

Abstract
Background
Cortical networks undergo large-scale switching between states of increased or decreased activity in normal sleep and cognition as well as in pathological conditions such as epilepsy. We previously found that focal hippocampal seizures in rats induce increased neuronal firing and cerebral blood flow in subcortical structures including the lateral septal area, along with frontal cortical slow oscillations resembling slow wave sleep. In addition, stimulation of the lateral septum in the absence of a seizure resulted in cortical deactivation with slow oscillations.
Hypothesis
We hypothesized that lateral septal activation might cause neocortical deactivation indirectly, possibly through impaired subcortical arousal. But how does subcortical stimulation cause slow wave activity in frontal cortex? How do arousal neurotransmitter levels (e.g. acetylcholine) change in cortex during the excitation of inhibitory projection nuclei?
Methods and Results
In the current study, we used simultaneous electrophysiology and enzyme- based amperometry in a rat model, and found a decrease in choline, along with slow wave activity in orbital frontal cortex during lateral septal stimulation in the absence of seizures. In contrast, the choline signal and local field potential in frontal cortex had no significant changes when stimulating the hippocampus, but showed increased choline and decreased slow wave activity with an arousal stimulus produced by toe pinch.
Conclusions
These findings indicate that the activation of subcortical inhibitory structures (such as lateral septum) can depress subcortical cholinergic arousal. This mechanism may play an important role in large-scale transitions of cortical activity in focal seizures, as well as in normal cortical function.
Keywords: Acetylcholine, Deactivation, Slow Oscillations, Lateral Septum, Default Mode Network
Introduction
Cortical networks undergo large-scale transitions in activity during normal cognition and sleep as well as in brain disorders. Investigation of the mechanisms of large-scale network switching in the brain is an important emerging area of neuroscience research [1-3]. Prior work has shown that the transition to slow-wave sleep involves massive changes in cortical network activity accompanied by a decrease in neurotransmitters such as acetylcholine arising from subcortical arousal systems [4]. We have recently found similarities between onset of slow-waves in deep sleep and cortical slow wave activity observed during partial limbic seizures [5-8]. The switching mechanism for these transitions is not well understood and still requires further investigation.
Circuits controlling subcortical arousal may play a critical role in regulating large-scale cortical transitions. Based on our findings in limbic seizures we proposed the “network inhibition hypothesis” [3, 9] in which increased activity in one region such as the hippocampus (HC) turns on inhibitory neurons in subcortical structures. GABAergic neurons in turn inhibit subcortical arousal leading to cortical slow-waves and decreased level of consciousness [10, 11]. We found using high-field functional magnetic resonance imaging (fMRI) that the lateral septal nuclei were strongly activated during hippocampal seizures, and that electrical stimulation of the lateral septal nuclei at 1-10 Hz produced a transition to cortical slow wave activity and behavioral arrest [7, 8]. Slow wave activity was prominent in multiple cortical areas including orbital frontal cortex (OFC).
But how does lateral septal activation cause cortical deactivation? Septal activation could contribute either via direct inhibition of neocortex or through diminished subcortical arousal. A long latency was detected from single septal stimuli to cortical Down states [8], consistent with a polysynaptic mechanism [12, 13]. In addition, the lateral septum (LS) does not have known direct projections to frontal cortex [14, 15]. The lateral septal nuclei receive their major inputs from the hippocampal formation, and then project to the medial septum, as well as to various hypothalamic areas, the mammillary complex, the ventral tegmental area, and other regions in the basal forebrain [14-17]. Therefore, GABAergic neurons in the lateral septum are poised to produce inhibitory projections to subcortical arousal regions including the nucleus basalis, which in turn provides the major cholinergic supply to the cortex [17-19]. Hence, we propose that lateral septal activation can cause neocortical deactivation indirectly, through decreased subcortical arousal. Therefore, a crucial question is whether septal activation and the transition to cortical slow waves are associated with decreased cortical cholinergic activity.
To investigate this question we used simultaneous electrophysiology and high time-resolution amperometry to measure cortical choline levels as a marker of cholinergic neurotransmission during lateral septal stimulation. We found that the transition to cortical slow waves during septal stimulation was accompanied by a marked decrease in cortical choline. These findings support the network inhibition hypothesis, with increased lateral septal activity and reduced cholinergic neurotransmission as possible mechanisms for switching cortical networks to a state of decreased arousal.
Materials and Methods
Animal preparation, surgery and histology
All procedures were in full compliance with approved institutional animal care and use protocols. A total of 18 adult female Sprague Dawley rats (Charles River Laboratories) weighting 230-300 g were used in the experiments. All surgeries were performed under anesthesia with ketamine (90 mg/kg) and xylazine (15 mg/kg, i.m.). Responsiveness was checked every 15 minutes by toe pinch. After surgery, animals were switched to a low-dose, “light-anesthesia” ketamine/xylazine (40/7 mg/kg) in preparation for stimulus initiation, as described previously [7].
At the end of experiments, animals were euthanized with Euthasol injection (Virbac, i.p.), and brains were harvested, and all electrode and amperometry probe assembly placements were verified based on histology. Brains were rapidly removed and transferred to 4% phosphate buffered paraformaldehyde, pH 7.4. After 1 week of fixation brains were transferred to 30% sucrose solution for at least 2 d until they sank. Brains were then sliced to 100 μm sections, transferred onto gelatin-coated glass slides, and stained with cresyl violet, and images were taken to ensure correct position of recording probes in the brain. Locations of the electrodes were mapped on coronal brain sections from a standard atlas [20]. All nonconfirmed experiments were discarded from the analysis.
Electrophysiology recordings
The recording configuration for simultaneous electrophysiological recordings, stimulation, and choline amperometry is shown in Fig. 1A. To enable performance of simultaneous electrophysiology and amperometry, we used relatively low impedance bipolar local field potential (LFP) recording electrodes without a separate electrophysiological ground to avoid shunting amperometry signals. Before recordings, a bipolar Teflon-coated stainless steel electrode (50-100 kΩ resistance, PlasticsOne, E363/2-2TW) was stereotactically placed in the left dorsal hippocampus [anteroposterior (AP),-3.8; mediolateral (ML), -2.5; superior–inferior (SI), −2.6] targeting CA1. A second bipolar electrode was implanted in the left OFC (AP, +4.2; ML, −2.2; SI, −2.4). Acrylic dental cement (Lang Dental Mfg. Co., Inc, USA) was used to affix the electrodes to a steel screw (0-80 × 3/32, PlasticsOne) implanted just caudal to the burr holes. These two electrodes were implanted with the tips lying in the coronal plane. A third bipolar electrode was placed in the sagittal plane in the septal area, targeting the intermediate part of the lateral septal nuclei (AP, +0.2; ML, +0.5; SI, −5.0) on the right side. The two tips of the electrodes were separated by approximately 0.5 mm and approximately 0.5 mm of insulation was shaved off. For choline recordings, a choline microelectrode (described below) was implanted using an angled approach at 20º to the midline, targeting the right OFC (AP, +4.2; ML, +2.2; SI, −2.4). All stereotactic coordinates were taken from Paxinos and Watson [20] and were measured in millimeters relative to bregma.
Figure 1.

Recording setup and examples of delta power changes in OFC. (A) Schematic diagram of animal preparation for experiments. Bipolar electrodes for stimulating and/or recording local field potential (LFP) were placed in hippocampus (HC) and in the orbital frontal cortex (OFC) on the left side, as well as in the lateral septum (LS) on the right side (left and right sides are not indicated on the diagram). In addition, a choline microelectrode was implanted into the OFC on the right side (homologous coordinates to left OFC LFP electrodes). (B) An example of LFP recordings during lateral septal stimulation under light anesthesia. LFP signals from LS, HC and OFC are shown. Electrical stimulation of LS at 3 Hz causes slow oscillations in the OFC, followed by return to fast activity during the recovery period. (C) An example of LFP recordings during hippocampal stimulation under light anesthesia. LFP signals from LS, HC and OFC are presented. Electrical stimulation of HC at 3 Hz causes no obvious changes in either LS or OFC. (D) Example of LFP recordings in LS, HC and OFC during toe pinch. Animal is under deep anesthesia with slow wave activity in OFC at baseline. Toe pinch causes a decrease in slow wave activity in OFC which then returns during recovery.
LFP recordings were acquired using a Microelectrode AC Amplifier (model 1800; A-M Systems) and broad-band filtered from 0.1 Hz to 500 Hz (x1000 gain). Signals were digitized and recorded using a CED Power 1401 and Spike2 software (CED). Signals from bipolar electrodes were digitized at 1,000 Hz.
Regional stimulation and toe pinch
After electrode placement for electrophysiology and choline measurements, the animals were initially in a state of relatively deep anesthesia. Toe pinches were administered to activate cortical arousal [21, 22] with toe pinch administered for 1 minute, released for 2 minutes, then administered a second time for 1 minute. Animals were then allowed to reach a light anesthesia state using ketamine/xylazine (40/7 mg/kg) as described previously [7], with the frequency of large, positively deflected delta oscillations on frontal cortex LFPs decreased to less than three per 10 s of recordings, compared with a frequency of ~12-15 delta waves per 10 s under deep anesthesia. Regional stimulations were performed from this light anesthesia state via the bipolar electrode implanted in the lateral septal nuclei or dorsal hippocampus (described above) using a 60 second, 3 Hz, 1 millisecond each phase biphasic pulse stimulation (50-100 μA). Stimulus intensity for lateral septal stimulation was increased until slow oscillations were induced in OFC, keeping the stimulus ≤ 100 μA. Any stimuli in which epileptiform discharges were seen in HC, OFC or LS electrodes were excluded from the analysis. Stimulus frequencies of lateral septum above 3 Hz (e.g. 10 Hz) had been used in prior studies [8] but were more prone to induce epileptiform activity so were not used here. Once slow activity was induced by the lateral septal stimulus, the same current was then given to the hippocampal area to detect the LFP and choline changes in OFC.
Analysis of delta frequency LFP power in the OFC
LFP data were analyzed by defining “baseline” (last 60 s of recordings before the stimulus or toe pinch), and “stimulation” (the whole 60 s recordings of the stimulus or toe pinch). LFP signals were subjected to fast Fourier transform (FFT) analysis in Spike2 to generate power spectra. Delta-range (1-4 Hz) LFP activity changes during the stimulation periods were used for the analysis as compared to baseline. Results were presented as percent change by calculation of 100 × (stimulation – baseline)/baseline.
Amperometry recordings
Choline-oxidase enzyme coated amperometric biosensors (Quanteon) were used to perform the choline measurements on a FAST system (FAST16- mkI, Quanteon), and this kind of enzyme-based amperometry allowed us to measure choline levels as a proxy for extracellular acetylcholine with sub-second timing [23]. Ceramic-based microelectrodes, with 4 rectangular (15 × 333 μm) platinum recording sites in side-by-side pairs, were purchased uncoated (Quanteon, S2) and prepared with choline oxidase enzyme coating in-house following the manufacturer’s instructions. Electrode preparation has been described elsewhere [23]. In brief, we used a cross-linked mesh of choline oxidase (Sigma-Aldrich, C5896-100UN), gluteraldehyde (Sigma-Aldrich, G5882 −10x1ML), and bovine serum albumin (Sigma-Aldrich, A3059-10g) to coat the lower two (“coated”) electrodes while the upper two (“sentinel”) electrodes were coated with the same mixture without choline oxidase. The electrodes were kept for 3-7 days at 4°C.
The microelectrodes were then coated with metaphenylenediamine (m-PD) (78450, Sigma-Aldrich) via electropolymerization in order to block electroactive interferents other than choline, such as ascorbic acid (AA) or dopamine (DA). After m-PD coating, calibrations were performed in vitro with fixed potential amperometry both before and after the experiment. A constant voltage of −0.7 V was applied versus Ag/AgCl reference electrode in a beaker containing 40 ml of 0.05 M PBS. Amperometric currents were digitized at 5 Hz. Following a stable baseline of current signal, aliquots of AA, choline, DA, and peroxide were added to achieve final concentrations of 250μM AA; 20, 40, and 60 μM choline, 2 μM DA, and 8.8 μM peroxide. Only electrodes passing the following criteria were included: > 4 pA/μM sensitivity for detecting choline on the coated electrodes; limit of detection (LOD) < 350 nM choline; ratio of selectivity for choline and AA, >180:1; linearity for detection of increasing analyte concentrations (20-60 μM) on coated electrodes, Pearson’s correlation (R2) > 0.99.
The microelectrode was slowly lowered targeting the right OFC (AP, +4.2; ML, +2.2; SI, −2.4) to find the choline signal. When the choline signal dramatically increased after lowering, we kept the electrode fixed. When the signal returned to baseline, toe pinches were administered under deep anesthesia. Electrical stimulations were then initiated when the animals came to light anesthesia. At least 10 minutes were allowed for recovery between successive stimuli. All electrode positions were confirmed by histology after completion of experiments.
Choline analysis
Coated electrodes were used for recordings in vivo by the in vitro calibration inclusion criteria (mentioned above). Sentinel electrodes were only excluded if they badly malfunctioned during the transfer between in vitro calibration and in vivo recordings. All the signals were first smoothed with a subtraction of a 10-point moving average. Choline signals were calculated as the difference between coated and sentinel electrodes. The last 5 seconds prior to electrical stimulation and the first 10 seconds of stimulation were removed for both display and statistical purposes in order to eliminate large artifacts generated by initiation of the stimulus. The choline data were analyzed by defining “baseline” (60 s), “stimulation” (50 s), and “recovery” (first 60 s following the end of stimulation). The recovery period of one animal was excluded from the hippocampal stimulation group because of an unusual artifact during the recovery period. For the toe pinch data, we defined “baseline” (last 60 s before toe pinch), “stimulation” (whole 60 s of toe pinch), and “recovery” (first 60 s after toe pinch off).
Statistical analysis
All LFP and choline data during electrical stimulation (or toe pinch) were compared with that in baseline, using a two-tailed one sample t test, and significance was assessed at P < 0.05 with Bonferroni correction. Results are reported as mean ± SEM. For most experiments, one recording was obtained per animal, but in any instances in which more than one sample was obtained, these were first pooled by averaging within animal and then subjected to group statistics across animals.
Results
Lateral septal stimulation and toe pinch cause reciprocal changes in cortical delta power
We stimulated the lateral septum and hippocampus in lightly anesthetized rats for 60 s using 3 Hz stimulus trains below seizure threshold. We observed that lateral septal stimulation could induce large-amplitude neocortical slow waves (Figure 1B). The slow wave activity had maximal power at about 1 Hz, which was not synchronous with the stimulus frequency (3 Hz). On average, stimulation of the lateral septum produced a significant elevation in cortical delta frequency LFP power compared to baseline (see Figure 3A, 11 animals, 139.4 ± 26.6%, two-tailed one sample t test Bonferroni corrected, P < 0.05). In contrast, no obvious changes could be seen in frontal cortical activity during identical stimulation of hippocampus (Figure 1C), and the delta frequency LFP power in the OFC during hippocampal stimulation also had no statistical difference compared with that in baseline (Figure 3A, 6 animals, 4.2 ± 6.5%, two-tailed one sample t test Bonferroni corrected, P > 0.05).
Figure 3.
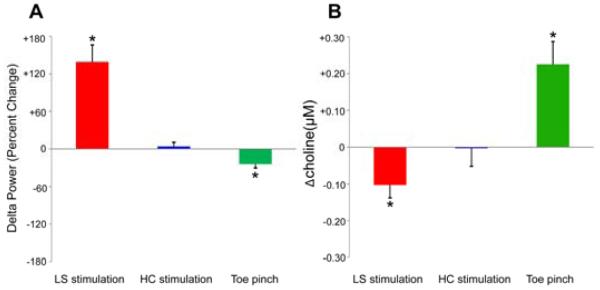
Reciprocal changes in delta power and choline signals with septal stimulation and toe pinch. Group data for orbital frontal cortical changes in LFP delta power and choline levels during electrical stimulation of lateral septum (LS) and hippocampus (HC) or during toe pinch. (A) LFP delta power changes in OFC during electrical stimulation in LS and HC or toe pinch compared to baseline. Stimulation of LS produced a statistically significant increase in cortical delta frequency LFP power, whereas stimulation of hippocampus caused no change in delta frequency LFP power. In addition, the delta frequency LFP power in OFC decreased significantly during toe pinch. Number of animals = 11 (LS stimulation); 6 (HC stimulation); 14 (toe pinch). (B) Mean ictal changes of choline levels in OFC relative to 60 s baseline are shown during LS or HC stimulation along with mean changes during toe pinch. Choline has a statistically significant decrease in OFC during lateral septal stimulation, whereas no significant difference can be detected during stimulation of HC. Toe pinch elicited a significant choline increase in OFC. Number of animals = 11 (LS stimulation); 6 (HC stimulation); 13 (toe pinch). Results are presented as mean (+/- SEM), two-tailed one sample t test Bonferroni corrected, *P < 0.05.
In contrast, toe pinch stimuli under deep anesthesia elicited a decrease in cortical delta power (Figure 1D). On average, toe pinch elicited a significant decrease of delta frequency LFP power as compared with baseline (Figure 3A, 14 animals, -24.5 ± 6.0%, two-tailed one sample t test Bonferroni corrected, P < 0.05).
Changes in cortical delta power are accompanied by reciprocal changes in cortical choline
To explore the relationship between cholinergic neurotransmission and neocortical physiological arousal during lateral septal stimulation and toe pinch, we measured choline levels using high-temporal resolution amperometry [23]. Our findings revealed that choline had a significant decrease in the OFC during lateral septal stimulations (Figure 2A, 3B; 11 animals, −0.102 ± 0.035 μM, two-tailed one sample t test Bonferroni corrected, P < 0.05). The choline signal declined during the stimulations and then gradually returned to baseline levels as the animals recovered (Figure 2A). Simultaneous recordings of the hippocampal and cortical LFP showed cortical slow oscillations without a hippocampal seizure during the lateral septal stimulation followed by normal cortical fast activity after recovery (Fig. 1B). For comparison, we also detected cholinergic transmission in the same area during stimulations in hippocampus with the same stimuli used for lateral septum. In contrast to lateral septal stimulations, the choline signal did not show any significant changes during the hippocampal stimulations as compared with baseline (Figure 2B, 3B; 6 animals, −0.003 ± 0.049 μM, two-tailed one sample t test Bonferroni corrected, P > 0.05).
Figure 2.

Septal stimulation and toe pinch cause opposite changes in cortical choline signals. (A) Choline recordings decrease in the OFC during lateral septal stimulation, with gradual recovery to baseline during the recovery period. (B) Choline signals have no obvious change when stimulating the hippocampus. (C) Choline signals during toe pinch have a large increase followed by recovery towards baseline. Data are presented as mean (± SEM). Number of animals = 11 (A), 6 (B), 13 (C).
Toe pinch is known to increase cortical arousal [21], and was therefore performed to evaluate the cholinergic transmission in OFC. When the animals were in deep anesthesia, toe pinch was administered for 60 seconds, and the choline signal had a large increase followed by a decrease after toe pinch ended (Figure 2C). On average, the increase of choline levels in OFC was significant during toe pinch as compared with baseline (Figure 3B; 13 animals, +0.225 ± 0.062 μM, two-tailed one sample t test Bonferroni corrected, P < 0.05).
Discussion
In the present study, we investigated choline signals in frontal cortex during lateral septal electrical stimulation in rats. We report, for the first time, a significant choline decrease along with synchronized slow oscillations in frontal cortex during lateral septal stimulation. These slow waves have been shown previously to include up and down states of neuronal firing similar to those seen in other states of depressed cortical function such as deep sleep, coma or limbic seizures [5-8, 24, 25]. The present findings suggest that septal activation is capable of switching the cortex into a state of decreased function and reduced cholinergic arousal. Based on our current findings and previous work [8]this pathway may play an important role in cortical network switching during the transition to impaired consciousness in limbic seizures. Similar mechanisms may also operate under normal conditions such as the transition to slow wave sleep or even during normal cognition. Indeed, previous work has shown that pharmacological blockade of cholinergic activity is capable of reducing cortical activation [26-30]; the main novel finding of the present work is to demonstrate that increased septal activity is capable of reducing cholinergic neurotransmission in conjunction with decreased cortical physiological arousal.
The network inhibition hypothesis posits that activation in one brain region, such as limbic hippocampal circuits, is capable of reducing activity in another region such as the fronto-parietal association cortex through activation of subcortical inhibitory structures which selectively inhibit subcortical arousal [3, 6, 9]. Briefly, the activation in the hippocampus could propagate (via fimbria-fornix and other pathways) to increase activity in inhibitory and sleep-promoting regions such as the anterior hypothalamus and lateral septum; as a consequence, subcortical arousal systems including the thalamus, basal forebrain, pedunculopontine tegmental nucleus, and other regions could show depressed activity, thus leading to deactivation and slow waves in the frontal cortex [11]. Interestingly, there is regional selectivity to the slow wave activity and depressed cortical metabolism observed in both limbic seizures and normal slow wave sleep, with the strongest changes occurring in the default mode network [31, 32]. We can thus speculate that in addition to promoting cortical slow waves during seizures or deep sleep, a milder level of septal activation and regional cholinergic withdrawal might contribute to switching of anti-correlated task-positive and default mode networks under normal conditions [1, 33].
As is known, the hippocampus and septum are intimately connected anatomically and functionally, and hippocampal seizures or stimulations can “propagate” to the LS [6-8]. However, in our current study, we applied a milder level of stimulation (≤ 100μA) to either HC or LS, and no obvious propagation between these structures was seen. Nevertheless, stimulation of LS (but not HC), produced robust slow oscillations in OFC (Figure 1B, C), suggesting the effect of LS stimulation is not via the HC.
Aside from the lateral septal nuclei, other subcortical inhibitory nuclei could potentially also contribute to network switching through modulation of subcortical arousal. For example, the anterior hypothalamus and ventrolateral pre-optic area are known to have GABAergic neurons projecting widely to subcortical arousal structures [34] and the thalamic reticular nucleus likewise has projections to midbrain arousal circuits [35]. In addition to acetylcholine, multiple other parallel neurotransmitter systems contribute to subcortical arousal and could play a role in network switching [34]. It has also been shown that stimulation of several other subcortical sites can produce cortical slow waves [36] although the results described here and previously suggest some regional selectivity since slow waves were elicited by stimulation of the lateral septum but not by identical stimuli to the hippocampus or mediodorsal thalamus [8]. Moreover, although cholinergic systems play an important role in arousal, it is likely that not only the cholinergic arousal system is modulated but other neuromodulatory systems as well, since there are several potential parallel pathways of cortical and subcortical excitation [34, 37]. Hence, much further work is needed to more fully identify and characterize the subcortical network mechanisms potentially contributing to normal and abnormal cortical network switching.
In conclusion, we found a decrease of choline levels in the cortex, along with cortical slow oscillations during lateral septal electrical stimulations. Together these findings suggest that subcortical cholinergic arousal is suppressed during lateral septal stimulation, which might play a critical role in the cortical deactivation including cortical slow oscillations and reduced cortical function. If confirmed through further studies, this could suggest a new mechanism underlying the initiation of cortical slow wave activity as well as normal long-range switching between anti- correlated cortical networks.
Highlights.
In this study we propose and investigate the novel hypothesis that lateral septal activation may depress cortical activity through inhibition of subcortical cholinergic arousal.
We used simultaneous electrophysiology and high time-resolution amperometry to measure cortical choline levels as a marker of acetylcholinergic neurotransmission during lateral septal stimulation.
We found a decrease of choline levels in the cortex, along with cortical slow oscillations during lateral septal stimulation.
Our findings offer a new potential mechanism for initiating transitions in cortical activity through inhibitory subcortical regions (e.g. lateral septum) decreasing subcortical arousal (e.g. acetylcholine) which may have broad implications for both normal and abnormal cortical network function.
Acknowledgements
We thank Quanteon LLC for technical support for the amperometry measurements. This work was supported by NIH R01 NS066974, the Swebelius Fund, and the Betsy and Jonathan Blattmachr Family (HB); NIH P30 NS052519 (FH); NIH F30 NS071628 and MSTP TG T32GM07205 (JEM), a China Scholarship Council Postgraduate Scholarship Program award CSC 201206190071 (WL); and a China Scholarship Council Postgraduate Scholarship Program award CSC 201206370075 (QZ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no competing financial interests.
References
- [1].Fox MD, Snyder AZ, Vincent JL, et al. The human brain is intrinsically organized into dynamic, anticorrelated functional networks. Proc Natl Acad Sci U S A. 2005;102:9673–8. doi: 10.1073/pnas.0504136102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature. 2006;441:589–94. doi: 10.1038/nature04767. [DOI] [PubMed] [Google Scholar]
- [3].Blumenfeld H. Impaired consciousness in epilepsy. Lancet Neurol. 2012;11:814–26. doi: 10.1016/S1474-4422(12)70188-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Steriade M, Amzica F, Nunez A. Cholinergic and noradrenergic modulation of the slow (approximately 0.3 Hz) oscillation in neocortical cells. J Neurophysiol. 1993;70:1385–400. doi: 10.1152/jn.1993.70.4.1385. [DOI] [PubMed] [Google Scholar]
- [5].Blumenfeld H, Rivera M, McNally KA, et al. Ictal neocortical slowing in temporal lobe epilepsy. Neurology. 2004;63:1015–21. doi: 10.1212/01.wnl.0000141086.91077.cd. [DOI] [PubMed] [Google Scholar]
- [6].Englot DJ, Yang L, Hamid H, et al. Impaired consciousness in temporal lobe seizures: role of cortical slow activity. Brain. 2010;133:3764–77. doi: 10.1093/brain/awq316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Englot DJ, Mishra AM, Mansuripur PK, et al. Remote effects of focal hippocampal seizures on the rat neocortex. J Neurosci. 2008;28:9066–81. doi: 10.1523/JNEUROSCI.2014-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Englot DJ, Modi B, Mishra AM, et al. Cortical deactivation induced by subcortical network dysfunction in limbic seizures. J Neurosci. 2009;29:13006–18. doi: 10.1523/JNEUROSCI.3846-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Norden AD, Blumenfeld H. The role of subcortical structures in human epilepsy. Epilepsy Behav. 2002;3:219–231. doi: 10.1016/s1525-5050(02)00029-x. [DOI] [PubMed] [Google Scholar]
- [10].Vanini G, Watson CJ, Lydic R, Baghdoyan HA. Gamma-aminobutyric acid-mediated neurotransmission in the pontine reticular formation modulates hypnosis, immobility, and breathing during isoflurane anesthesia. Anesthesiology. 2008;109:978–88. doi: 10.1097/ALN.0b013e31818e3b1b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Englot DJ, Blumenfeld H. Consciousness and epilepsy: why are complex-partial seizures complex? Prog Brain Res. 2009;177:147–70. doi: 10.1016/S0079-6123(09)17711-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lee WL, Hablitz JJ. Initiation of epileptiform activity by excitatory amino acid receptors in the disinhibited rat neocortex. J Neurophysiol. 1991;65:87–95. doi: 10.1152/jn.1991.65.1.87. [DOI] [PubMed] [Google Scholar]
- [13].Racine RJ, Chapman CA, Teskey GC, Milgram NW. Post-activation potentiation in the neocortex. III. Kindling-induced potentiation in the chronic preparation. Brain Res. 1995;702:77–86. doi: 10.1016/0006-8993(95)01024-9. [DOI] [PubMed] [Google Scholar]
- [14].Colom LV, Garcia-Hernandez A, Castaneda MT, Perez-Cordova MG, Garrido-Sanabria ER. Septo-hippocampal networks in chronically epileptic rats: potential antiepileptic effects of theta rhythm generation. J Neurophysiol. 2006;95:3645–53. doi: 10.1152/jn.00040.2006. [DOI] [PubMed] [Google Scholar]
- [15].Risold PY, Swanson LW. Connections of the rat lateral septal complex. Brain Res Brain Res Rev. 1997;24:115–95. doi: 10.1016/s0165-0173(97)00009-x. [DOI] [PubMed] [Google Scholar]
- [16].Irle E, Markowitsch HJ. Afferent connections of the substantia innominata/basal nucleus of Meynert in carnivores and primates. J Hirnforsch. 1986;27:343–67. [PubMed] [Google Scholar]
- [17].Mesulam MM, Mufson EJ. Neural inputs into the nucleus basalis of the substantia innominata (Ch4) in the rhesus monkey. Brain. 1984;107:253–74. doi: 10.1093/brain/107.1.253. Pt 1. [DOI] [PubMed] [Google Scholar]
- [18].Mesulam MM. Principles of behavioral and cognitive neurology. 2nd. Oxford University Press; Oxford ; New York: 2000. p. 540. xviii. [Google Scholar]
- [19].Varoqueaux F, Poulain P. Projections of the mediolateral part of the lateral septum to the hypothalamus, revealed by Fos expression and axonal tracing in rats. Anat Embryol (Berl) 1999;199:249–63. doi: 10.1007/s004290050226. [DOI] [PubMed] [Google Scholar]
- [20].Paxinos G, Watson C. The Rat Brain: In Stereotaxic Coordinates. 4th Academic Press, Incorporated; 1998. [Google Scholar]
- [21].Mena-Segovia J, Sims HM, Magill PJ, Bolam JP. Cholinergic brainstem neurons modulate cortical gamma activity during slow oscillations. J Physiol. 2008;586:2947–60. doi: 10.1113/jphysiol.2008.153874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang H, Lin SC, Nicolelis MA. Acquiring local field potential information from amperometric neurochemical recordings. J Neurosci Methods. 2009;179:191–200. doi: 10.1016/j.jneumeth.2009.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Parikh V, Pomerleau F, Huettl P, et al. Rapid assessment of in vivo cholinergic transmission by amperometric detection of changes in extracellular choline levels. Eur J Neurosci. 2004;20:1545–54. doi: 10.1111/j.1460-9568.2004.03614.x. [DOI] [PubMed] [Google Scholar]
- [24].Steriade M, Contreras D, Curro Dossi R, Nunez A. The slow (< 1 Hz) oscillation in reticular thalamic and thalamocortical neurons: scenario of sleep rhythm generation in interacting thalamic and neocortical networks. J Neurosci. 1993;13:3284–99. doi: 10.1523/JNEUROSCI.13-08-03284.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Haider B, Duque A, Hasenstaub AR, McCormick DA. Neocortical network activity in vivo is generated through a dynamic balance of excitation and inhibition. J Neurosci. 2006;26:4535–45. doi: 10.1523/JNEUROSCI.5297-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Levin ED, McClernon FJ, Rezvani AH. Nicotinic effects on cognitive function: behavioral characterization, pharmacological specification, and anatomic localization. Psychopharmacology (Berl) 2006;184:523–39. doi: 10.1007/s00213-005-0164-7. [DOI] [PubMed] [Google Scholar]
- [27].Bushnell PJ, Oshiro WM, Padnos BK. Detection of visual signals by rats: effects of chlordiazepoxide and cholinergic and adrenergic drugs on sustained attention. Psychopharmacology (Berl) 1997;134:230–41. doi: 10.1007/s002130050446. [DOI] [PubMed] [Google Scholar]
- [28].Rezvani AH, Bushnell PJ, Levin ED. Effects of nicotine and mecamylamine on choice accuracy in an operant visual signal detection task in female rats. Psychopharmacology (Berl) 2002;164:369–75. doi: 10.1007/s00213-002-1221-0. [DOI] [PubMed] [Google Scholar]
- [29].Rezvani AH, Kholdebarin E, Cauley MC, Dawson E, Levin ED. Attenuation of pharmacologically-induced attentional impairment by methylphenidate in rats. Pharmacol Biochem Behav. 2009;92:141–6. doi: 10.1016/j.pbb.2008.11.005. [DOI] [PubMed] [Google Scholar]
- [30].McGaughy J, Sarter M. Effects of chlordiazepoxide and scopolamine, but not aging, on the detection and identification of conditional visual stimuli. J Gerontol A Biol Sci Med Sci. 1995;50:B90–6. doi: 10.1093/gerona/50a.2.b90. [DOI] [PubMed] [Google Scholar]
- [31].Danielson NB, Guo JN, Blumenfeld H. The default mode network and altered consciousness in epilepsy. Behav Neurol. 2011;24:55–65. doi: 10.3233/BEN-2011-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Nir Y, Massimini M, Boly M, Tononi G. Neuroimaging of Consciousness. Springer-Verlag Berlin Heidelberg; 2013. Sleep and consciousness; pp. 133–182. [Google Scholar]
- [33].Dosenbach NU, Fair DA, Miezin FM, et al. Distinct brain networks for adaptive and stable task control in humans. Proc Natl Acad Sci U S A. 2007;104:11073–8. doi: 10.1073/pnas.0704320104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–63. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- [35].Parent A, Steriade M. Midbrain tegmental projections of nucleus reticularis thalami of cat and monkey: a retrograde transport and antidromic invasion study. J Comp Neurol. 1984;229:548–58. doi: 10.1002/cne.902290408. [DOI] [PubMed] [Google Scholar]
- [36].Steriade MM, McCarley RW. Brain Control of Wakefulness and Sleep. 2nd Springer; 2005. [Google Scholar]
- [37].Espana RA, Scammell TE. Sleep neurobiology from a clinical perspective. Sleep. 2011;34:845–58. doi: 10.5665/SLEEP.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]