

Abstract
The mechanistic details of many polyketide synthases (PKSs) remain elusive due to the instability of transient intermediates that are not accessible via conventional methods. Here we report an atom replacement strategy that enables the rapid preparation of polyketone surrogates by selective atom replacement, thereby providing key substrate mimetics for detailed mechanistic evaluations. Polyketone mimetics are positioned on the actinorhodin acyl carrier protein (actACP) to probe the underpinnings of substrate association upon nascent chain elongation and processivity. Protein NMR is used to visualize substrate interaction with the actACP, where a tetraketide substrate is shown not to bind within the protein, while heptaketide and octaketide substrates show strong association between helix II and IV. To examine the later cyclization stages, we extended this strategy to prepare stabilized cyclic intermediates and evaluate their binding by the actACP. Elongated monocyclic mimics show much longer residence time within actACP than shortened analogs. Taken together, these observations suggest ACP-substrate association occurs both before and after ketoreductase action upon the fully elongated polyketone, indicating a key role played by the ACP within PKS timing and processivity. These atom replacement mimetics offer new tools to study protein and substrate interactions and are applicable to a wide variety of PKSs.
Synthetic mimics of complex natural products are important tools to evaluate the mechanisms of natural product biosynthesis and to access syntheses that diversify the natural product templates. For example, the advent of the Stork–Eschenmoser hypothesis1 was a defining moment in terpene biosynthesis that enabled synthetic mimickry2,3 to help elucidate the mechanisms of terpene biosynthesis.4 Comparable intermediates in aromatic polyketide biosynthesis have not been possible due to the instability of nascent polyketones, hindering advancement in the study of PKS mechanisms, timing, and processivity.5 Here we describe an approach for generating polyketide intermediates via atom replacement to mimic elongation intermediates in iterative (type II) polyketide synthases. We demonstrate application of these probes to evaluate the biosynthesis of actinorhodin,6 showing that these materials can mimic partially elongated and cyclized substrates attached to the actACP7 through analysis of intermediate association.
Figure 1 depicts the proposed biosynthesis of actinorhodin (1). Few intermediates within this pathway have been identified,5 leading to uncertainties in both the succession and process of elongation, reduction, and ring closure. Current structural evidence suggests that the process is dictated by stabilizing residues within binding pockets of corresponding synthases,8 but this remains hypothetical. Important questions remain to be addressed, including: Why must chain elongation reach full length before reduction and cyclization? How is site-specific carbonyl reduction achieved? Are linear polyketones sequestered by the ACP during chain elongation? Access to elongated, polar mimics of ketide intermediates, such as 4–6 (Figure 1), could enlighten these uncertainties by their study in complex with PKS proteins for binding, stabilization, and chain transfer.
Figure 1.
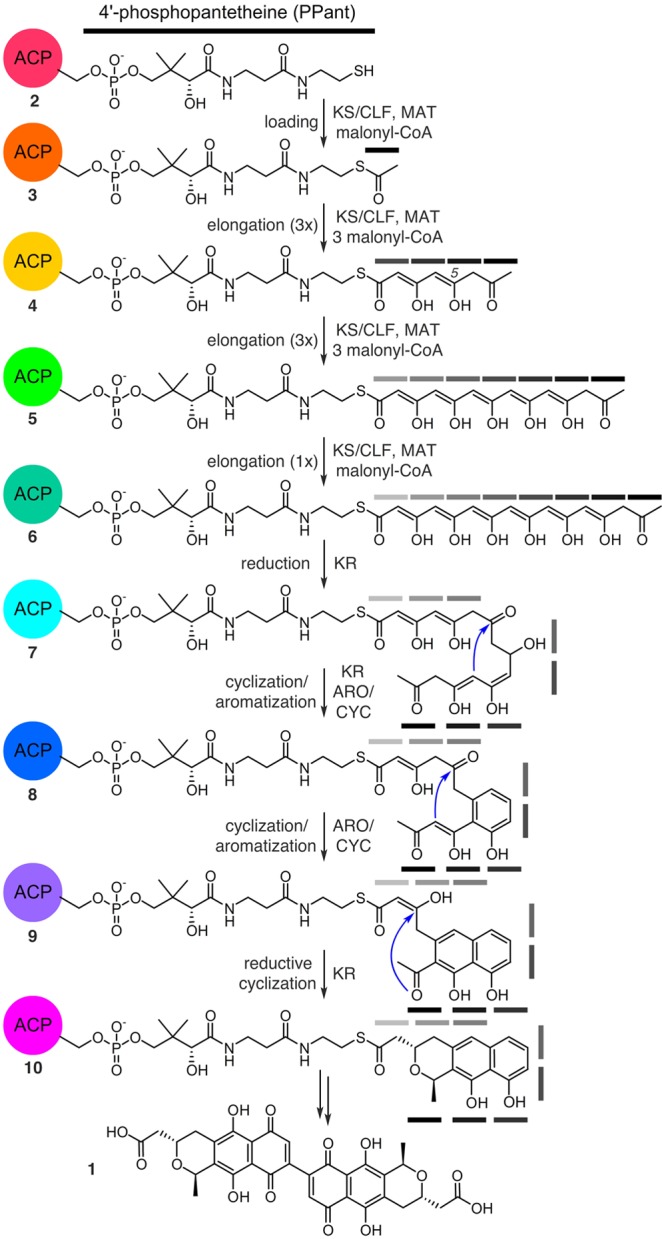
Proposed biosynthesis of 1 from holo-actACP (2). One depiction of the enol/ketol tautomerization states has been shown, in reality, multiple states likely exist. Gray bars denote ketide units.
Although triketone units can be adapted synthetically, as demonstrated by Barrett9a−9c and Harris,9d,9e preparation and study of these and longer polyketones have been impeded by inherent reactivity through spontaneous intra- and intermolecular Aldol/Claisen condensations. We identified a practical solution to this problem by replacing select carbonyls with sulfur atoms, thereby thwarting spontaneous condensation and at the same time providing probes to study the effects of length, polarity, and hydrophobicity upon the PKS elongation and cyclization process. We strategically substituted carbonyl groups nonessential for cyclization and/or reduction with sulfur atoms or isoxazole rings and validated their design via docking simulations of the probes upon actACP (Supporting Information). As shown in Figure 2, the corresponding thioethers would serve as a chemical “knockout” of the reactive carbonyl moieties. These probes differ from previously published analogs with the inclusion of both polyketide mimics and the full 4′-phosphopantetheine moiety, thus allowing us to evaluate the significance of ACP–probe interactions. In addition, this atom replacement design offers the added benefit of facile transformation to sulfoxides or sulfones, offering the ability to install greater polarity into the same parent compound.
Figure 2.
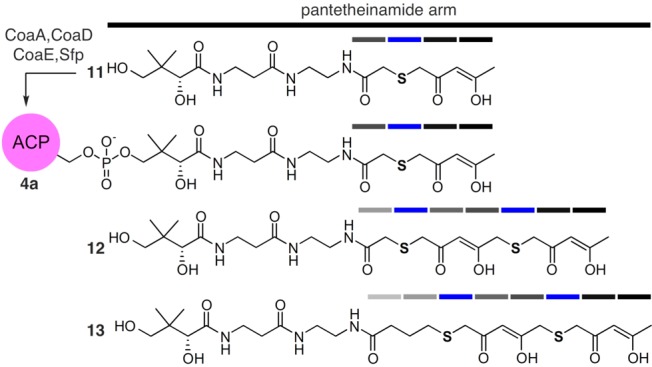
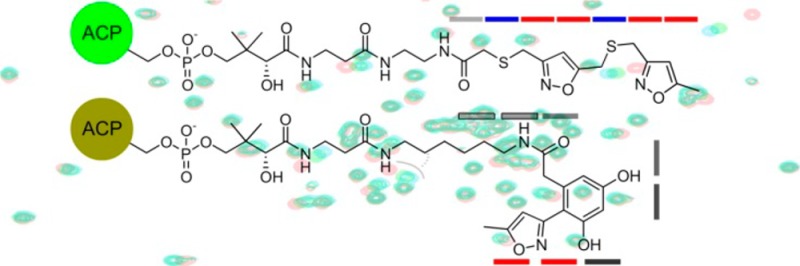
Structures of S atom replaced crypto-C8-actACP (4a). In this design, select carbonyl groups are replaced by S atoms, therein reducing number of potential Aldol/Claisen condensations. Access to 4a was achieved by chemoenzymatic conversion of mimetic 11 to its corresponding CoA analog by the action of three E. coli enzymes CoaA, CoaD, CoaE followed by loading on the actACP by Sfp (see Supporting Information).10−12 Mimetics 12 and 13 and their corresponding crypto-loaded ACPs were not achieved due to instability. While the installation of the thioethers in 12 and 13 eliminated select Aldol/Claisen condensations, it did not ablate it. Ketide units are denoted by bars and are colored according to replacement of the carbonyl group with S atom (blue).
We began by targeting an atom replacement mimetic of the ACP species bearing tetraketide unit 4 (Figure 1). Of the four carbonyl groups, the third ketide, as shown in 4a (blue bar, Figure 2), was selected for replacement. This involved the preparation of tetraketide mimetic 11 (Figure 2), which would be appended onto the actACP using chemoenzymatic methods developed in our laboratories.11,12 After exploring multiple approaches, we identified a route using an isoxazole as a tool to install a protected diketone unit.13 As shown in Scheme 1, mimetic 11 was prepared through an eight-step route that began with commercially available isoxazole 14. Reduction of 14 and subsequent mesylation afforded 15, which was converted to thioacetate 16 over the three steps.
Scheme 1. Synthesis of Tetraketide Mimetic 11.

Ar = 4-methoxybenzyl.
Thioacetate 16 was then converted into acid 17 by a three-step one-pot reaction sequence that began with deacetylation of the thioacetal, alkylation of the incipient thiolate, and ester hydrolysis (Scheme 1). Alternatively, addition of thioglycolic acid to mesylate 15 also provided acid 17 (not shown, see Supporting Information). Acid 17 was then coupled to protected-pantetheinamine 18 using conventional EDAC coupling conditions to afford amide 19. The synthesis of mimetic 11 was completed in three steps by removal of p-methoxyphenylacetal using aq. AcOH, opening of the isoxazole in 20 by treatment with Mo(CO)6 in refluxing aq. CH3CN, and conversion of 21 to 11 by treatment with AcOH:H2O:CH3CN (2:2:1). Using this route, we prepared 11 from isoxazole 14 in 10% overall yield over eight steps.
We then turned our attention to examination of mimetics bearing chain-elongated ketides as illustrated by heptaketide 12 and octaketide 13 (Figure 2). Our goal was to compare the short crypto-C8-actACP 4a to the longer crypto-C14 or C16 actACPs as a means to validate the chain length specificity during extension of the ketide chain. We began by evaluating the synthesis of 12 and 13 (Figure 2). As depicted in Scheme 2, the synthesis of 12 began by converting isoxazole 24 to its mesylate 25 and coupling it to 16 using our tandem three-step coupling process, as illustrated for the elaboration of 16 to 17 (Scheme 1). The resulting bis-isoxazole-acid 26 was then converted into its corresponding thioacetate 27 in four steps. Again using the tandem three-step coupling process, we were able to access acid 28 in high yield from 27. The resulting acid 28 was coupled to protected pantetheinamine 18, which after deprotection, afforded heptaketide 22 in nine steps from 16 and 24 in 19% overall yield. Unfortunately, while multiple conditions and methods were screened, the conversion of 22 (Scheme 1) to 12 (Figure 2) via isoxazole ring opening underwent rapid condensation providing an intractable mixture.
Scheme 2. Synthesis of Chain Elongated Mimetics, Heptaketide 22 and Octaketide 23.

Ar = 4-methoxybenzyl.
Comparable complications arose in efforts to prepare mimetic 13 (Figure 2). Using thioacetate 27 as a central intermediate, preparation of 13 required only modification of the alkylating agent. Conversion of 27 to 29 was readily achieved using t-butyl 4-bromobutanoate (Scheme 2). Coupling acid 29 to 18, followed by deprotection, completed access to octaketide mimetic 23 in nine steps from 16 and 24 in 28% overall yield. Isoxazole opening in 23 again led to an intractable mixture of products. This indicated that stable mimetics required additional atom replacement.
Fortunately, the solution arose from 20 (Scheme 1), 22 (Scheme 2), and 23 (Scheme 2). Here, the isoxazole motif not only served as a synthetic tool but also provided a second type of atom replacement (Figure 3). In this dual atom replaced model, both carbonyl and dicarbonyl units are replaced with thioether and isoxazole units, respectively. Unlike mimetics 12 and 13, long chain analogs such as 22 and 23 are readily accessed and are stable, due to their inability to undergo intramolecular Aldol/Claisen condensations.
Figure 3.
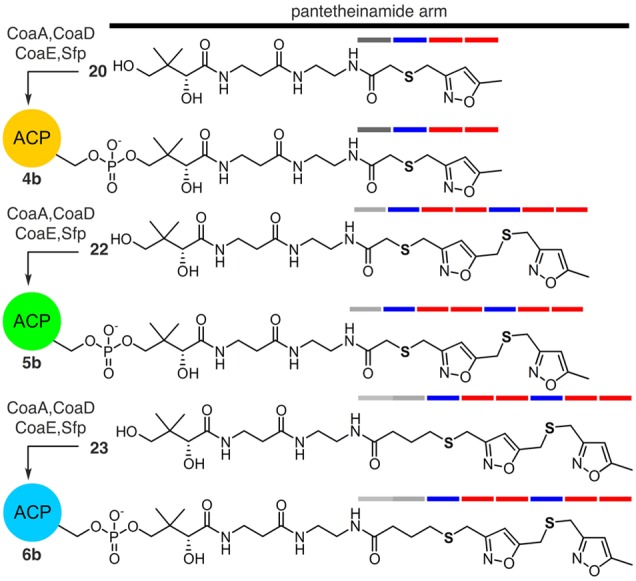
Structures of dual atom replaced crypto-C8-actACP 4b, crypto-C14-actACP 5b, and crypto-C16-actACP 6b. Access to 4b, 5b, and 6b was accomplished by chemoenzymatic attachment of 20 (Scheme 1), 22 (Scheme 2), and 23 (Scheme 2), respectively, to the actACP.10−12 Ketide units are denoted by bars and are colored according to replacement of a carbonyl with a sulfur atom (blue) or replacement of diketide unit with isoxazole (red).
We applied these atom replacement mimetics to study actACP substrate sequestration as detected by solution-phase protein NMR.12,14 We appended mimetics 11, 20, 22, and 23 to apo-actACP forming the corresponding crypto-C8-actACP 4a (Figure 2), crypto-C8-actACP 4b (Figure 3), crypto-C14-actACP 5b (Figure 3), and crypto-C16-actACP 6b (Figure 3).12 We then examined samples of proteins 4a, 4b, 5b, and 6b by solution-phase 1H,15N-HSQC NMR (Figure 4a,c).10 Comparison of crypto-C8-actACP 4a and isoxazole-containing crypto-C8-actACP 4b to holo-actACP via chemical shift perturbation (CSP) returned only very slight perturbations, indicative of a substrate that is mostly solvent exposed and not sequestered on the NMR time scale (Figure 4b).12 These results compare favorably to known analog sequestration, where early intermediates (C6 and smaller) have been shown to not sequester.14c
Figure 4.

Sequestration analysis of atom replaced mimetics. (a) 1H,15N-HSQC overlays depicting the holo-actACP (2, red) and the crypto-C8-actACP (4a, pink), crypto-C8-actACP (4b, orange) bearing 11 and 20, respectively. (b) CSP plots obtained from the spectra shown in panel (a). (c) 1H,15N-HSQC traces depicting overlays of the holo-actACP (2, red), crypto-C14-actACP (5b, green) bearing heptaketide mimetic 22 and crypto-C16-actACP (6b, blue) bearing the octaketide mimetic 23. (d) CSP plots obtained from the spectra shown in panel (c). The docked actACP structure with these probes can be found in Figure S34.
Octaketide 6b shows ∼30% larger CSPs over 5b in these key residues, suggesting that the longer substrate, which mimics the fully mature octaketide polyketone in 6, displays longer residency within the hydrophobic cleft. This pocket is delineated by CSPs found in the same residues of actACP previously reported to stabilize aromatic product analogs.12 Minimal CSPs for 4a and 4b indicate that tetraketide intermediates are not stabilized by interaction with the actACP, likely favoring further elongation by the actKS/CLF. This supports the hypothesis that sequestration of the elongating polyketone is driven by the comparative energetic stability between the ACP sequestered substrate and the ACP-KS/CLF complex. These studies suggest that the mature polyketone is stabilized through association by the hydrophobic cleft formed by movement of helix III of actACP during chain elongation.
Next we turned to evaluate the latter, cyclized intermediates, 7–10 (Figure 1), proposed in the biosynthesis of actinorhodin (1). We realized that this atom replacement methodology could be uniquely extended for the study of the cyclization events that occur in type II PKS pathways. These transformations have long been unclear, as the intractable and spontaneous cyclization of ketide intermediates (Figure 1) have prevented their preparation for detailed studies.8a,15 In particular, we were interested in mimetics that would allow us to probe ACP sequestration as a potential regulating force of substrate transport between elongation and initial cyclization, as illustrated by the conversion of 7 to 8 (Figure 1) by the actinorhodin ketoreductase (KR) as well as the processivity between KR and the aromatase/cyclase (ARO/CYC).15b
A central mystery in type II PKS ACP arises during the processing of highly reactive polyketone intermediates to cyclized products, as exemplified by the conversion of 6 to 10 (Figure 1). This process includes steps of cyclization, reduction, and aromatization. We have hypothesized that intermediate stabilization through sequestration of elongated intermediates within the ACP may play a role during this transformation. To test this hypothesis, we recently demonstrated that actACP binds mature tricyclic product analogs between helices II and IV.12 Crump and co-workers decisively showed that simple di- and triketide analogs are not sequestered by actACP,7a,17 but linear hydrocarbons do bind within the protein. Taken together, these findings indicate that identity of the intermediates likely plays a major role in substrate translocation between enzymes. As demonstrated above, the presence of polar residues such as D62 and D63 on helix III may stabilize the polar nature of polyketone intermediates and may be better suited to the environment of the actACP interior cavity. But what about the monocyclic product of the KR? Is this species directly routed to the ARO/CYC domain or does it return to the actACP pocket between enzymatic transformations? To address these questions, the preparation of elongated monocyclic probes is critical for further insight into the role of sequestration in type II PKS biosynthesis.
To this end, analogs 8a–8c (Figure 5) were proposed as probes that mimic the structural identity of putative intermediates as a means of evaluating actACP sequestration. Analog 8a offered the most simplified mimetic by providing a single aromatic ring with six ketide units. Analog 8b and 8c more closely resembled the putative intermediate 8 (Figure 1) with the addition of an upstream diketide, where the ketide units extended from six in 8a and 8b to eight in 8c, respectively. Our first goal in this effort was the preparation of 8a–8c for phosphopantetheine attachment to 15N-enriched actACP for solution-phase protein NMR studies of substrate sequestration.
Figure 5.
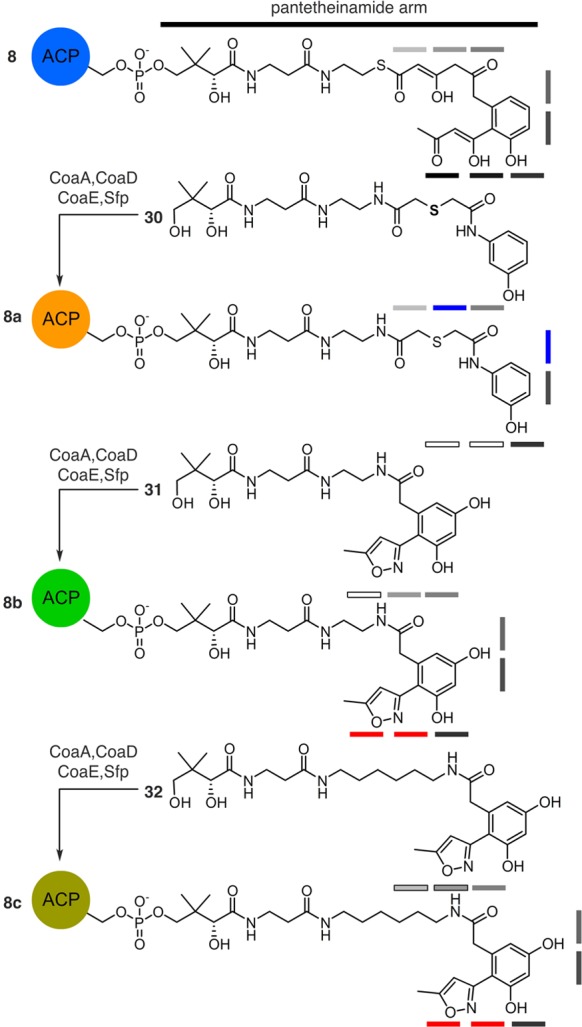
Structures of atom replaced crypto-actACP 8a, 8b, and 8c. Access to 8a, 8b, and 8c was achieved by chemoenzymatic conversion of mimetics 30, 31, and 32, respectively, to their corresponding CoA analogs by the action three E. coli enzymes CoaA, CoaD, CoaE,10 followed by loading on the actACP by Sfp (see Supporting Information).11 Mimetic 30 contains N and S atom replacement (blue bars) and bears structural truncation (black-outlined bars). In mimetics 31 and 32, 1,3-dicarbonyl units are replaced by isoxazole (red bars). Two different length substrates were examined. Shaded gray bars denote ketide units.
We began with the preparation of 8a by application of hexaketide mimic 30. A general strategy for the synthesis of 30 involves the treatment of m-aminophenol (33) with thiodiglycolic anhydride (34) to provide aromatic acid 35 (Scheme 3). Acid 35 was then coupled to protected pantetheinamine 18 to afford 36. Amide 36 was then deprotected by using aqueous acetic acid to deliver mimetic 30 in 19% overall yield from 33. While 30 was readily accessed, it lacks two of the terminal ketides (black-outlined boxes, Figure 5). We next explored mimetics 31 and 32 that provide approximate isosteric placement of this additional diketide moiety. As depicted in Figure 5, these materials expanded on our use of an isoxazole as a diketide mimetic.
Scheme 3. Synthesis of Mimetic 30.

Ar = 4-methoxyphenyl.
We then turned to the preparation of 8b and 8c from the respective mimetics 31 and 32 (Figure 5). The synthesis of mimetics 31 (Scheme 4) and 32 (Scheme 5) arose through preparation of acid 41 as a central intermediate. Application of a Vilsmeier–Haak formylation to 37(18) produced crystalline 38 in excellent yield. Aldehyde 38 was converted to its corresponding oxime 39, which was then subjected to a 1,3-dipolar cycloaddition with isopropenyl acetate to afford isoxazole 40. Preparation of acid 41 was then completed by hydrolysis under basic conditions.
Scheme 4. Synthesis of Mimetic 31.

Ar = 4-methoxyphenyl.
Scheme 5. Synthesis of Mimetic 32.

Ar = 4-methoxyphenyl.
Two additional steps were required to complete the synthesis of mimetic 31 (Scheme 4). Acid 41 was coupled to protected-pantetheinamine 18 to deliver amide 42. Global deprotection under conventional hydrogenation conditions provided 31 in 15% overall yield from 37. While mimetic 31 contained the missing diketide unit from 30 (Figure 5), its chain length did not match that of the putative intermediate 8 (Figure 5). We therefore prepared a third mimetic 32 (Scheme 5) through the addition of a six-carbon spacer.
Advantageously, acid 41 could be used to prepare a chain-elongated mimetic 32 (Scheme 5). This began by preparing amide 43 by coupling N-Boc-1,6-hexanediamine to 41. Boc-deprotection followed by coupling of amine 44 to protected pantothenic acid 45 afforded amide 46. A two-step sequence involving deprotection of the benzyl ethers in 46, followed by removal of the PMP acetal in 47 under acidic conditions, provided mimetic 32 in 13% overall yield from 41.
We then loaded mimetics 30-32 to 15N-enriched actACP chemoenzymatically as previously reported10−12 to generate crypto-actACP 8a, 8b, and 8c, respectively. Next, we subjected crypto-actACP 8a–8c to solution-phase protein NMR studies (Figure 6a). CSPs were observed (Figure 6b), which compare the crypto-actACP 8a–8c to holo-actACP. We found that the crypto-actACP loaded with cyclized hexaketide mimetic 30 showed slight CSPs; the crypto-actACP loaded with cyclized mimetic 31 showed moderate CSPs; and the crypto-actACP loaded with cyclized mimetic 32 showed large CSPs in residues important to the interior sequestration cavity of actACP (Figure 6).12
Figure 6.

Sequestration analysis of atom replaced cyclic intermediates. (a) 1H,15N-HSQC traces depicting overlays of the holo-actACP (2, red) and the crypto-actACP (8a, orange) bearing hexaketide mimetic 30 (Figure 5). (b) 1H,15N-HSQC traces depicting overlays of the holo-actACP (2, red), crypto-actACP (8b, green) bearing hexaketide mimetic 31 and crypto-actACP (8c, brown) bearing octaketide mimetic 32. (c) CSP plots obtained from the spectra shown in panels (a and b).
On the NMR time scale, the fully elongated, cyclized octaketide 32 had much higher residence time in the actACP interior cavity than 30 or 31. The strongest perturbations were observed in residues located at the end of helix III and the following loop, which corroborate with the sequestration residues that we previously reported in emodin-crypto-ACP.12 Interestingly, only species 8c showed moderate periodic perturbations throughout residues in helix II and those immediately preceding it. This information mirrored our study on linear polyketide mimetics (Figure 4), which showed the strongest sequestration of the fully elongated mimetic 32.
In this study, we have shown that selective replacement of carbonyl groups with heteroatoms facilitates access to a diverse class of polyketide mimetics that can be used to interrogate polyketide biosynthetic enzymes. Inherent to this methodology, these atom replacement mimetics can be chemoenzymatically conjugated to ACP through the corresponding CoA analogs, which provide the native scaffold to study these processes. While no probe could ever exactly mimic the properties of natural polyketone intermediates, this work demonstrates that molecules that are much more polar than fatty acids14c or tricyclic aromatics12 do associate with actACP, that longer chains experience more residence time, and that cyclic intermediates demonstrate the longest residence times. Our data, combined with docking simulation of these probes (Figure S34), strongly support that binding of both linear and cyclized polyketide mimetics is not observed unless the mimetics are of sufficient length. Taken together, these observations suggest that until the polyketide has reached its terminal length, no preferential sequestration by the actACP stabilizes the intermediate. However, at full elongation, polyketide binding with the actACP could facilitate release from the ketosynthase and assist transfer to the KR for the first cyclization and reduction steps.15b Binding of the first cyclized, reduced intermediate by the actACP would then occur to facilitate release from the KR, followed by delivery to the ARO/CYC.
In this study, the goal of these atom replacement mimetics was to help understand the comparative binding and stabilization of polyketone intermediates by ACPs. These mimetics were not designed to serve as functional substrates of PKS enzymes but instead to mimic the length, polarity, and hydrophobicity found in natural intermediates. As with all mimetics, the match was not perfect. In these examples, the C–S bonds are considerably longer than those of their anticipated ketides. The actual enol-keto states of natural polyketones remain unknown, and the isoxazole moiety in part restricts conformational freedom. These caveats aside, the establishment of polyketide mimietics provides an excellent tool for interrogating the iterative processes polyketide biosynthesis.
Finally, the involvement of the ACP in these pathways adds significant complexity when compared to non-templated biosynthetic pathways, and the creation of new tools to understand this role merits special attention. Most critically, this study defines new methods of atom replacement that can be used to rapidly assemble both linear and cyclic polyketide mimetics for structural and functional applications. Studies on the protein–protein interactions of these species are ongoing.
Acknowledgments
We thank Dr. X. Huang (UCSD), Dr. A. Mrse (UCSD), Dr. Y. Su (UCSD), and Dr. P. Dennison (ICI) for assistance with collection of NMR and MS data. We also thank Prof. C. A. Townsend (Johns Hopkins), Prof. F. Ishikawa (UCSD), Prof. C. D. Vanderwal (UCI), and Prof. A. R. Chamberlin (UCI) for helpful advice. We also thank Prof. M. P. Crump for your advices from full structural determinations as well as helpful discussions. Funding was provided by National Institutes of Health GM100305 and GM095970.
Supporting Information Available
Copies of spectral data, synthetic methods, and experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ G.S. and H.R. contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Sadler P. A.; Eschenmoser A.; Scheinz H.; Stork G. Helv. Chem. Acta 1957, 40, 2191. [Google Scholar]; b Stork G.; Burgstahler A. W. J. Am. Chem. Soc. 1955, 77, 5068. [Google Scholar]; c Ruzicka L.; Eschenmoser A.; Heusser H. Experientia 1953, 9, 357.13116962 [Google Scholar]; d Ruzicka L. Experientia 1954, 50, 1. [Google Scholar]
- Abe I.; Rohmer M.; Prestwich G. D. Chem. Rev. 1993, 93, 2189. [Google Scholar]
- a Johnson W. S.; Semmelhack M. F.; Sultanbawa M. U.; Dolak L. A. J. Am. Chem. Soc. 1968, 90, 2994. [Google Scholar]; b Schmidt R.; Huesmann P. L.; Johnson W. S. J. Am. Chem. Soc. 1980, 102, 5122. [Google Scholar]
- a Pichersky E.; Noel J. P.; Dudareva N. Science 2006, 311, 808. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yoder R. A.; Johnston J. N. Chem. Rev. 2005, 105, 4730. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Poralla K. Chem. Biol. 2004, 11, 12. [DOI] [PubMed] [Google Scholar]; d Xu R.; Fazio G. C.; Matsuda S. P. Phytochemistry 2004, 65, 261. [DOI] [PubMed] [Google Scholar]
- a Akey D. L.; Gehret J. J.; Khare D.; Smith J. L. Nat. Prod. Rep. 2012, 29, 1038. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Crawford J. M.; Townsend C. A. Nat. Rev. Microbiol. 2010, 8, 870. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Khosla C. J. Org. Chem. 2009, 74, 6416. [DOI] [PubMed] [Google Scholar]; d Das A.; Khosla C. Acc. Chem. Res. 2009, 42, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Bumpus S. B.; Kelleher N. L. Curr. Opin. Chem. Biol. 2008, 12, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Korman T. P.; Hill J. A.; Vu T. N.; Tsai S. C. Biochemistry 2004, 43, 14529. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rudd B. A.; Hopwood D. A. J. Gen. Microbiol. 1979, 114, 35. [DOI] [PubMed] [Google Scholar]; c Fernández-Moreno M. A.; Martínez E.; Caballero J. L.; Ichinose K.; Hopwood D. A.; Malpartida F. J. Biol. Chem. 1994, 269, 24854. [PubMed] [Google Scholar]
- a Crump M. P.; Crosby J.; Dempsey C. E.; Parkinson J. A.; Murray M.; Hopwood D. A.; Simpson T. J. Biochemistry 1997, 36, 6000. [DOI] [PubMed] [Google Scholar]; b Revill W. P.; Bibb M. J.; Hopwood D. A. J. Bacteriol. 1996, 178, 5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Teufel R.; Miyanaga A.; Michaudel Q.; Stull F.; Louie G.; Noel J. P.; Baran P. S.; Palfey B.; Moore B. S. Nature 2013, 503, 552. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ames B. D.; Lee M. Y.; Moody C.; Zhang W.; Tang Y.; Tsai S. C. Biochemistry 2011, 50, 8392. [DOI] [PMC free article] [PubMed] [Google Scholar]; b1 Tsai S. C.; Lu H.; Cane D. E.; Khosla C.; Stroud R. M. Biochemistry 2002, 41, 12598. [DOI] [PubMed] [Google Scholar]; c Pan H.; Tsai S. C.; Meadows E. S.; Miercke L. J.; Keatinge-Clay A. T.; O’Connell J.; Khosla C.; Stroud R. M. Structure 2002, 10, 1559. [DOI] [PubMed] [Google Scholar]
- a Fouché M.; Rooney L.; Barrett A. G. M. J. Org. Chem. 2012, 77, 3060. [DOI] [PubMed] [Google Scholar]; b Calo F.; Richardson J.; Barrett A. G. M. Org. Lett. 2009, 11, 4910. [DOI] [PubMed] [Google Scholar]; c Navarro I.; Basset J. F.; Hebbe S.; Major S. M.; Werner T.; Howsham C.; Bräckow J.; Barrett A. G. M. J. Am. Chem. Soc. 2008, 130, 10293. [DOI] [PubMed] [Google Scholar]; d Harris T. M.; Murray T. P.; Harris C. M.; Gumulka M. J. Chem. Soc. Chem. Comm. 1974, 10, 362. [Google Scholar]; e Harris T. M.; Harris C. M. Tetrahedron 1977, 33, 2159. [Google Scholar]
- a Worthington A. S.; Burkart M. D. Org. Biomol. Chem. 2006, 4, 44. [DOI] [PubMed] [Google Scholar]; b Kosa N. M.; Haushalter R. W.; Smith A. R.; Burkart M. D. Nat. Methods 2012, 9, 981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a La Clair J. J.; Foley T. L.; Schegg T. R.; Regan C. M.; Burkart M. D. Chem. Biol. 2004, 11, 195. [DOI] [PubMed] [Google Scholar]; b Meier J. L.; Burkart M. D. Methods Enzymol. 2009, 458, 219. [DOI] [PubMed] [Google Scholar]; c Foley T. L.; Burkart M. D. Curr. Opin. Chem. Biol. 2007, 11, 12. [DOI] [PubMed] [Google Scholar]
- Haushalter R. W.; Filipp F. V.; Ko K. S.; Yu R.; Opella S. J.; Burkart M. D. ACS Chem. Biol. 2011, 6, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Stork G.; Hagedorn A. A. III J. Am. Chem. Soc. 1978, 100, 3609. [Google Scholar]; b Stork G.; La Clair J. J.; Spargo P.; Nargund R. P.; Totah N. J. Am. Chem. Soc. 1996, 118, 5304. [Google Scholar]; c Wright P. M.; Myers A. G. Tetrahedron 2011, 67, 9853. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Sun C.; Wang Q.; Brubaker J. D.; Wright P. M.; Lerner C. D.; Noson K.; Charest M.; Siegel D. R.; Wang Y. M.; Myers A. G. J. Am. Chem. Soc. 2008, 130, 17913. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Charest M. G.; Siegel D. R.; Myers A. G. J. Am. Chem. Soc. 2005, 127, 8292. [DOI] [PubMed] [Google Scholar]
- a Roujeinikova A.; Simon W. J.; Gilroy J.; Rice D. W.; Rafferty J. B.; Slabas A. R. J. Mol. Biol. 2007, 365, 135. [DOI] [PubMed] [Google Scholar]; b Płoskoń E.; Arthur C. J.; Kanari A. L.; Wattana-amorn P.; Williams C.; Crosby J.; Simpson T. J.; Willis C. L.; Crump M. P. Chem. Biol. 2010, 17, 776. [DOI] [PubMed] [Google Scholar]; c Evans S. E.; Williams C.; Arthur C. J.; Płoskoń E.; Wattana-amorn P.; Cox R. J.; Crosby J.; Willis C. L.; Simpson T. J.; Crump M. P. J. Mol. Biol. 2009, 389, 511. [DOI] [PubMed] [Google Scholar]
- a Nguyen C.; Haushalter R. W.; Lee D. J.; Markwick P. R.; Bruegger J.; Caldara-Festin G.; Finzel K.; Jackson D. R.; Ishikawa F.; O’Dowd B.; McCammon J. A.; Opella S. J.; Tsai S. C.; Burkart M. D. Nature 2014, 505, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Javidpour P.; Bruegger J.; Srithahan S.; Korman T. P.; Crump M. P.; Crosby J.; Burkart M. D.; Tsai S. C. Chem. Biol. 2013, 20, 1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revill W. P.; Bibb M. J.; Hopwood D. A. J. Bacteriol. 1996, 178, 5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsini M. A.; Pettus T. R. R.; Wenderski T. A. Org. Lett. 2011, 13, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.