

Abstract
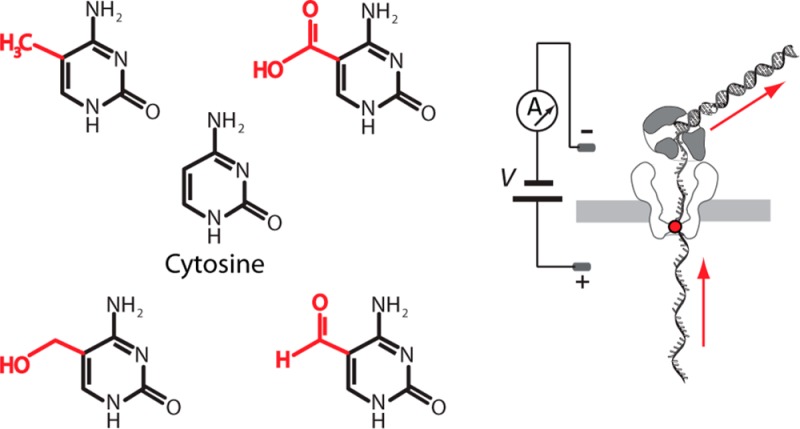
Individual DNA molecules can be read at single nucleotide precision using nanopores coupled to processive enzymes. Discrimination among the four canonical bases has been achieved, as has discrimination among cytosine, 5-methylcytosine (mC), and 5-hydroxymethylcytosine (hmC). Two additional modified cytosine bases, 5-carboxylcytosine (caC) and 5-formylcytosine (fC), are produced during enzymatic conversion of hmC to cytosine in mammalian cells. Thus, an accurate picture of the cytosine epigenetic status in target cells should also include these C5-cytosine variants. In the present study, we used a patch clamp amplifier to acquire ionic current traces caused by phi29 DNA polymerase-controlled translocation of DNA templates through the M2MspA pore. Decision boundaries based on three consecutive ionic current states were implemented to call mC, hmC, caC, fC, or cytosine at CG dinucleotides in ∼4400 individual DNA molecules. We found that the percentage of correct base calls for single pass reads ranged from 91.6% to 98.3%. This accuracy depended upon the identity of nearest neighbor bases surrounding the CG dinucleotide.
Introduction
Epigenetic modifications of DNA contribute to gene regulation in biological cells.1 For example, 5-methylcytosine (mC) and 5-hydroxymethylcytosine (hmC) influence mammalian embryonic stem cell maintenance,2,3 angiogenesis,4 and development.5 Loss of proper epigenetic regulation has been associated with cancers.6−8 Recently, the family of Ten-Eleven Translocation (TET) proteins have been shown to oxidize mC into hmC and further oxidize hmC into 5-formylcytosine (fC) and 5-carboxylcytosine (caC).9,10 5-Formylcytosine is measurable in mouse embryonic stem cells,11 and initial studies have shown that fC and caC can reduce the rate of RNA polymerase II transcription.12
To date, genome-scale methylome analysis has primarily been based on bisulfite sequencing13 or on variations of that technique designed to distinguish mC from hmC14 and fC.15 An alternative fluorescence-based technique for DNA methylation detection has been implemented by Pacific Biosciences.16,17 It builds upon single-molecule real-time (SMRT) DNA sequencing. The Pacific Biosciences instrument detects base-specific fluorescent leaving groups during nucleotide incorporation by a DNA polymerase at the base of a zero mode waveguide. Modified cytosine bases on the template strand are detected by monitoring the interpulse duration as guanine nucleotides are incorporated into the daughter strand.16,17
Two groups have demonstrated that a nanopore device can discriminate among cytosine (C), mC, and hmC at CG dinucleotides within single synthetic DNA molecules as they translocate processively through nanopores.18,19 Advances that enabled this technology were enzymatic control at single-nucleotide precision using DNA polymerases coupled to the pore20 and reading 3 to 4 nucleotide “words” using a mutant form of the Mycobacterium smegmatis porin (M2MspA).21
Here we used a nanopore device combining wild-type phi29 DNA polymerase (phi29 DNAP) and M2MspA to directly read and discriminate among all five C5-cytosine variants known to occur in mammalian genomes (Figure 1). Decision boundaries based on a random forest of trees algorithm gave single pass call accuracies that ranged from 91.6% to 98.3% depending upon nearest neighbor base identities.
Figure 1.
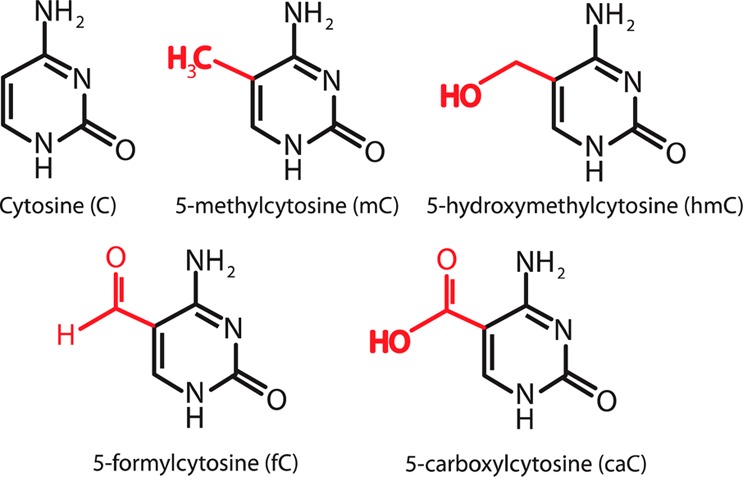
Five C5-cytosine variants examined in this study. Distinguishing functional groups are highlighted in red.
Experimental Section
Proteins
The M2MspA protein22 was expressed in E. coli using the SUMOpro Expression System (LifeSensors) and purified as described previously.18 It was stored at −20 °C in 10 mM Tris (pH 7.8), 150 mM NaCl, 50% glycerol. Wild-type bacteriophage phi29 DNAP was obtained from Enzymatics Corporation (833 000 U/mL; specific activity 83 000 U/mg) and stored at −20 °C in 10 mM Tris-HCl (pH 7.4), 100 mM KCl, 0.1 mM EDTA, 1 mM DTT, 50% glycerol.
DNA
Oligonucleotides were purchased from the Stanford Peptide and Nucleic Acid Facility and then purified using denaturing PAGE. The mC, fC, caC, and hmC phosphoramidites were purchased from Glen Research (Sterling, VA). DNA hybrids added to the cis well contacting the nanopore were composed of three strands (Figure 2a): an 82nt template strand that was read by the nanopore (Figure S1), a 55nt hairpin primer strand, and a 15nt blocking oligomer strand bearing an abasic tail. Template strands were constructed using a CAT trinucleotide repeat as a background sequence. Within this sequence we replaced an 'A' with four nucleotides (Figure 2a, red letters) of the form XnCGY, where X and Y composed all pairs of canonical bases (16 permutations). For each XnCGY context, we synthesized five template strands, each bearing one C5-cytosine variant at nC. To allow for concurrent analysis of all C5-cytosine variants for a given XnCGY context, we paired each C5-cytosine variant with a unique downstream label on the same strand (Figure 2a, blue letters).
Figure 2.

Strategy for reading C5-cytosine variants along individual DNA templates using a nanopore. (a) DNA hybrids used in this study. Each DNA hybrid is composed of three strands: an 82nt template strand (black), a 55nt hairpin primer (gray), and a blocking oligomer (yellow). Each template strand is composed mainly of a CAT trinucleotide repeat. Within that repeat, we replaced an ‘A’ with 1 of 80 possible XnCGY 4mers where X = G, A, T, or C, Y = G, A, T, or C, and nC (red dot) = C, mC, hmC, fC, or caC. 10nt toward the 5′ terminus of the template strand, we replaced three nucleotides with a label sequence (blue letters and dot) to independently identify the cytosine variant present on the strand. In the blocking oligomer strand (yellow), “a” represents an abasic residue and “b” represents a C3P spacer used to prevent phi29 DNAP exonuclease activity. (b) A cartoon representation of strand translocation through the nanopore, and associated ionic current trace for a single event. This particular event is for a DNA template bearing GfCGC at the XnCGY position and the associated TTT label. (i) Open channel ionic current. A constant 180 mV (trans-side positive) is applied across the nanopore. (ii) A DNA-phi29 DNAP complex is captured by the pore electric field. This results in a partial current blockade. (iii) The applied voltage pulls the template DNA strand (black arrow) through the pore forcing the DNA polymerase to act as a wedge that unzips the blocking oligomer. The C5-cytosine variant (red dot) moves into the pore constriction thus influencing the ionic current level in several segments. (iv) The blocking oligomer is completely removed, exposing the 3′-OH group of the hairpin primer strand. DNA synthesis by phi29 DNAP begins pulling the template strand back through the pore (red arrow). (v) The C5-cytosine variant (red dot) moves back into the constriction of the pore and is read (red horizontal lines in the trace denote the ionic current segments that were the focus of this study). The inset at right is an expanded view of that portion of the trace where all eight quantified ionic current segments are denoted by red lines. (vi) The label (blue dot) moves into the pore causing an ionic current signature (blue lines in trace) that identifies which cytosine variant was present on the strand. (i′) Synthesis continues to pull the template strand through the pore until it can no longer be retained by the pore electric field thus releasing the phi29 DNAP-DNA complex to the cis well, and restoring the open channel current. In the trace, the vertical dashed line marks a change in time scale.
The hybrids were made by combining the template, primer, and blocking oligomer at a 5:6:6 ratio in 0.1 M KCl, 10 mM Tris (pH 7.6), and 1 mM EDTA, followed by incubation at 95 °C for 2.5 min and snap cooling in an ice water bath. The presence of a single mC, hmC, fC, or caC in the GnCGC and CnCGG context was confirmed by liquid chromatography/mass spectrometry.
Nanopore Experiments
Single M2MspA pores were inserted into lipid bilayers in 0.3 M KCl, 10 mM HEPES/KOH (pH 8.00 ± 0.05) at 23 °C as previously described.21,22 Channels had open currents of ∼113 pA at 180 mV (constant), trans-side positive (Figure 2bi). Each experiment contained 5 DNA hybrids (1 μM each) that had identical template strands except for the C5-cytosine variant at position nC (C, mC, hmC, fC, or caC). The cis well also contained 1 mM DTT, 1 mM EDTA, 10 mM MgCl2, 1 mM of each dNTP, and 3.75 μM phi29 DNAP.
Data Collection
Ionic current detection and voltage control were performed using an integrating patch clamp amplifier (Axopatch 200B, Molecular Devices) in voltage clamp mode. Data were sampled at 100 kHz using an analog-to-digital converter (Digidata 1440A, Molecular Devices) in whole-cell configuration filtered at 5 kHz using a low-pass Bessel filter. Analysis of events was semiautomated using clampfit 10.4 software (Molecular Devices).
We analyzed “on pathway” events (Figure 2b) defined as follows: (1) the event must start from the open channel with a drop to about 40 pA (Figure 2bii), followed by the unzipping regime; (2) each of the eight quantified ionic current segments must be present in the synthesis regime (Figure 2b, inset); (3) the label (Figure 2bvi) must also be present following the eight quantified segments and a distinctive three ionic current segment caused by translocation of C-A-T nucleotides in series. The absence of required ionic current segments (criteria 2 and 3) was predominantly because the rate of nucleotide displacement past the nanopore sensor exceeded the rate of data acquisition.20 An on pathway event could contain an ionic current drop to near 0 pA as long as it did not occur during the eight quantified segments or the label segments. These criteria yielded ∼4400 events which were extracted and analyzed. They composed ∼69% of all events at least 1 s in duration. Total events for individual XnCGY contexts are listed in Table 1.
Table 1. Accuracies for Calling C5-Cytosine Variants Using a Random Forest Classifiera.
| context | count | accuracy |
|---|---|---|
| AnCGA | 250 | 96.6 (±0.54) |
| AnCGC | 250 | 95.9 (±0.72) |
| AnCGG | 251 | 91.9 (±0.57) |
| AnCGT | 250 | 92.7 (±0.93) |
| CnCGA | 287 | 94.0 (±0.55) |
| CnCGC | 251 | 97.0 (±0.36) |
| CnCGG | 330 | 93.3 (±0.61) |
| CnCGT | 250 | 93.4 (±0.94) |
| GnCGA | 250 | 93.6 (±0.75) |
| GnCGC | 513 | 98.3 (±0.22) |
| GnCGG | 250 | 94.2 (±0.69) |
| GnCGT | 250 | 96.5 (±0.65) |
| TnCGA | 250 | 95.6 (±0.81) |
| TnCGC | 250 | 95.0 (±0.66) |
| TnCGG | 250 | 95.6 (±0.61) |
| TnCGT | 259 | 91.6 (±1.19) |
Twenty iterations of 5-fold cross-validation were performed for each XnCGY context. For each iteration, an accuracy measurement was made (% correct across entire data set of C, mC, hmC, fC, and caC for that context). Column 3 is the mean and standard deviation of those 20 measurements. Column 2 is the total number of events quantified for each XnCGY context.
Machine Learning
The machine learning methods we employed were described in detail previously.18 Briefly, we used a forest of extremely randomized trees23 to select the ionic current segments whose mean average importance across all contexts was above that of a uniform importance model. It was found that segments 4, 5, and 6 were above this threshold. We used the mean of the ionic current values from the other five segments (1, 2, 3, 7, and 8) on a per-event basis to normalize against drift in ionic current over time. The accuracy of the random forest classifier for each XnCGY context was established using 5-fold cross-validation of the data for the three most important ionic current segments (see Figure 4). This cross-validation was performed 20 times, giving a distribution of error rates for each XnCGY context.
Figure 4.

Identification of ionic current segments important for discriminating among C5-cytosine variants. Each row represents an XnCGY 4mer, and each column represents one of the eight ionic current segments that we quantified. The “heat” of a cell is the relative importance of that segment in making a correct C5-cytosine variant classification. The relative importance of each segment was estimated using a forest of extremely randomized trees fit to each 4mer context. Each row was normalized to sum to 1.
Confusion Matrices
Confusion matrices for each XnCGY context (Figure S5) were generated by comparing C5-cytosine variant predictions (“calls”) made by the random forest classifier with the identifying labels for each C5-cytosine variant. To generate the merged confusion matrix (Table 2), the conditional probabilities for the 16 XnCGY contexts (approximately 4400 total translocation events) were summed for each cell and divided by 16.
Table 2. Merged Confusion Matrix for C5-Cytosine Variant Callsa.
| calls | |||||
|---|---|---|---|---|---|
| labels | C | mC | hmC | fC | caC |
| TGG | 0.90 | 0.04 | 0.05 | 0.01 | 0.00 |
| TCA | 0.02 | 0.96 | 0.01 | 0.00 | 0.01 |
| TCa | 0.03 | 0.01 | 0.93 | 0.02 | 0.00 |
| TTT | 0.01 | 0.01 | 0.06 | 0.92 | 0.00 |
| aaa | 0.00 | 0.02 | 0.01 | 0.00 | 0.97 |
Each entry is the fraction of events in a given category averaged over all 16 XnCGY 4mer contexts. The fraction of calls that were correct (as determined by the label) is highlighted in bold font. Incorrect calls (as determined by the label) are in standard font. The total number of events is the sum of those enumerated in Table 1. The overall mean error rate for these experiments is the average of the diagonal of this confusion matrix, weighted by the proportion of events bearing each cytosine variant. See Figure 2 for description of labels.
Mass Spectrometry
We previously used liquid chromatography-tandem mass spectrometry (LC-MS/MS) to verify the presence of C, mC, and hmC in CnCGG and GnCGC bearing oligomers run on the nanopore by analyzing oligonucleotides digested to nucleoside monophosphate components.18 In this study, we extended our analysis to fC- and caC-containing strands using a protocol that analyzed deoxynucleosides. 2 μg of each of the ten 82mer oligonucleotides (5 for each context: CnCGG and GnCGC) were digested to deoxynucleosides using 2 units of DNA Degradase Plus (Zymo) in 25 μL of 1X degradase buffer at 37 °C for 12 h. These reactions were diluted with 50 μL of filtered water and run through NanoSep3K Omega spin columns (Pall Corporation) to separate the deoxynucleosides from the enzyme and undigested DNA. Standard deoxynucleosides were obtained from Sigma (dC), Berry and Associates (hmC, fC, caC), and US Biological (mC). A standard cocktail was prepared containing 10 μM each deoxycytidine, mC, hmC, fC, and caC in 1X degradase buffer. 20 μL of standards or 500 ng of digested sample DNA were analyzed by LC-MS/MS on a Thermo Finnigan LTQ mass spectrometer (Thermo) at the University of California Santa Cruz Mass Spectrometry Facility. Reversed-phase HPLC was performed exactly as described in ref (17) and is quoted verbatim as follows: “Reversed-phase HPLC was done with a Synergi Hydro 4-μm Fusion-RP 80A column (150 mm × 2.00 mm diameter; 4-μm particle size) (Phenomenex). Solvent A was 0.1% formic acid in water. Solvent B was 0.1% formic acid in methanol. The gradient was as follows: time (t) = 0–3 min, 100% solvent A; t = 3–5 min, 70% solvent A, 30% solvent B; t = 5–10 min, 10% solvent A, 90% solvent B; t = 10–20 min, 100% solvent B. The flow rate for chromatography was 200 μL/min.” Following HPLC the eluant was processed by MS in positive mode over a full scan range of m/z 225–295, followed by MS/MS scans from the global time scheduled list of five compounds: m/z = 228.2 (C), 242.2 (mC), 256.2 (hmC), 272.2 (fdC), 258.2 (caC). The electrospray voltage was 4.5 kV. The collision-induced dissociation at a normalized collision energy of 35% was used for MS/MS. XCalibur software (Thermo) was used to analyze the data. MS/MS for the modified dCs gave the distinct breakdown products with the same values as reported by others.24 The presence of C, mC, hmC, fC, and caC in our template samples was confirmed in MS/MS by the presence of 112, 126, 142, 140, 156 m/z ions, respectively. All 10 oligonucleotides tested contained the expected cytosine variant.
Results and Discussion
Comparison of Ionic Current Patterns for Five C5-Cytosine Variants in the GnCGC 4mer Context
A representative synthetic DNA hybrid used in this study is shown in Figure 2a. It is composed of three annealed synthetic DNA strands. Briefly, the 82nt template strand (Figure 2a, black) contains the CG dinucleotide that is the focus of this study. The cytosine of the dinucleotide (nC) can be C, mC, hmC, caC, or fC as is the case in the example (Figure 2a, red letters). The template bases that neighbor the CG dinucleotide (X and Y, Figure 2a) can each be any of the four canonical bases (in the example, G and C, respectively). Beginning 10nt toward the 5′ end of the CG dinucleotide, each template strand bears a 3nt marker (Figure 2a, blue letters) that causes an ionic current signature as it passes through the nanopore that independently identifies the cytosine variant (Figure 2b, blue lines). For example, the fC within the template shown in Figure 2a is identified by the ionic current caused by a TTT marker. The section of the template strand that is read by the nanopore is otherwise composed of CAT trinucleotide repeats to simplify analysis. The blocking oligomer (Figure 2a, yellow letters) anneals to the template strand and prevents phi29 DNAP from accessing the ssDNA/dsDNA primer–template junction in bulk phase. When the DNA template is captured in the nanopore, the blocking oligomer is unzipped and removed thus allowing polymerase-catalyzed synthesis to proceed.18,20 The hairpin primer (Figure 2a, gray letters) provides the 3′-OH that initiates DNA synthesis by phi29 DNAP.
A diagram of all steps during DNA translocation through the M2MspA pore is shown in Figure 2b along with a corresponding ionic current trace below. The trace was due to translocation of the DNA template bearing the target GfCGC 4mer (Figure 2a). Because our quantitative analysis focused on the ionic current readout during elongation of the daughter strand, the following discussion focuses in Figure 2b steps iv–vi. Briefly, once the blocking oligomer had been completely removed, the 3′-OH of the hairpin primer was positioned at the enzyme active site (Figure 2biv). At this stage, strand synthesis began which pulled the template strand through the pore (against the applied voltage) in single nucleotide steps. Initially, a series of three ionic current segments was repeatedly observed. This series corresponded to translocation of CAT trinucleotide repeats within the template.18,21 As synthesis proceeded, the target GfCGC 4mer entered and stepped through the pore constriction (Figure 2bv, red dot), and a series of eight distinct ionic current segments was observed (Figure 2bv and inset). Ten nucleotides further toward the 5′-terminus of the template strand, the three-nucleotide label passed through the pore sensor (Figure 2bvi, blue dot). In the example, the label is TTT. This resulted in distinct ionic current segments (Figure 2bvi, blue horizontal lines) some of which were used to establish the correct identity of the cytosine variant within the captured template (for example, the ∼25 pA step; Figure 2bvi, blue line). Similar eight-segment ionic current traces corresponding to each of the five cytosine variants in the target GnCGC 4mer are shown in Figure 3a along with their respective label traces (Figure 3b). Previously we showed that these labels did not alter the ionic current pattern arising from translocation of the target XnCGY 4mer.18 This was independently confirmed by label swap experiments (Figure S2).
Figure 3.
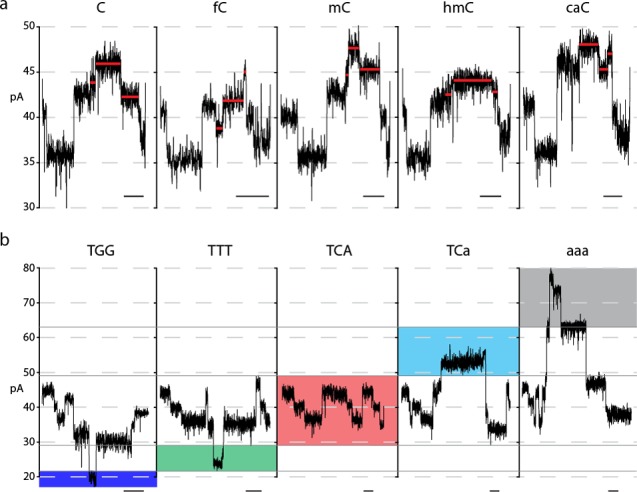
Representative ionic current traces for C5-cytosine variants in the GnCGC 4mer context. (a) Each panel shows the eight consecutive ionic current segments that we quantified. The cytosine variant present on the strand is denoted above the trace. Segments 4, 5, and 6, which differed the most between C5-cytosine variants, are highlighted with horizontal red lines. (b) Representative ionic current traces caused by translocation of the label sequence through the pore. The ionic current amplitude range used to identify the label (and by association, the C5-cytosine variant on the same strand) is highlighted by the shaded boxes: blue for TGG (C), green for TTT (fC), red for TCA (mC), cyan for TCa (hmC), and gray for aaa (caC) (‘a’ denotes an abasic residue). Traces for each panel of (a) and the panel directly below it (b) are from the same event. Time scale bars are 30 ms. We note that the labels were designed to give easily discernible ionic current readouts. Nonetheless, there could have been relatively small errors caused by label misreads. This means that the accuracies we give for C5-cytosine variant calls are conservative.
Ionic Current Patterns Caused by the Five C5-Cytosine Variants for All Nearest Neighbor Contexts
We acquired ionic current traces for all five possible C5-cytosine variants within all 16 possible nearest neighbor combinations in the target XnCGY context. Approximately 250 strands were captured and analyzed for each of these 16 DNA contexts giving a total of ∼4400 translocation events. Eight ionic current segments were extracted and quantified for each event. Representative traces for all XnCGY target 4mers are shown in the supplement (Figure S3).
In a prior study, we found that only three of the eight quantified ionic current segments were needed to achieve discrimination among C, mC, and hmC using the synthetic DNA template strand employed here.18 Initial inspection of the ionic current traces in this work (Figure 2bv, Figure 3, Figure S3, red lines) suggested that this would also be the case when all five cytosine variants were considered.
To test this quantitatively, we implemented a forest of extremely randomized trees.23 Briefly, a forest of extremely randomized trees is a set of decision tree classifiers where each classifier uses only a limited number of features among many that have been quantified. Features that are important for making a correct classification will be present in classifiers that perform well. Among the eight quantified segments, we found that segments 1–3, 7, and 8 had little importance for classifying C5-cytosine variants, while segments 4, 5, and 6 were consistently important (Figure 4).
Error Estimates for Calling C5-Cytosine Variants
Graphs of ionic current segment 4, 5, and 6 for each event in each XnCGY context revealed good separation between C5-cytosine variants (Figure 5, Figure S4). To establish this quantitatively, error estimates for calling C5-cytosine variants were generated using a random forest classifier and stratified 5-fold cross validation.18 Briefly, 80% of the data for a given XnCGY context were used to train a classifier and thus establish decision boundaries between the five C5-cytosine variants. These boundaries were then used to classify the remaining 20% of the data for that XnCGY context. Whether or not a given call was correct could then be established by comparison to its associated label. We repeated this process four times by holding out a different 20% data block and training on the other 80%. The sum of correct calls over the total number of calls gave a percent accuracy. To ensure that our accuracy estimates were not anomalously high or low due to an unrepresentative training set, this process was repeated 20 times using shuffled data for each XnCGY context.
Figure 5.
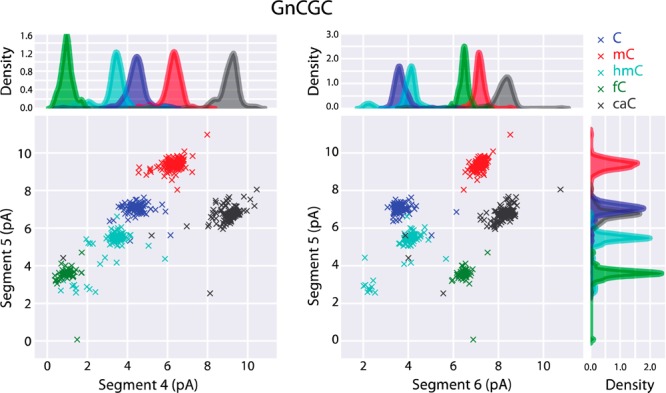
Graphical comparison of important ionic current segments that discriminate among C5-cytosine variants. In this representative case, each template strand contained the GnCGC 4mer context, where nC could be C, mC, hmC, fC, or caC. Because a mixture of all five cytosine variants was added to the cis well bathing a single M2MspA nanopore for each experiment, the downstream label on each template was used to establish the true variant identity at nC. In both panels, X’s represent normalized ionic current values for an individual DNA strand read one time. The color key for the C5-cytosine variant and its paired label is as follows: blue, cytosine/TGG; red, mC/TCA; cyan, hmC/TCa; green, fC/TTT; and gray, caC/aaa. For both panels, the Y-axis is normalized ionic current for segment 5. In the panel at left, the X-axis is normalized ionic current for segment 4; in the panel at right the X-axis is normalized ionic current for segment 6. Marginal histograms using kernel densities are shown for each of the three segments used in classification. Histogram colors correspond to colors assigned to X’s.
The percent accuracies for calling the five C5-cytosine variants are presented in Table 1. For the 16 XnCGY contexts, these accuracies ranged from 91.6% (TnCGT) to 98.3% (GnCGC) with an average accuracy of 94.7%. This accuracy is essentially equal to what we observed previously when we examined only three of the variants (C, mC, and hmC).18
It was useful to establish which cytosine variants were miscalled as one another. To this end, we compiled confusion matrices (Table 2, Figure S5) that compared strand labels (rows) against C5-cytosine variant calls (columns).
The merged data indicate that the largest errors occurred because C was frequently miscalled as mC or hmC (Table 2, row 1) or because fC was frequently miscalled as hmC (Table 2, row 4). Examination of confusion matrices for individual XnCGY contexts agreed (Figure S5). The 32 highest off-diagonal entries (10% of the 320 possible) included 21 of these three miscall classes. This is consistent with superposition of ionic current values for C5-variants in related 2-D plots (e.g., plots for TnCGA, GnCGA, TnCGC, CnCGT, and CnCGG, Figure S4).
Conclusions
We have shown that the identity of the five C5-cytosine bases known to occur in mammalian cells can be discriminated from one another when individual DNA strands are analyzed using a nanopore device. The single-pass call accuracy ranged from 91.6% to 98.3% depending upon neighboring nucleobases.
We note that this accuracy was measured for XnCGY 4mers inserted into a DNA template strand where the nucleobases at X and Y were the same within a given experiment, and where CAT repeats were present on either side of the 4mer in all cases. In other words, the accuracies we observed were for C5-cytosine variants within a controlled reference DNA sequence. This underestimates the error frequency that would be observed during de novo sequencing. It follows that early progress on nanopore-based epigenetic analysis of biological samples will be achieved using genomic DNA from organisms with well-documented reference sequences (e.g., mouse, mustard weed (Arabidopsis thaliana), and human).
Nanopore-based epigenetic analysis should be a useful complement to bisulfite-based methods due to several unique features of this emerging technology: (1) Genomic DNA is read directly by the nanopore, therefore errors caused by copying do not occur. (2) Genomic DNA can be retained in nanopores indefinitely; therefore, rereads of a captured DNA fragment could be performed and thus deliver high accuracy base calls provided the errors are random. This is likely to be essential when an unambiguous base call is needed at a specific position on one DNA strand. (3) Long reads of genomic DNA (>10kb) are plausible using nanopores; therefore, linkages between modified cytosines may be revealed that are biologically significant and otherwise difficult to discern. And, (4) all five C5-cytosine variants known to occur at CpG dinucleotides in mammalian genomes can be detected in one assay.
Acknowledgments
This work was supported by National Human Genome Research Institute Grant HG006321-02 (to M.A.).We thank Peter Walker at the Stanford Peptide and Nucleic Acid facility for oligonucleotide synthesis; Jeff Nivala for expression of the M2MspA protein; Kevin Karplus for advice on machine learning methods; Li Zhang, Robin Abu-Shumays, and Baldandorj Baatar for assistance with mass spectrometry analysis; and Hugh Olsen for editing the final draft.
Supporting Information Available
Figure S1 (nucleotide sequences of DNA template strands); Figure S2 (label swap experiment); Figure S3 (representative examples of the eight ionic current segments quantified for five C5-cytosine variants within each XnCGY context); Figure S4 (graphs comparing normalized ionic current values used to classify C5-cytosine variants for each XnCGY context); Figure S5 (confusion matrices for each XnCGY context). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): M.A. is a consultant to Oxford Nanopore Technologies, Oxford, U.K.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Medvedeva Y. A.; Khamis A. M.; Kulakovskiy I. V.; Ba-Alawi W.; Bhuyan M. S. I.; Kawaji H.; Lassmann T.; Harbers M.; Forrest A. R.; Bajic V. B. BMC Genomics 2014, 15, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wossidlo M.; Nakamura T.; Lepikhov K.; Marques C. J.; Zakhartchenko V.; Boiani M.; Arand J.; Nakano T.; Reik W.; Walter J. Nat. Commun. 2011, 2, 241. [DOI] [PubMed] [Google Scholar]
- Booth M. J.; Branco M. R.; Ficz G.; Oxley D.; Krueger F.; Reik W.; Balasubramanian S. Science 2012, 336, 934–937. [DOI] [PubMed] [Google Scholar]
- Shiva Shankar T. V.; Willems L. Vasc. Pharmacol. 2014, 60, 57–66. [DOI] [PubMed] [Google Scholar]
- Fu Y.; He C. Curr. Opin. Chem. Biol. 2012, 16, 516–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacolla A.; Cooper D.; Vasquez K. Genes 2014, 5, 108–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian C. G.; Xu Y. F.; Ceol C.; Wu F. Z.; Larson A.; Dresser K.; Xu W. Q.; Tan L.; Hu Y. G.; Zhan Q.; Lee C. W.; Hu D.; Lian B. Q.; Kleffel S.; Yang Y. J.; Neiswender J.; Khorasani A. J.; Fang R.; Lezcano C.; Duncan L. M.; Scolyer R. A.; Thompson J. F.; Kakavand H.; Houvras Y.; Zon L. I.; Mihm M. C.; Kaiser U. B.; Schatton T.; Woda B. A.; Murphy G. F.; Shi Y. J. G. Cell 2012, 150, 1135–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmfeldt L.; Mullighan C. G. Cancer Cell 2011, 20, 1–2. [DOI] [PubMed] [Google Scholar]
- Ito S.; Shen L.; Dai Q.; Wu S. C.; Collins L. B.; Swenberg J. A.; He C.; Zhang Y. Science (New York, N.Y.) 2011, 333, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Zhang Y. Genes Dev. 2011, 25, 2436–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffeneder T.; Hackner B.; Truβ M.; Munzel M.; Muller M.; Deiml C. A.; Hagemeier C.; Carell T. Angew. Chem., Int. Ed. Engl. 2011, 50, 7008–7012. [DOI] [PubMed] [Google Scholar]
- Kellinger M. W.; Song C. X.; Chong J.; Lu X. Y.; He C.; Wang D. Nat. Struct. Mol. Biol. 2012, 19, 831–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommer M.; McDonald L. E.; Millar D. S.; Collis C. M.; Watt F.; Grigg G. W.; Molloy P. L.; Paul C. L. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 1827–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth M. J.; Ost T. W. B.; Beraldi D.; Bell N. M.; Branco M. R.; Reik W.; Balasubramanian S. Nat. Protoc. 2013, 8, 1841–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth M. J.; Marsico G.; Bachman M.; Beraldi D.; Balasubramanian S. Nat. Chem. 2014, 6, 435–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flusberg B. A.; Webster D. R.; Lee J. H.; Travers K. J.; Olivares E. C.; Clark T. A.; Korlach J.; Turner S. W. Nat. Methods 2010, 7, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark T. A.; Lu X.; Luong K.; Dai Q.; Boitano M.; Turner S. W.; He C.; Korlach J. BMC Biol. 2013, 11, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber J.; Wescoe Z. L.; Abu-Shumays R.; Vivian J. T.; Baatar B.; Karplus K.; Akeson M. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 18910–18915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laszlo A. H.; Derrington I. M.; Brinkerho H.; Langford K. W.; Nova I. C.; Samson J. M.; Bartlett J. J.; Pavlenok M.; Gundlach J. H. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 18904–18909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherf G. M.; Lieberman K. R.; Rashid H.; Lam C. E.; Karplus K.; Akeson M. Nat. Biotechnol. 2012, 30, 344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manrao E. A.; Derrington I. M.; Laszlo A. H.; Langford K. W.; Hopper M. K.; Gillgren N.; Pavlenok M.; Niederweis M.; Gundlach J. H. Nat. Biotechnol. 2012, 30, 349–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler T. Z.; Pavlenok M.; Derrington I. M.; Niederweis M.; Gundlach J. H. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 20647–20652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurts P.; Ernst D.; Wehenkel L. Machine Learning 2006, 63, 3–42. [Google Scholar]
- Shen L.; Zhang Y.. Enzymatic Analysis of Tet Proteins: Key Enzymes in the Metabolism of DNA Methylation. In Methods in Enzymology; Wu C., Allis C. D., Eds.; Academic Press: 2012; Vol. 512, pp 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.