaccine encoding myelin basic protein for multiple sclerosis. Ann Neurol 2008;63:611–620. [DOI] [PubMed] [Google Scholar]
- 8.Kanter JL, Narayana S, Ho PP, et al. Lipid microarrays identify key mediators of autoimmune brain inflammation. Nat Med 2006;12:138–143. [DOI] [PubMed] [Google Scholar]
- 9.Quintana FJ, Farez MF, Izquierdo G, Lucas M, Cohen IR, Weiner HL. Antigen microarrays identify CNS-produced autoantibodies in RRMS. Neurology 2012;78:532–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quintana FJ, Farez MF, Viglietta V, et al. Antigen microarrays identify unique serum autoantibody signatures in clinical and pathologic subtypes of multiple sclerosis. Proc Natl Acad Sci U S A 2008;105:18889–18894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quintana FJ, Basso AS, Iglesias AH, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008;453:65–71. [DOI] [PubMed] [Google Scholar]
- 12.Robinson WH, Fontoura P, Lee BJ, et al. Protein microarrays guide tolerizing DNA vaccine treatment of autoimmune encephalomyelitis. Nat Biotechnol 2003;21:1033–1039. [DOI] [PubMed] [Google Scholar]
- 13.Banwell B, Bar-Or A, Arnold DL, et al. Clinical, environmental, and genetic determinants of multiple sclerosis in children with acute demyelination: a prospective national cohort study. Lancet Neurol 2011;10:436–445. [DOI] [PubMed] [Google Scholar]
- 14.Polman CH, Reingold SC, Edan G, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria.” Ann Neurol 2005;58:840–846. [DOI] [PubMed] [Google Scholar]
- 15.De Jager PL, Chibnik LB, Cui J, et al. Integration of genetic risk factors into a clinical algorithm for multiple sclerosis susceptibility: a weighted genetic risk score. Lancet Neurol 2009;8:1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Probstel AK, Dornmair K, Bittner R, et al. Antibodies to MOG are transient in childhood acute disseminated encephalomyelitis. Neurology 2011;77:580–588. [DOI] [PubMed] [Google Scholar]
- 17.Lehmann PV, Forsthuber T, Miller A, Sercarz EE. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature 1992;358:155–157. [DOI] [PubMed] [Google Scholar]
- 18.Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol 2002;2:85–95. [DOI] [PubMed] [Google Scholar]
- 19.Goebels N, Hofstetter H, Schmidt S, Brunner C, Wekerle H, Hohlfeld R. Repertoire dynamics of autoreactive T cells in multiple sclerosis patients and healthy subjects: epitope spreading versus clonal persistence. Brain 2000;123:508–518. [DOI] [PubMed] [Google Scholar]
- 20.Bailey SL, Schreiner B, Mcmahon EJ, Miller SD. CNS myeloid DCs presenting endogenous myelin peptides “preferentially” polarize CD4+ TH-17 cells in relapsing EAE. Nat Immunol 2007;8:172–180. [DOI] [PubMed] [Google Scholar]
- 21.Mcmahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med 2005;11:335–339. [DOI] [PubMed] [Google Scholar]
- 22.Astier AL, Meiffren G, Freeman S, Hafler DA. Alterations in CD46-mediated Tr1 regulatory T cells in patients with multiple sclerosis. J Clin Invest 2006;116:3252–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balint B, Haas J, Schwarz A, et al. T-cell homeostasis in pediatric multiple sclerosis: old cells in young patients. Neurology 2013;81:784–792. [DOI] [PubMed] [Google Scholar]
- 24.Viglietta V. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med 2004;199:971–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Venken K, Hellings N, Hensen K, et al. Secondary progressive in contrast to relapsing-remitting multiple sclerosis patients show a normal CD4+CD25+ regulatory T-cell function and FOXP3 expression. J Neurosci Res 2006;83:1432–1446. [DOI] [PubMed] [Google Scholar]
- 26.Dhaunchak AS, Becker C, Schulman H, et al. Implication of perturbed axoglial apparatus in early pediatric multiple sclerosis. Ann Neurol 2012;71:601–613. [DOI] [PubMed] [Google Scholar]
- 27.Quintana FJ, Murugaiyan G, Farez MF, et al. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 2010;107:20768–20773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yeste A, Nadeau M, Burns EJ, Weiner HL, Quintana FJ. Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 2012;109:11270–11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.