

Abstract
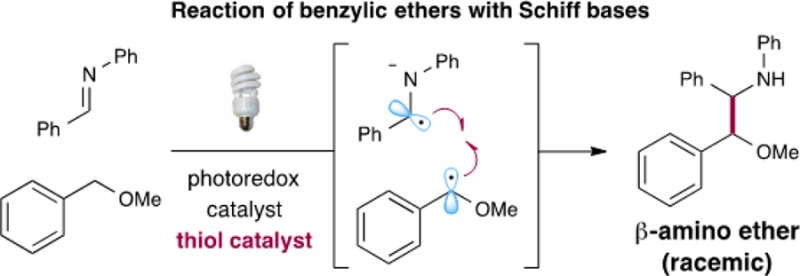
The photoredox-mediated coupling of benzylic ethers with Schiff bases has been accomplished. Direct benzylic C–H activation by a combination of a thiol catalyst with an iridium photocatalyst and subsequent radical–radical coupling with secondary aldimines affords a variety of β-amino ether products in good to excellent yields. Mechanistic studies suggest that a reductive quenching pathway of the photocatalyst is operable.
The direct and controlled C–H functionalization of sp3-hybridized carbons has become a challenging goal in modern synthetic organic chemistry.1 In this context, our laboratory has recently introduced a protocol that allows the activation and subsequent arylation of benzylic C–H bonds via the cooperative action of a photoredox catalyst and a thiol organocatalyst (eq 1).2 This transformation relies on the coupling of two catalytically generated radicals: an arene radical anion formed by reduction of an arylnitrile, and a benzyl ether radical generated by thiyl-mediated hydrogen atom abstraction from benzylic ethers. Moreover, we postulate that the thiol organocatalyst undergoes oxidative proton-coupled electron transfer (PCET)3 in the presence of photoexcited catalysts to generate the requisite thiyl radical4 that can selectively cleave the C–H bond of benzylic ethers.5 This photoredox–organocatalysis C–H functionalization mechanism exploits several established physical properties (e.g., bond dissociation energies (BDEs),6 hydrogen atom transfer exchange constants,7 and oxidation potentials) that are predictable across a wide range of reaction classes. Herein we translate this general C–H functionalization concept to the direct coupling of benzyl ethers with secondary aldimines to afford β-amino ethers in one step (eq 2). This new radical–radical coupling protocol employs readily available substrates, a visible light source, and the combination of a thiol catalyst and photocatalyst.
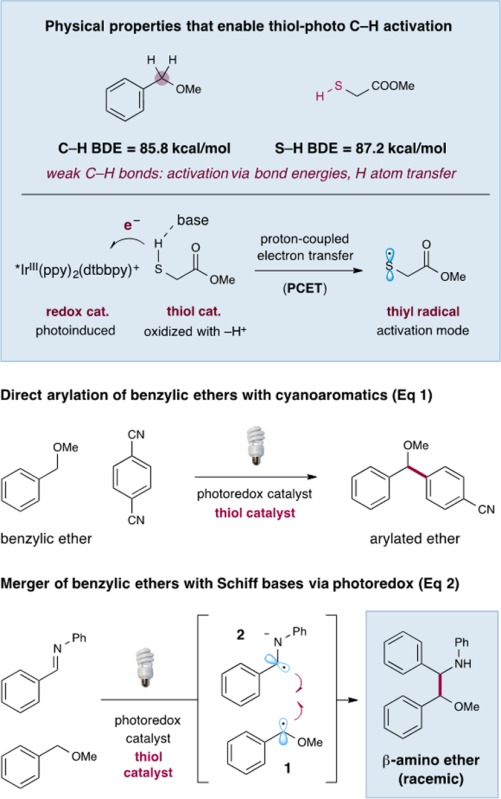
The β-amino ether functionality is a widespread motif in synthetic organic chemistry. However, the synthesis of these structural units is often challenging and typically requires multiple steps, organometallic reagents, and immoderate reaction conditions.8 With this in mind, we recently hypothesized that a one-step C–C bond coupling of benzylic ether radicals and an α-amino radical would allow direct access to this important synthon using simple and mild reagents and catalysts. Seminal studies of Roberts in the 1990s demonstrated that electrophilic thiyl radicals are excellent systems to abstract H• from aldehydes to generate acyl radicals.9 We recently demonstrated that similar open-shell thiyl species, generated via proton-coupled electron transfer, are superior catalysts that generate α-oxy radicals.2 With this in mind, we questioned whether photoredox catalysis would allow the concurrent generation of α-benzylic ether radical 1 (via oxidation) and α-amino radical anion 2 (via reduction) prior to a critical hetero-coupling of these two highenergy species (eq 2). Importantly, this new multi-catalysis reaction mechanism would be organocatalytic, redox-neutral, as well as atom-economical and should provide an unprecedented and non-classical C–C disconnection that will find utility in the fields of medicinal chemistry and organic molecule construction.
Design Plan. The proposed catalytic cycle of our new light-driven redox transformation is depicted in Scheme 1.10 It has long been established that the photoredox catalyst IrIII(ppy)2(dtbbpy)PF6 (3) will be photoexcited to the *IrIII state 4 by visible light irradiation with a household light bulb or blue LEDs. Excited species 4 can function as an oxidant (E1/2*III/II = +0.66 V vs SCE in MeCN)11 and can be quenched by the thiol catalyst methyl thioglycolate (6) to produce IrII(ppy)2(dtbbpy) (5), as well as thiyl radical 7.
Scheme 1. Proposed Mechanism of the Heterocoupling Reaction.
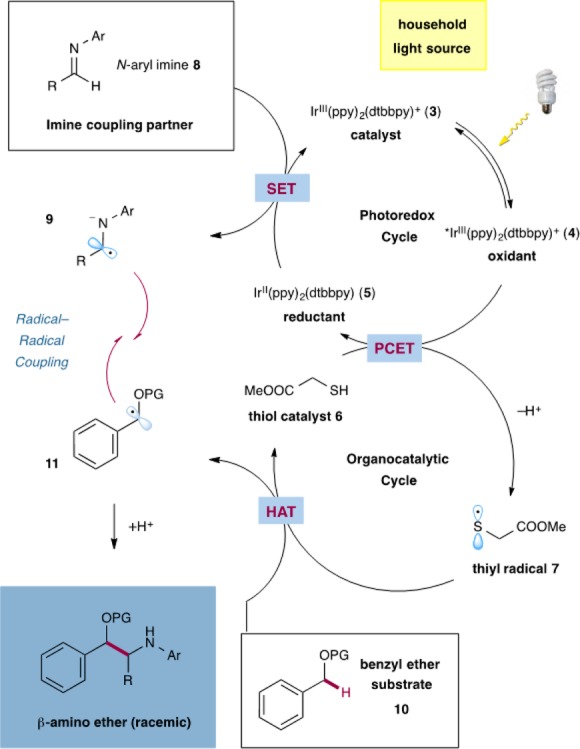
Although the oxidation potential of typical thiols (E1/2 = +0.85 V vs SCE for cysteine)12 should render this electron transfer unfavorable, we expect that the addition of a weakly basic additive (K2HPO4 or LiOAc) would facilitate the formation of thiyl radical 7 via a concerted PCET event. In order to close the photoredox cycle, IrII species 5 (E1/2III/II = −1.51 V vs SCE)11 should be able to reduce the N-aryl imine 8 (E1/2red = −1.98 V vs SCE for R = Ar = Ph)13 under the reaction conditions via a single-electron transfer (SET) event to afford the α-amino radical anion 9. With respect to BDEs, thiyl radical 7 should readily abstract a hydrogen atom from benzyl ether substrate 10 (α-C–H BDE = 85.8 kcal/mol for benzyl methyl ether)14 in a hydrogen atom transfer (HAT) process to regenerate the thiol catalyst 6 (S–H BDE = 87.2 kcal/mol)15 and to complete the organocatalytic cycle.1 We assume the resulting benzylic radical 11 will rapidly combine with α-amino radical anion 9 to afford the desired β-amino ether product. This critical C–C bond-forming step should be facilitated by the persistent radical effect, wherein species 9 is a long-lived open-shell species that does not readily homodimerize, thereby enabling a highly selective radical–radical heterocoupling event.16 Although a radical–radical coupling is proposed we are aware that an addition of benzylic radical 11 into imine 8 followed by reduction of the N-centered radical is also feasible. However, we consider this pathway as unlikely since imine dimer and imine reduction byproducts have been observed in certain cases.
Table 1. Initial Studies and Reaction Optimization.
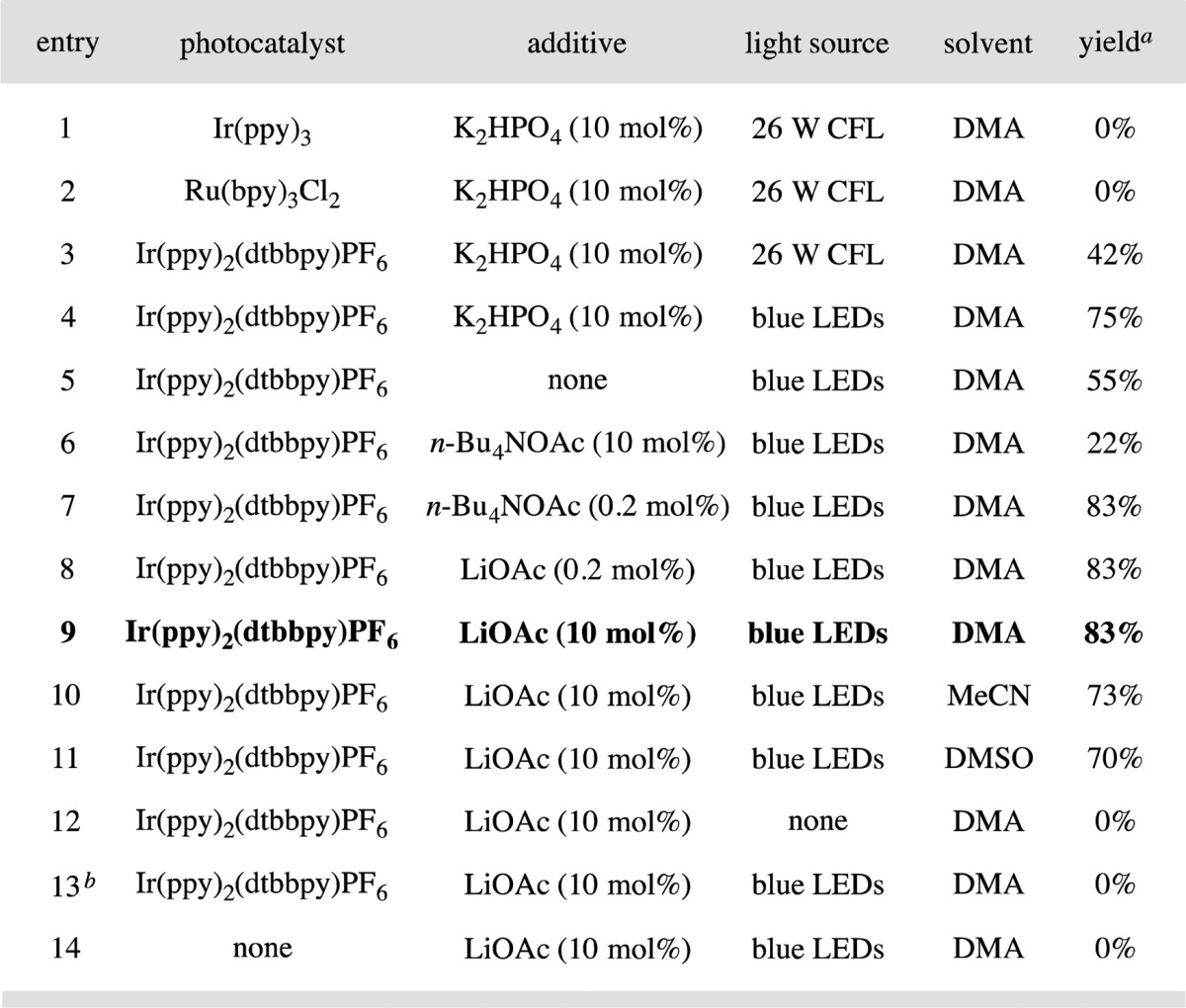
Yield determined by 1H NMR analysis using 1-bromo-3,5-bis(trifluoromethyl)benzene as the internal standard.
Reaction performed in the absence of methyl thioglycolate.
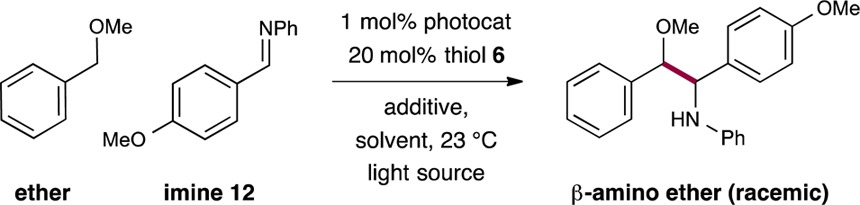
Results. The proposed ether–imine coupling was first evaluated with a series of established photoredox catalysts (Table 1). Initially we found that irradiation of benzyl methyl ether, imine 12, methyl thioglycolate (6), and K2HPO4 with visible light in the presence of Ir(ppy)3 or Ru(bpy)3Cl2 resulted in no observable yield of the desired 1,2-amino ether product (entries 1 and 2). Fortunately, however, use of the heteroleptic catalyst Ir(ppy)2(dtbbpy)PF6 (3) provided the desired β-amino ether in 42% yield (entry 3). The efficiency of this heteroradical coupling was further improved by irradiating the photocatalyst 3 (λmax = 460 nm) with light adjusted to the catalyst’s absorption maximum via the implementation of blue LEDs (entry 4, 75% yield). Next, we determined that the base additive had a significant influence on the reaction outcome. More specifically, while the lack of a basic additive resulted in a moderate yield (entry 5, 55% yield), we found that the use of the soluble n-Bu4NOAc base in trace quantities provided a significant improvement (entry 7, 0.2 mol%, 83% yield). Notably, the use of n-Bu4NOAc at the 10 mol% level resulted in diminished yields (entry 6, 22% yield). These results are consistent with the requirement of a soluble base that enables a concerted PCET mechanism for the formation of thiyl radical 7 (Scheme 1). We recognize that a stepwise pathway may also be operable, wherein deprotonation of the thiol catalyst could precede the oxidation of the resulting thiolate anion (mechanistic studies are ongoing to delineate between these two pathways). As an effective alternative to the hygroscopic n-Bu4NOAc, we have established LiOAc (entry 8) as a more operationally convenient additive at 10 mol% loading (entry 9).17 In addition, we have found that DMA is the superior medium for this process while solvents such as MeCN or DMSO are also competent (entries 10 and 11, 73% and 70% yield). Finally, control experiments verified the requirement of light, an organocatalyst, and a photocatalyst, as no product was observed in the absence of these components (entries 12–14).
Reaction Scope. With the optimal coupling conditions in hand, we next sought to determine the scope of aldimines that can be employed in this new radical–radical heterocoupling protocol. As shown in Table 2, electronically “neutral” imines (entry 1, 77% yield), as well as electron-rich arenes, increase the overall coupling efficiency (entries 2–4, 79–85% yield). We rationalized that electron-rich α-amino radicals are more nucleophilic and thereby more rapidly react with benzylic radical 11, resulting in generally higher yields. In addition, bicyclic arenes, such as naphthalene (entry 5, 85% yield) the heteroaromatic quinoline (entry 6, 58% yield) are well tolerated. With respect to the aniline-derived moiety of the imine, we found that strongly electron-donating and -withdrawing groups have a deleterious effect on overall reaction yield (entries 7 and 8, 61% and 56% yield); yet, high efficiency is achieved when halogen atoms are introduced at the para-positions (entries 9 and 10, both 84% yield). Also, aldimines that incorporate a CF3 group or pyridine-based heteroaromatics were found to be viable coupling partners for this redox process (entries 11–14, 58–75% yield).
Table 2. Scope of the Imine Coupling Partnera.
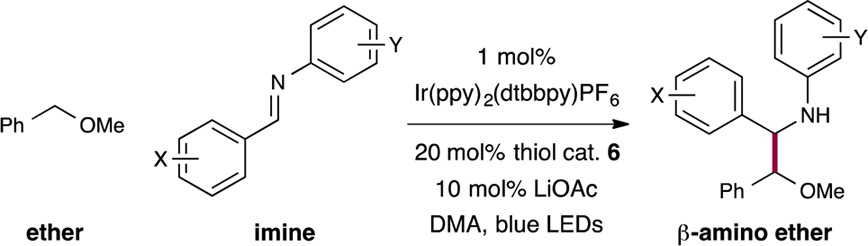
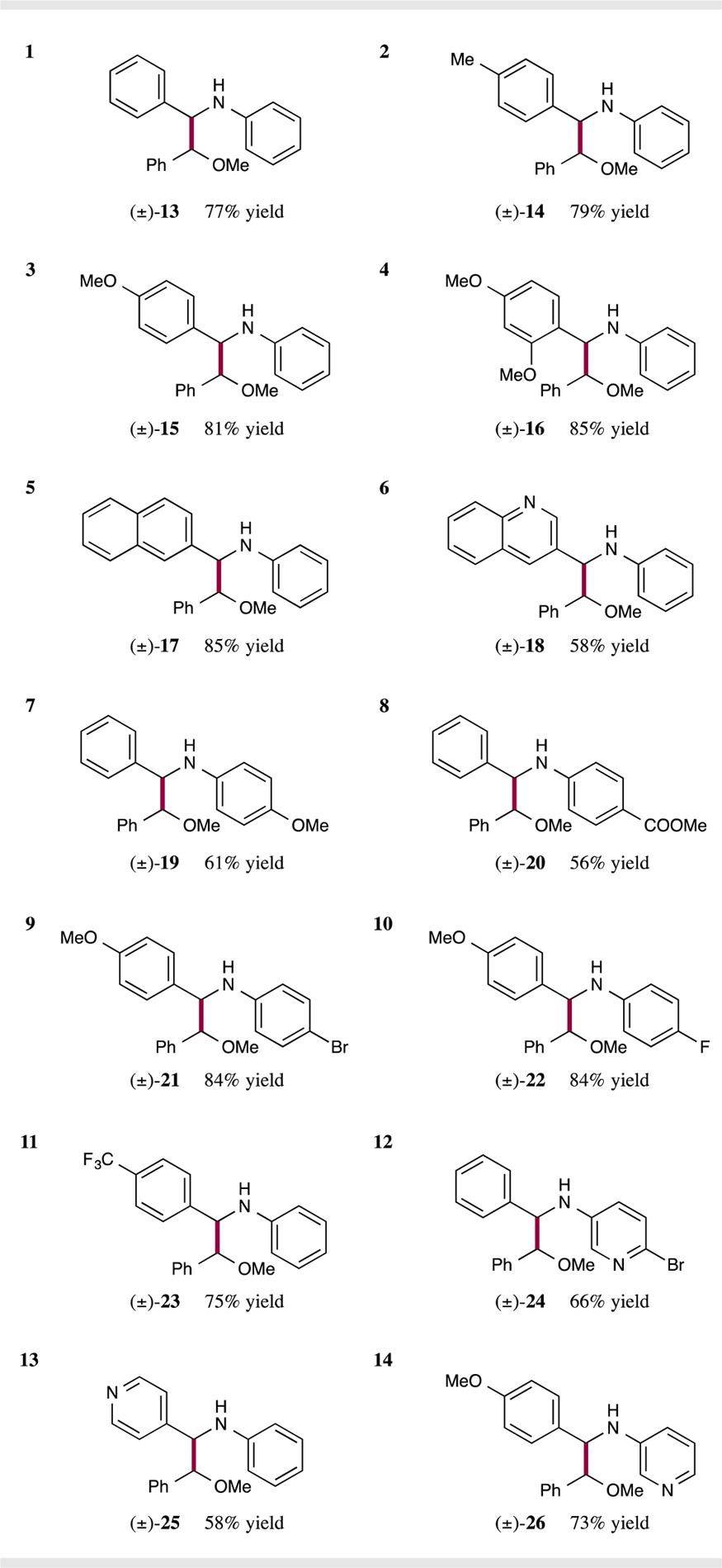
Isolated yields; see Supporting Information for experimental details. Diastereomeric ratios (d.r.) ≥ 1.4 : 1, determined by 1H NMR analysis.
Next we focused on expanding the range of benzylic ethers in this dual catalysis protocol (Table 3). To our delight we found that electron-rich ethers give the desired products in high yield (entry 1, 85% yield), along with para-, meta-, and ortho-substituted arenes (entries 1–4, 73–85% yield). With respect to the potential for broad application, it is important to recognize that a variety of protecting groups are well tolerated in this C–C bond forming process (entries 5–8, 70–82% yield). Moreover, cyclic ethers such as phthalane and isochromane are useful substrates, further demonstrating the versatility of this photoredox reaction (entries 9–11, 62–75% yield). Perhaps most notably, a wide range of heteroaromatic-containing ethers can be readily implemented using these reaction conditions (entries 12–14, 45–54% yield), an important consideration with respect to medicinal chemistry applications.
Table 3. Scope of the Ether Coupling Partnera.
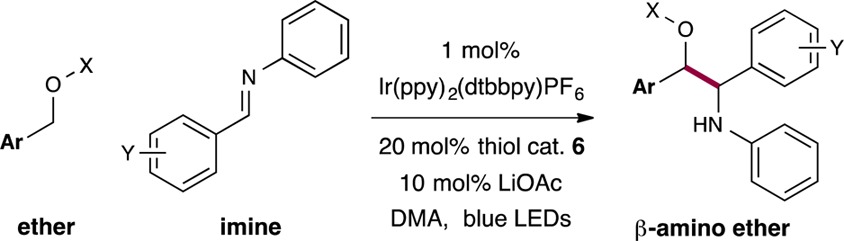
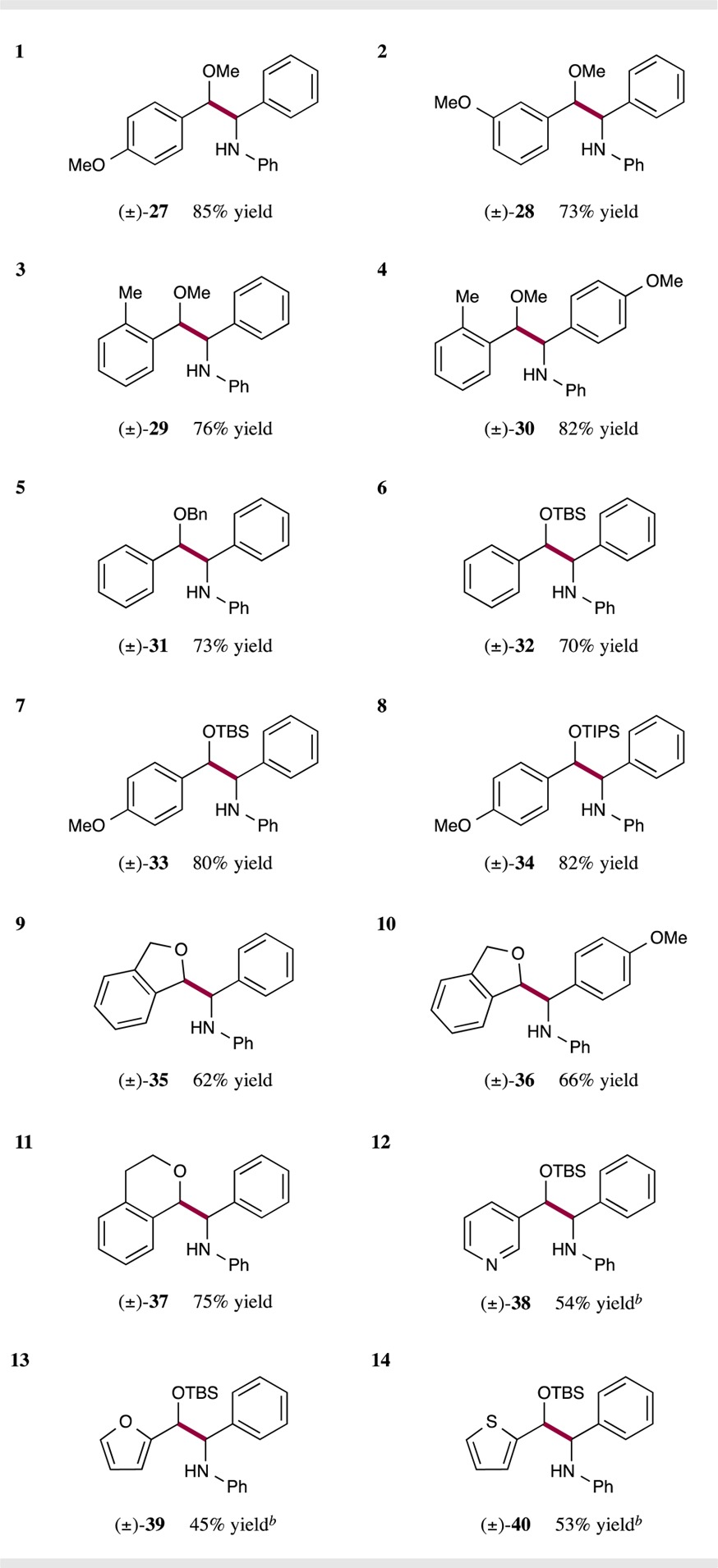
Isolated yields; see Supporting Information for experimental details. Diastereomeric ratios between 1:1 and 1.5:1, determined by 1H NMR analysis.
Reaction performed with 10 equiv of aromatic ether.
During the course of these studies, we rationalized that the rate of the reaction is likely dependent on the number of photons penetrating the reaction vessel and is therefore dependent on the illumination surface area. Thus, we investigated the progress of this transformation with respect to the size and nature of the reaction containers employed.17 It was shown that the typical reaction with N-phenyl imine 12 and benzyl methyl ether was slowest (22 h) in a standard reaction vial (as used in Tables 1–3). Indeed, when the same transformation was conducted in an NMR tube, the reaction proceeded with a significantly faster reaction rate with complete product formation determined at 14 h. However, the fastest protocol we have achieved thus far for this β-amino ether formation (6 h to completion) has been accomplished by maximizing the surface area via the use of PFA tubing as a reaction vessel.18 These results clearly demonstrate that (i) this transformation as currently performed is photon-limited with respect to the overall rate and (ii) it will likely be well suited to reaction scale-up via flow protocols.
Last, to provide further mechanistic insight into the pathway of this new photoredox coupling, we have conducted a series of Stern–Volmer quenching studies19 with Ir(ppy)2(dtbbpy)PF6 (3), benzalaniline, butyl thioglycolate (41),20 and n-Bu4NOAc as a soluble additive.21 As shown in Figure 1, these investigations clearly demonstrate the thiol catalyst 41 can indeed quench the excited photocatalyst, but only in the presence of the acetate base (no quenching was observed by the thiol in the absence of acetate). In addition, benzalaniline quenches the excited photocatalyst only to a minor extent,19 suggesting that a reductive quenching is the predominant pathway.
Figure 1.
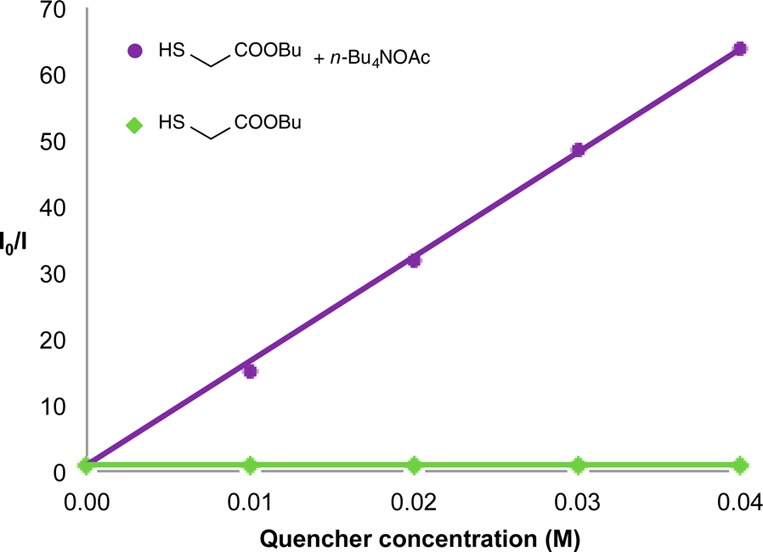
Stern–Volmer quenching studies with butyl thioglycolate (41) and n-Bu4NOAc.
Acknowledgments
Financial support was provided by NIHGMS (R01 GM103558-03), and kind gifts from Merck and Amgen. D.H. is grateful for financial support from the German Research Foundation (DFG).
Supporting Information Available
Procedures and spectral data are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Bergman R. G. Nature 2007, 446, 391. [DOI] [PubMed] [Google Scholar]; b Godula K.; Sames D. Science 2006, 312, 67. [DOI] [PubMed] [Google Scholar]; c Labinger J. A.; Bercaw J. E. Nature 2002, 417, 507. [DOI] [PubMed] [Google Scholar]
- Qvortrup K.; Rankic D. A.; MacMillan D. W. C. J. Am. Chem. Soc. 2014, 136, 626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mayer J. M. Annu. Rev. Phys. Chem. 2004, 55, 363. [DOI] [PubMed] [Google Scholar]; b Huynh M. H. V.; Meyer T. J. Chem. Rev. 2007, 107, 5004. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Warren J. J.; Tronic T. A.; Mayer J. M. Chem. Rev. 2010, 110, 6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tyson E. L.; Ament M. S.; Yoon T. P. J. Org. Chem. 2013, 78, 2046. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nguyen T. M.; Manohar N.; Nicewicz D. A. Angew. Chem., Int. Ed. 2014, 53, 6198. [DOI] [PMC free article] [PubMed] [Google Scholar]; c DeForest C. A.; Anseth K. S. Angew. Chem., Int. Ed. 2012, 51, 1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For some related examples on thiyl mediated hydrogen atom abstraction catalysis in organic synthesis, see:Feray L.; Kuznetsov N.; Renaud P. Sibi M. P. In Radicals in Organic Synthesis, Vol. 2; Renaud P., Sibi M. P., Eds.; Wiley-VCH: Weinheim, 2001; p 256. [Google Scholar]
- Luo Y.-R.Handbook of Bond Dissociation Energies in Organic Compounds; CRC Press LLC: Boca Raton, FL, 2003. [Google Scholar]
- Mayer J. M. Acc. Chem. Res. 2011, 44, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Arrasate S.; Lete E.; Sotomayor N. Tetrahedron: Asymmetry 2002, 13, 311. [Google Scholar]; b Bergmeier S. C. Tetrahedron 2000, 56, 2561. [Google Scholar]; c Reddy K. S.; Solà L.; Moyano A.; Pericàs M. A.; Riera A. J. Org. Chem. 1999, 64, 3969. [Google Scholar]; d Cho G. Y.; Ko S. Y. J. Org. Chem. 1999, 64, 8745. [Google Scholar]
- a Roberts B. P. Chem. Soc. Rev. 1999, 28, 25. [Google Scholar]; b Dang H.-S.; Roberts B. P. J. Chem. Soc., Perkin Trans. 1 1998, 67. [Google Scholar]; c Dang H.-S.; Roberts B. P. Tetrahedron Lett. 1999, 404, 8929. [Google Scholar]
- a Nicewicz D. A.; MacMillan D. W. C. Science 2008, 322, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nagib D. A.; Scott M. E.; MacMillan D. W. C. J. Am. Chem. Soc. 2009, 131, 10875. [DOI] [PMC free article] [PubMed] [Google Scholar]; c McNally A.; Prier C. K.; MacMillan D. W. C. Science 2011, 334, 1114. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Nagib D. A.; MacMillan D. W. C. Nature 2011, 480, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Rono L. J.; Yayla H. G.; Wang D. Y.; Armstrong M. F.; Knowles R. R. J. Am. Chem. Soc. 2013, 135, 17735. [DOI] [PubMed] [Google Scholar]
- Slinker J. D.; Gorodetsky A. A.; Lowry M. S.; Wang J.; Parker S.; Rohl R.; Bernhard S.; Malliaras G. G. J. Am. Chem. Soc. 2004, 126, 2763. [DOI] [PubMed] [Google Scholar]
- Shaidarova L. G.; Ziganshina S.-A.; Budnikov G. K. J. Anal. Chem. 2003, 58, 640. [Google Scholar]
- Root D. K.; Smith W. H. J. Electrochem. Soc. 1982, 129, 1231. [Google Scholar]
- Ochiai M.; Yamane S.; Hoque M. M.; Saito M.; Miyamoto K. Chem. Commun. 2012, 48, 5280. [DOI] [PubMed] [Google Scholar]
- Escoubet S.; Gastaldi G.; Vanthuyne N.; Gil G.; Siri D.; Bertrand M. P. J. Org. Chem. 2006, 71, 7288. [DOI] [PubMed] [Google Scholar]
- a Studer A. Chem.—Eur. J. 2001, 7, 1159. [DOI] [PubMed] [Google Scholar]; b Studer A. Chem. Soc. Rev. 2004, 33, 267. [DOI] [PubMed] [Google Scholar]; c Fischer H. J. Am. Chem. Soc. 1986, 108, 3925. [Google Scholar]; d Rüegge D.; Fischer H. Int. J. Chem. Kinet. 1989, 21, 703. [Google Scholar]; e Kothe T.; Marque S.; Martschke R.; Popov M.; Fischer H. J. Chem. Soc., Perkin Trans. 2 1998, 1553. [Google Scholar]
- See Supporting Information (SI) for experimental details.
- Nguyen J. D.; Reiß B.; Dai C.; Stephenson C. R. J. Chem. Commun. 2013, 49, 4352. [DOI] [PubMed] [Google Scholar]
- See SI for fluorescence quenching experiments.
- Due to easier handling, butyl thioglycolate (41) was the preferred reagent in Stern–Volmer studies.
- In contrast to LiOAc, n-Bu4NOAc is fully soluble in DMA and was therefore applied in Stern–Volmer studies.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.