

Abstract
Allenes are important 2π building blocks in organic synthesis and engage as 2-carbon components in many metal-catalyzed reactions. Wender and co-workers discovered that methyl substituents on the terminal allene double bond counterintuitively change the reactivities of allenes in [Rh(CO)2Cl]2-catalyzed intermolecular (5 + 2) cycloadditions with vinylcyclopropanes (VCPs). More sterically encumbered allenes afford higher cycloadduct yields, and such effects are also observed in other Rh(I)-catalyzed intermolecular cycloadditions. Through density functional theory calculations (B3LYP and M06) and experiment, we explored this enigmatic reactivity and selectivity of allenes in [Rh(CO)2Cl]2-catalyzed intermolecular (5 + 2) cycloadditions with VCPs. The apparent low reactivity of terminally unsubstituted allenes is associated with a competing allene dimerization that irreversibly sequesters rhodium. With terminally substituted allenes, steric repulsion between the terminal substituents significantly increases the barrier of allene dimerization while the barrier of the (5 + 2) cycloaddition is not affected, and thus the cycloaddition prevails. Computation has also revealed the origin of chemoselectivity in (5 + 2) cycloadditions with allene-ynes. Although simple allene and acetylene have similar reaction barriers, intermolecular (5 + 2) cycloadditions of allene-ynes occur exclusively at the terminal allene double bond. The terminal double bond is more reactive due to the enhanced d−π* backdonation. At the same time, insertion of the internal double bond of an allene-yne has a higher barrier as it would break π conjugation. Substituted alkynes are more difficult to insert compared with acetylene, because of the steric repulsion from the additional substituents. This leads to the greater reactivity of the allene double bond relative to the alkynyl group in allene-ynes.
Introduction
A preeminent goal of organic synthesis is to achieve structural complexity with functional value in a safe, simple, environmentally acceptable and step-, atom-, and time-economical fashion.1 As exemplified by the Diels–Alder reaction, cycloadditions are uniquely powerful processes to achieve this goal. They proceed in one operation with the convergent assembly of often commercially or readily available simple molecular components and produce a new ring system with generally up to four new stereocenters, enabling a rapid buildup of target relevant complexity. Prompted by the exceptional and growing importance of natural and designed targets based on seven-membered rings,2 such as tumor promoting phorbol esters and latency activating prostratin analogues, the latter uniquely important leads for HIV/AIDS eradication,3 Wender et al. reported in 1995 the first examples of transition-metal-catalyzed (5 + 2) cycloadditions of vinylcyclopropanes (VCPs) and π-systems.4 Subsequent contributions from this and other groups have advanced the (5 + 2) cycloaddition to a versatile, practical, and efficient route to various functionalized seven-membered rings.5−7 Among the current transition metal catalysts, rhodium complexes are found to exhibit high catalytic activity and provide often exceptional chemo-, regio- and enantioselectivity. Rhodium catalysts are thus far the only systems to effect intermolecular (5 + 2) cycloadditions.
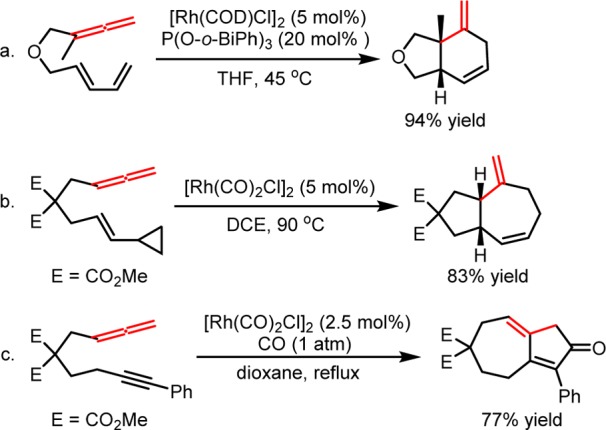
Allenes, one of the most common 2π components in cycloadditions, have been widely employed in Rh(I)-catalyzed intramolecular (m + n) and (m + n + o) cycloadditions.8,9 For example, Wender and co-workers reported the Rh(I)-catalyzed intramolecular (4 + 2) cycloadditions of 1, 3-dienes and allenes, affording the 6,5-, 6,6-, and 6,7-fused ring systems in an efficient fashion (Scheme 1a).10 The same group also reported the Rh(I)-catalyzed intramolecular (5 + 2) cycloadditions of VCPs and allenes (Scheme 1b).11 The reaction works with mono-, di-, tri- and tetra-substituted allenes, producing the seven-membered ring products with an exocyclic double bond that cannot otherwise be accessed through the corresponding cycloaddition of alkenes or alkynes. The intermolecular (5 + 2) cycloaddition reaction with allenes is however limited, prompting the development of a general allene equivalent exploiting the reactivity of alkynes.12 More generally, Brummond,13 Mukai,14 and Wender15 have independently studied the Rh(I)-catalyzed intramolecular Pauson–Khand type (2 + 2 + 1) cycloadditions with allenes, providing effective routes to functionalized cyclopentanones and cyclopentenones (Scheme 1c).16
Scheme 1. Selected Examples of Rh(I)-Catalyzed Intramolecular (m + n) and (m + n + o) Cycloadditions with Allenes.
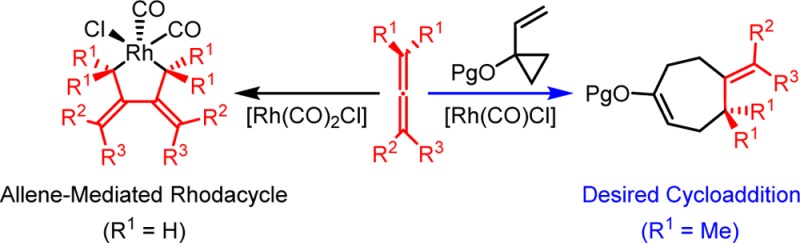
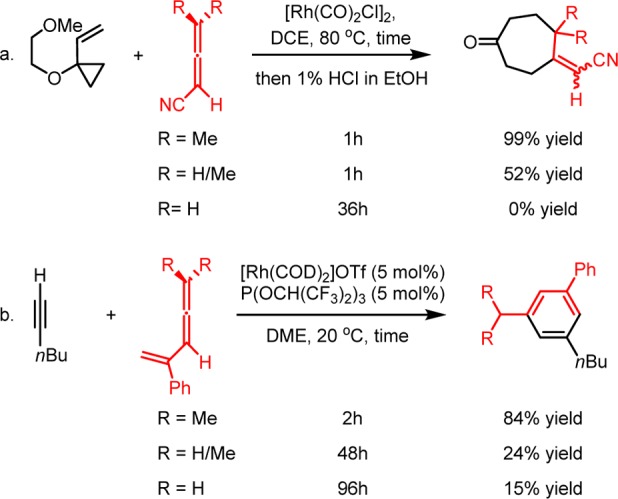
In 2005, Wegner, de Meijere and Wender reported the first [Rh(CO)2Cl]2-catalyzed intermolecular (5 + 2) cycloadditions of VCPs with allenes containing alkynyl, alkenyl, cyano, and cyanoalkyl substituents.5j In that work, the sterically encumbering methyl substituents on the terminal allene double bond were found necessary to achieve the intermolecular (5 + 2) cycloaddition, while terminally mono- and unsubstituted allenes produced the cycloadduct much less efficiently (Scheme 2a). More importantly, this “terminal methyl effect” is not only observed in the Rh(I)-catalyzed intermolecular (5 + 2) cycloadditions. It also is encountered in Rh(I)-catalyzed intermolecular (4 + 2) cycloadditions of allene-enes and alkenes (Scheme 2b).17 The methyl substituents on the terminal allene double bond are again necessary for efficient (4 + 2) cycloadditions, while terminally mono- and unsubstituted allene-enes reacted with much lower efficiencies. Murakami and co-workers have also observed a similar trend in Rh(I)-catalyzed (4 + 1) cycloadditions between allene-enes and carbon monoxide, where terminally mono- and disubstituted allene-enes underwent the desired cycloaddition, and terminally unsubstituted allene-enes formed an unreactive rhodium complex with the catalyst.18 Similarly, in cases of Rh(I)-catalyzed carbonylative rearrangements of allenyl ethers,19 (6 + 1) cycloadditions of allenylcyclobutanes,20 and even iridium-catalyzed (5 + 1) cycloadditions of allenylcyclopropanes,21 terminally unsubstituted or monosubstituted allene substrates gave lower yields than their disubstituted counterparts or were altogether unreactive. This enigmatic methyl effect limits the intermolecular use of allenes. Interestingly, this effect is not encountered in many Rh(I)-catalyzed intramolecular (m + n) and (m + n + o) cycloadditions, perhaps due to reduced reaction concentrations that suppress intermolecular side reactions and/or the higher formal concentration that favors an intramolecular process. However, in some cases even intramolecular rhodium-catalyzed cycloadditions have a discernible reactivity preference for terminally disubstituted allenes over their unsubstituted counterparts.22 Our DFT calculations also indicate that allenes with or without terminal methyl substituents have similar activation barriers for (5 + 2) cycloaddition (see discussions below). Therefore, it was thought that methyl substituents on allenes might favor the (5 + 2) and other cycloadditions by suppressing side reactions or catalyst inhibition.
Scheme 2. Examples of Terminal Methyl Effects on Allene Reactivities in Rh(I)-Catalyzed Intermolecular Cycloadditions5j,17.
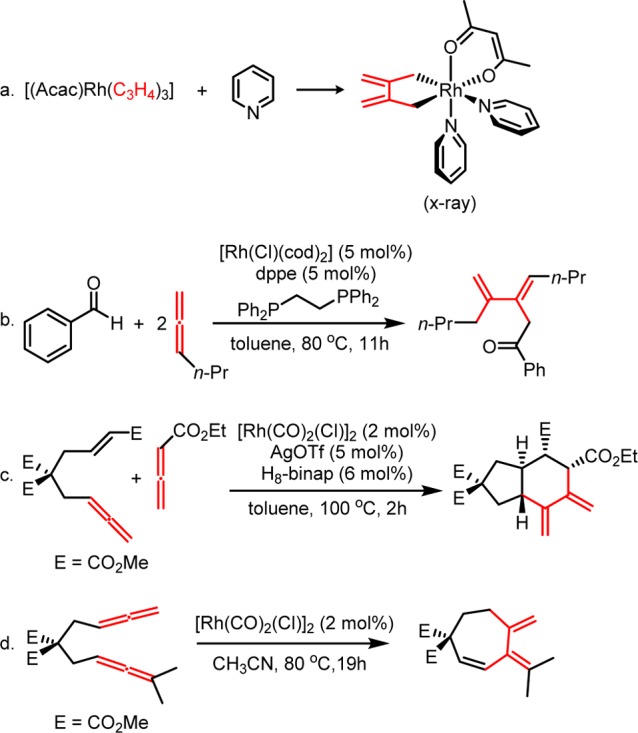
Allene dimerization is well-known. For example, 1,1-dimethylallene itself slowly dimerizes under mild conditions to form cyclobutane derivatives.23 Johnson’s calculations show that the dimerization of allene (uncatalyzed) occurs via a stepwise mechanism with a significant activation barrier of 34.5 kcal/mol (Scheme 3).24 Pertinent to our study, rhodium complexes have been found to catalyze several processes related to allene dimerization. In 1973, Ingrosso discovered that addition of a pyridine ligand induces the allene dimerization in the [(acac)Rh(C3H4)3] complex to form [(acac)Rh(C6H8)(pyridine)2], which has been characterized by X-ray crystallography (Scheme 4a).25 Murakami recently reported a Rh(I)-catalyzed 1:2 coupling between aldehydes and allenes (Scheme 4b),26 and Himo’s computational study confirms that this reaction occurs via an initial dimerization of allene with the rhodium catalyst.27 Alexanian reported a Rh(I)-catalyzed ene-allene–allene (2 + 2 + 2) cycloaddition that proceeds through an allene dimerization and alkene insertion, giving direct access to stereochemically rich six-membered carbocycles (Scheme 4c).28 Additionally, Ma reported that [Rh(CO)2Cl]2, the same catalyst used in the (5 + 2) cycloaddition, catalyzes the intramolecular dimerization of allene, followed by β-hydride elimination to yield a seven-membered ring product (Scheme 4d).29 On the basis of these previous studies of Rh(I)-catalyzed cycloadditions and allene dimerizations, we propose that the allene dimerization could be a hidden catalyst-poisoning pathway in Rh(I)-catalyzed intermolecular (5 + 2) and possibly many other (m + n) and (m + n + o) cycloadditions involving allenes.
Scheme 3. Experimental and Theoretical Studies of Uncatalyzed Thermal Dimerization of Allenes (Gibbs Free Energies in kcal/mol)23,24.

Scheme 4. Selected Examples of Rh(I)-Catalyzed Dimerization of Allenes and Related Methodologies.
The competing (5 + 2) cycloaddition and allene dimerization pathways with allene-ynes are described in Scheme 5. Starting from the precatalyst [Rh(CO)2Cl]2 (1), the (5 + 2) cycloaddition pathway proceeds with the active catalyst [Rh(CO)Cl] (2), formed via dissociation of the dimeric rhodium catalyst and elimination of a CO ligand.30 Initial cyclopropane cleavage gives a metallacyclohexene intermediate 3, and subsequent allene insertion produces a metallacyclooctene intermediate 5. From 5, C–C reductive elimination gives the seven-membered ring product. Alternatively, the [Rh(CO)xCl] (x = 1, 2) active catalyst 6 could catalyze the allene dimerization, leading to rhodium complex 8, which could be stable enough to inhibit the catalytic activity for the (5 + 2) cycloaddition.
Scheme 5. Proposed Mechanisms for Competing Rh(I)-Catalyzed (5 + 2) Cycloaddition and Allene Dimerization with Allene-ynes.
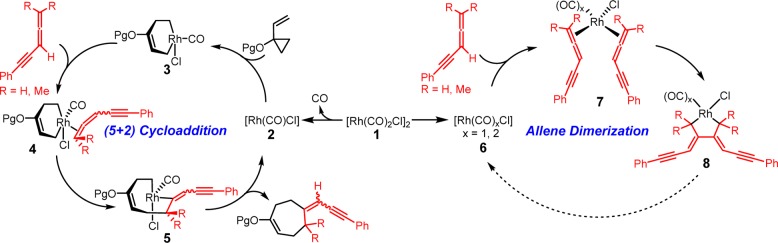
Besides the unique reactivity observed for the (5 + 2) cycloaddition of VCPs with variously substituted allene-ynes, the chemoselectivity is also intriguing. Although alkynes are typically more reactive than allenes in (5 + 2) cycloadditions, as documented in all prior work, reactions with conjugated allene-ynes occur exclusively at the terminal double bond of the allene (Scheme 6).5j To understand the origins of the unique reactivity and selectivity of these allene-based reactions, a subject pertinent to many other metal catalyzed reactions of allenes, we initiated DFT calculations and experimental studies on the contrasting behavior of allenes in intermolecular Rh(I)-catalyzed (5 + 2) cycloadditions with VCPs.
Scheme 6. Chemoselectivity of [Rh(CO)2Cl]2-Catalyzed Intermolecular (5 + 2) Cycloaddition of VCP 9 and Allene-yne 10.
Computational Methods
Geometry optimizations, vibrational frequencies, and thermal energy corrections were performed with the B3LYP functional, 6-31G(d) basis set for all main group elements and SDD basis set for rhodium implemented in Gaussian 09.31 Energies were evaluated with the M06 method,32 the 6-311+G(d,p) basis set for all main group elements, and SDD basis set for rhodium. All reported free energies involve zero-point vibrational energy corrections and thermal corrections to Gibbs free energy at 298 K. The solvation free energy corrections were computed with the CPCM model33 on gas-phase optimized geometries, and dichloroethane was chosen as the solvent for consistency with the experiment. Extensive conformational searches for intermediates and transition states have been conducted, and only the most stable conformers and isomers are discussed.
Results and Discussion
1. Origins of Reactivity
Reactions with Terminally Unsubstituted Allenes
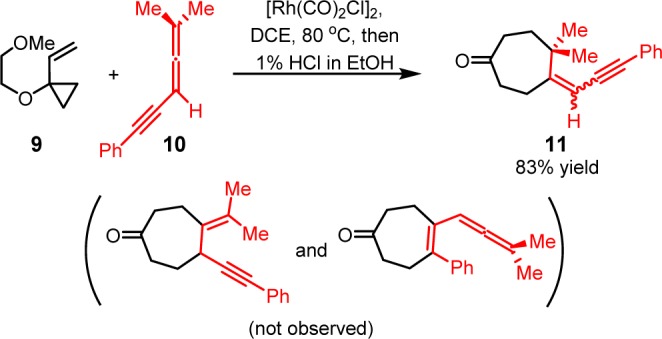
We first studied the (5 + 2) cycloaddition and allene dimerization pathways with allene-yne 12, which lacks the terminal allene substituents and experimentally yields no (5 + 2) cycloaddition product (Scheme 7). The 2-methoxyethoxy group on the VCP was replaced with a methoxy group in the calculations. With the [Rh(CO)Cl] active catalyst, the (5 + 2) cycloaddition of 1-methoxy-1-vinylcyclopropane and 12 gives the seven-membered ring product 13. Alternatively, the [Rh(CO)xCl] (x = 1, 2) complex can catalyze the dimerization of 12 and form the rhodacyclopentane complex 14. The calculated free energy changes for both pathways is shown in Figure 1 and optimized structures of selected intermediates and transition states are shown in Figure 2.
Scheme 7. Computational Model of [Rh(CO)2Cl]2-Catalyzed (5 + 2) Cycloaddition and Dimerization of the Terminally Unsubstituted Allene-yne 12.
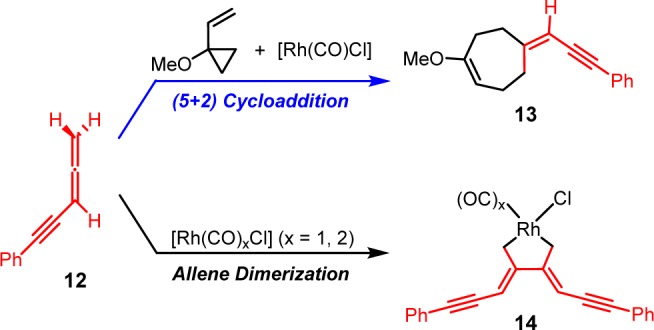
Figure 1.

Free energy profile of [Rh(CO)2Cl]2-catalyzed (5 + 2) cycloaddition and allene dimerization pathways with terminally unsubstituted allene-yne 12. Gibbs free energies (298 K) with respect to 15 are shown in kcal/mol.
Figure 2.

Structures of selected intermediates and transition states of the [Rh(CO)2Cl]2-catalyzed (5 + 2) cycloaddition and allene dimerization pathways with terminally unsubstituted allene-yne 12 (only the α-carbon of phenyl group is shown).
On the basis of previous computational studies,34,35 the active catalyst for the (5 + 2) cycloaddition is [Rh(CO)Cl], which is formed via dissociation of the dimeric [Rh(CO)2Cl]2 precatalyst and elimination of CO. From the [Rh(CO)Cl]-VCP complex 15, cyclopropane cleavage occurs to give a metallacyclohexene intermediate 16,36 and subsequent exergonic coordination of the allene-yne produces π complex 17. Insertion of the terminal double bond of the allene-yne via TS18 requires an activation barrier of 15.0 kcal/mol with respect to 17 and produces a metallacyclooctene intermediate 19.37 Insertion of the internal allene double bond or the alkyne requires a higher barrier (see later for discussions on chemo- and regioselectivity). Subsequent reductive elimination via TS20 gives the product-coordinated complex 21. Therefore, the 2π insertion of allene-yne via TS18 is the rate-determining step of the (5 + 2) cycloaddition pathway.
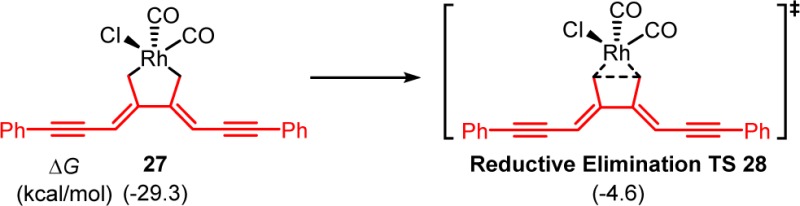
Because the exact mechanism of allene dimerization is still unclear, we computed both mechanisms involving [Rh(CO)2Cl] and [Rh(CO)Cl] as the active catalyst (shown in solid and dashed black lines, respectively, in Figure 1). If the [Rh(CO)Cl] complex is the active catalyst, substitution of the coordinated VCP by two allene-ynes converts 15 to intermediate 22. From 22, the oxidative cyclization of allene-ynes via TS23 requires an 18.5 kcal/mol barrier and the formed intermediate 24 is very stable. Alternatively, oxidative cyclization of allene-ynes with [Rh(CO)2Cl] occurs from intermediate 25. Although 25 is less stable than 22, TS26 is more favorable than TS23. This indicates that allene dimerization occurs more rapidly with [Rh(CO)2Cl]. The resultant intermediate 27 is very stable as compared to the Rh-allene complex 25, making the allene dimerization irreversible.38 Comparing the two reaction pathways, the overall barrier of allene dimerization (TS26) is only 1.3 kcal/mol higher than the (5 + 2) cycloaddition (TS18). These calculations suggest that the (5 + 2) cycloaddition and allene dimerization pathways are competitive with terminally unsubstituted allene-ynes. Therefore, the competitive and irreversible allene dimerization pathway “poisons” the rhodium catalyst by preventing its participation in the desired (5 + 2) cycloaddition.
Reactions with Dimethyl-Substituted Allene
We also studied the (5 + 2) cycloaddition and allene dimerization pathways with terminally dimethyl-substituted allene-yne 10. The computed free energy surface is shown in Figure 3. From [Rh(CO)Cl]-VCP complex 15, cyclopropane cleavage gives metallacyclohexene intermediate 16 and subsequent endergonic allene-yne coordination produces intermediate 29. The 2π insertion of the terminal double bond of the allene-yne occurs via TS30, and the formed metallacyclooctene intermediate 31 undergoes a facile C–C reductive elimination to generate the product-coordinated complex 33. Similar to the unsubstituted allene-yne 12, the rate-determining step of the (5 + 2) cycloaddition pathway with methyl-substituted allene-yne 10 is 2π insertion, and the overall barrier is 15.4 kcal/mol. The overall barriers of (5 + 2) cycloaddition pathway with allene-ynes are not affected by the terminal methyl substituents (15.0 kcal/mol with allene-yne 12 and 15.4 kcal/mol with methyl-substituted allene-yne 10). Despite their similar reactivities in the (5 + 2) cycloaddition, the methyl-substituted allene-yne 10 has a much higher barrier for the allene dimerization. The dimerization transition state TS35 is 4.0 kcal/mol higher in free energy than the 2π insertion transition state TS30. Therefore, the competing allene dimerization pathway in this case is disfavored relative to the experimentally observed (5 + 2) cycloaddition.
Figure 3.

Free energy changes of [Rh(CO)2Cl]2-catalyzed (5 + 2) cycloaddition and allene dimerization pathways with dimethyl-substituted allene-yne 10. Gibbs free energies (298 K) with respect to 15 are shown in kcal/mol.
Terminal Methyl Effects on Allene Dimerization
In order to understand the terminal methyl effects on the allene dimerization reactivity, all possible dimerization transition states with allene-ynes 12 and 10 are located and shown in Figure 4. Allene-ynes have a terminal double bond and an internal double bond, so there are three types of dimerization transition states. Allene-yne 12 can dimerize with both the terminal double bonds via TS26, one terminal and one internal double bond via TS37, or both internal double bonds via TS38. Computation indicates that TS26 is the most favorable transition state. Dimerization with the internal double bond (TS37 and TS38) will break the conjugation with the alkynyl group and raise the activation barrier. Therefore, the dimerization of allene-yne 12 occurs with the two terminal double bonds and requires an activation barrier of 13.3 kcal/mol compared to the [Rh(CO)Cl]-VCP complex 15.
Figure 4.

Optimized structures and relative free energies (with respect to 15) of transition states of [Rh(CO)2Cl]-mediated dimerization of allene-ynes 12 and 10 (only the α-carbon of phenyl group is shown for simplicity).
For dimethyl-substituted allene-yne 10, the preference between the three possible dimerization transition states is altered by steric repulsion of the methyl substituents (Figure 4). The dimerization with the two terminal double bonds via TS39 is no longer favored and the barrier is 25.5 kcal/mol. On the other hand, dimerization of one terminal and one internal double bond via TS35 is now the most favorable transition state and the activation barrier is 17.0 kcal/mol. Still, steric repulsion of the methyl substituents leads to a higher barrier for dimerization compared to the unsubstituted allene-yne (TS37, 15.1 kcal/mol). These results indicate that the high barrier of dimerization of terminally substituted allenes is due to steric repulsions between the terminal allene substituents and the rhodium catalyst.
Experimental Observations
To experimentally explore our computational prediction of a catalyst-poisoning allene dimerization pathway, a series of reactions were conducted to examine the interaction between des-dimethyl allene-yne 12 and the proposed active catalytic species. We hypothesized that if allene-yne 12 does not act as a catalyst poison, then a mixture of it and catalytically competent dimethyl allene-yne 10 in the presence of vinylcyclopropane 9 and precatalyst ([Rh(CO)2Cl]2) should provide yields of cycloadduct comparable to those originally reported (Scheme 6). However, if allene-yne 12 is indeed acting as a catalytic poison, addition of even a small (catalytic) amount to the cycloaddition of allene-yne 10 and vinylcyclopropane should shut down the reaction and no cycloadduct product would be observed.
First, 1-(2-methoxyethoxy)-1-vinylcyclopropane, dimethyl allene-yne 10, and [Rh(CO)2Cl]2 in DCE were mixed for 5 min at room temperature (Scheme 8, Experiment 1). An aliquot of one-half of the reaction mixture was removed and added to a vial containing a substoichiometric quantity (10 mol %) of the suspected catalyst “poison” precursor 12. Both mixtures were heated to 80 °C for 55 min. As predicted by theory, the mixture that had been “poisoned” contained no isolable quantities of the expected (5 + 2) cycloadduct. The original poison-free reaction mixture, however, provided the expected cycloadduct in 72% yield and an (E)/(Z) ratio of 1.0:1.5, consistent with the reported literature values. This result suggests that the des-dimethyl allene-yne, when added early on to an otherwise reactive mixture of allene-yne 10, VCP and catalyst, kills the rhodium catalyst irreversibly.
Scheme 8. Poisoning of the Cycloaddition between Vinylcyclopropane and Dimethyl Alleneyne.

The above experiment does not indicate whether the catalyst poison blocks conversion of precatalyst to active catalyst or irreversibly captures the active catalyst. To address this point, as in the first experiment, VCP 9, allene-yne 10, and precatalyst in DCE were combined, this time for 20 min, before an aliquot was taken and added to a substoichiometric amount (10 mol %) of allene-yne 12 (Scheme 8, Experiment 2). Both mixtures were heated to 80 °C for the remaining 40 min. After workup and purification, the nonpoisoned sample once again provided the cycloadduct with a yield (69%) and (E)/(Z) ratio (1.0:1.7) comparable to those previously reported. However, unlike in our previous experiment, the initiated but then poisoned reaction also showed formation of product, albeit in reduced yield (54%). These experiments indicate not only that the des-dimethyl allene-yne is unreactive toward Rh-catalyzed cycloaddition, but also that it can irreversibly capture a functioning catalyst, as predicted computationally.
2. Origins of Chemoselectivity
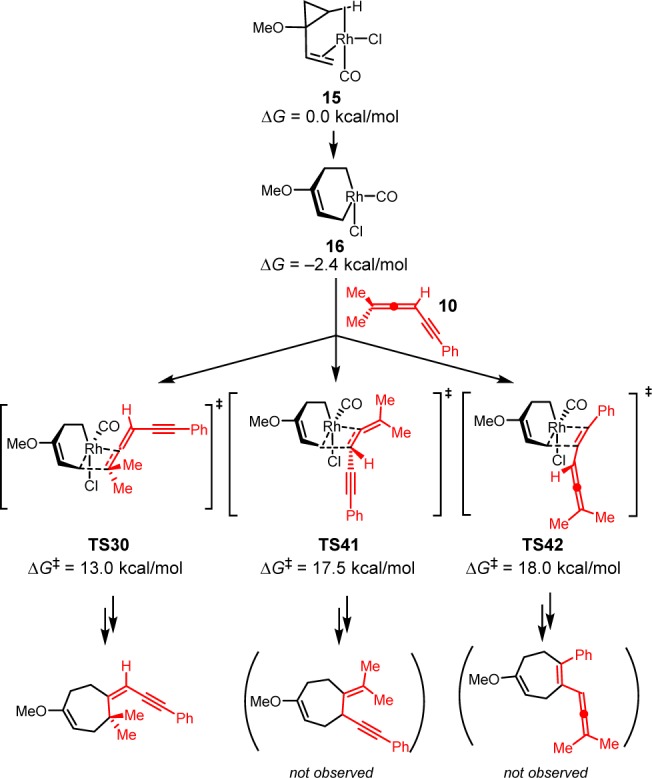
Although allene-ynes have three possible π-bonds that could engage in a (5 + 2) cycloaddition, the reaction occurs exclusively with the terminal double bond of the allene. The chemoselectivity is determined in the 2π insertion step, which is rate-limiting and irreversible. We calculated the three possible insertion transition states with allene-yne 10. The most favorable TS isomers in each pathway and their activation barriers are shown in Scheme 9. Consistent with the experiment, insertion of the terminal double bond via TS30 requires a barrier of 13.0 kcal/mol relative to the [Rh(CO)Cl]-VCP complex 15. The 2π insertion of the internal double bond via TS41 requires a 17.5 kcal/mol barrier and insertion of the triple bond via TS42 requires an 18.0 kcal/mol barrier, both are much higher than the 2π insertion barrier of terminal double bond. In line with this analysis, the (5 + 2) cycloadditions occur exclusively with the terminal double bond of allene-ynes.
Scheme 9. Possible 2π Insertion Transition States in the (5 + 2) Cycloaddition Pathway with Allene-yne 10.
In order to understand the origins of chemoselectivity, we compared the insertion barrier of allene-yne 10 with those of simple allene and acetylene to explore the steric and electronic effects of substituents on the insertion barriers with different π bonds (Scheme 10). Scheme 10a shows the substituent effects on the reactivity of the terminal double bond of allene-ynes. Insertion with simple allene requires a 13.8 kcal/mol barrier via TS43 with respect to the [Rh(CO)Cl]-VCP complex 15. The terminal double bond in allene-yne 12 is more reactive in 2π insertion than simple allenes. In TS18, the phenylalkynyl substituent on allene-yne 12 lowers the 2π insertion barrier by 1.8 kcal/mol. This is presumably due to the increased d−π* back-donation from rhodium to the conjugated π* orbital of the allene-yne. Interestingly, the terminal methyl substituents show only moderate steric repulsions around the forming C–C bond in 2π insertion. TS30 is only 1.0 kcal/mol higher in energy than the unsubstituted TS18. This indicates the terminal allene substituents prevent the substrates from dimerizing but have only minor effects on the rates of (5 + 2) cycloadditions.
Scheme 10. 2π Insertion Barriers (5 + 2) Cycloaddition Pathway with Different Allenes, Alkynes and Allene-ynes.

In contrast to the activation of the terminal double bond, a conjugated alkynyl group leads to higher 2π insertion barriers in reactions with the internal allenyl double bond (Scheme 10b). The 2π insertion with internal double bonds of allene-ynes via TS44 and TS41 requires about 4 kcal/mol higher barriers than the insertion of simple allene via TS43. The low reactivity of the internal double bond of allene-ynes is due to the π conjugation between the allene and the alkyne. Insertion into the internal double bonds breaks the conjugation and raises the barrier dramatically.
Although acetylene and simple allene have similar 2π insertion barriers (15.1 kcal/mol, TS45 versus 13.8 kcal/mol, TS43), in line with our previous computational studies, substituted alkynes in general have much higher barriers for 2π insertion. The 2π insertion with the triple bond of allene-yne 12 requires 18.0 kcal/mol (TS42, Scheme 10c), which is comparable to that of 2-butyne (18.5 kcal/mol, TS46). The lower reactivity of internal alkynes in the 2π insertion is because of the steric repulsions from the additional substituents of the alkynes. These calculations indicate that the 2π insertion with alkynes is more sensitive to steric effects than the reaction with substituted allenes.
In summary, the terminal double bond of allene-yne is selectively activated in the 2π insertion step due to the electronic effects of the conjugated alkynyl group, while the internal double bond and the triple bond are deactivated. This leads to exclusive formation of the (5 + 2) cycloadduct with the terminal double bond of the allene-ynes.
Conclusions
DFT calculations and experiments have revealed the mechanism and origins of substituent effects on reactivity and chemoselectivity of allene-ynes in Rh(I)-catalyzed intermolecular (5 + 2) cycloadditions with VCP. The Rh(I)-catalyzed (5 + 2) cycloaddition and allene dimerization are found to be competitive when allene-ynes lack methyl substituents on the terminal double bond. The competing allene dimerization is irreversible, generating a stable rhodium complex, thereby effectively poisoning the rhodium catalyst and shutting down the desired (5 + 2) cycloaddition. With the terminal methyl substituents, the barrier for allene dimerization of allene-ynes increases significantly while that of the (5 + 2) cycloaddition pathway is not affected, so the allene-ynes with terminal methyl substituents are able to undergo the (5 + 2) cycloaddition. Intramolecular (5 + 2) cycloadditions of allenes and VCPs are not similarly affected due to the expected higher formal concentration of the two reactive components relative to dimerization. The competing allene dimerization pathway explains the enigmatic reactivities of allenes in Rh(I)-catalyzed inter- and intramolecular cycloadditions and provides mechanistic insights into many other Rh(I)-catalyzed (m + n) and (m + n + o) cycloadditions.
The exclusive chemoselectivity for the terminal allene-yne double bond in the (5 + 2) cycloaddition has also been addressed. The rate-limiting 2π insertion step with the terminal double bond of an allene-yne requires a much lower barrier than for the 2π insertion with the internal double bond or the triple bond. Compared to the insertion of a simple allene, the insertion of the terminal double bond of an allene-yne has a stronger d−π* interaction between rhodium and the enyne from allene-yne, so the insertion barrier is lower. On the other hand, the internal double bond of the allene-yne is conjugated with the alkynyl group, and insertion into this π-bond breaks its conjugation and significantly increases the barrier. In addition, the insertion of the triple bond of allene-ynes is more difficult than that of acetylene, because of the additional steric repulsions. Therefore, the substituent effects differentiate the similar intrinsic reactivities of simple allene and acetylene, leading to the exclusive (5 + 2) cycloaddition with the terminal double bond of allene-ynes. These studies provide a comprehensive theoretical analysis of the reactions of allenes in rhodium-catalyzed cycloadditions that is consistent with experimental observations and provides a theoretical and experimental framework for the analysis of other related metal catalyzed allene-based cycloadditions.
Acknowledgments
We are grateful to the National Science Foundation (CHE- 1361104 (KNH), CHE-1265956 (PAW)) and the National Institutes of Health (CA031845 (PAW)) for financial support of this research. Additional funding was provided by the NSF Graduate Research Fellowship (MCS). Calculations were performed on the Hoffman2 Cluster at UCLA and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF (OCI-1053575).
Supporting Information Available
Energies and coordinates of DFT-computed stationary points. General information for experiments. Detailed results of experiment 1 and 2. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For a definition of the “ideal synthesis” incorporating the increasingly important issues of safety, step and atom count, waste stream, environmental, operational and time considerations, see the following and references contained therein:; a Wender P. A.; Miller B. L. Nature 2009, 460, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wender P. A. Nat. Prod. Rep. 2014, 31, 433. [DOI] [PubMed] [Google Scholar]; c Wender P. A.; Miller B. L. In Organic Synthesis: Theory & Applications; Hudlicky T., Ed.; JAI: Greenwich, 1993; Vol. 2, pp 27–66. [Google Scholar]; d Sheldon R. A. Chem. Commun. 2008, 3352. [DOI] [PubMed] [Google Scholar]; e Anastas P. T.Green Chemistry: Theory and Practice; Oxford University Press: Oxford, 1998. [Google Scholar]; For discussion of step and time economy, see:; f Wender P. A.; Handy S. T.; Wright D. L. Chem. Ind. 1997, 765. [Google Scholar]; g Wender P. A.; Croatt M. P.; Witulski B. Tetrahedron 2006, 62, 7505.and references therein. [Google Scholar]; h Wender P. A.; Verma V. A.; Paxton T. J.; Pillow T. H. Acc. Chem. Res. 2008, 41, 40. [DOI] [PubMed] [Google Scholar]; For a discussion of atom economy, see:; i Trost B. M. Science 1991, 254, 1471. [DOI] [PubMed] [Google Scholar]; j Trost B. M. Angew. Chem., Int. Ed. 1995, 34, 259. [Google Scholar]
- a Yet L. Chem. Rev. 2000, 100, 2963. [DOI] [PubMed] [Google Scholar]; b Battiste M. A.; Pelphrey P. M.; Wright D. L. Chem.—Eur. J. 2006, 12, 3438. [DOI] [PubMed] [Google Scholar]; c Wender P. A.; Croatt M. P.; Deschamps N. M. In Comprehensive Organometallic Chemistry III; Crabtree R. H., Mingos D. M. P., Eds.; Elsevier: Oxford, 2007; Vol. 10, pp 603–648. [Google Scholar]; d Butenschön H. Angew. Chem., Int. Ed. 2008, 47, 5287. [DOI] [PubMed] [Google Scholar]; e Pellissier H. Adv. Synth. Catal. 2011, 353, 189. [Google Scholar]; f Ylijoki K. E. O.; Stryker J. M. Chem. Rev. 2013, 113, 2244. [DOI] [PubMed] [Google Scholar]
- For recent studies and lead references, see:; a Wender P. A.; Kee J.-M.; Warrington J. M. Science 2008, 320, 649. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Beans E. J.; Fournogerakis D.; Gauntlett C.; Heumann L. V.; Kramer R.; Marsden M. D.; Chun T.-W.; Zack J. A.; Wender P. A. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 11698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wender P. A.; Takahashi H.; Witulski B. J. Am. Chem. Soc. 1995, 117, 4720. [Google Scholar]
- For rhodium catalysts, see:; a Wender P. A.; Rieck H.; Fuji M. J. Am. Chem. Soc. 1998, 120, 10976. [Google Scholar]; b Wender P. A.; Sperandio D. J. Org. Chem. 1998, 63, 4164. [Google Scholar]; c Gilbertson S. R.; Hoge G. S. Tetrahedron Lett. 1998, 39, 2075. [Google Scholar]; d Wender P. A.; Dyckman A. J.; Husfeld C. O.; Scanio M. J. C. Org. Lett. 2000, 2, 1609. [DOI] [PubMed] [Google Scholar]; e Wang B.; Cao P.; Zhang X. Tetrahedron Lett. 2000, 41, 8041. [Google Scholar]; f Wender P. A.; Barzilay C. M.; Dyckman A. J. J. Am. Chem. Soc. 2001, 123, 179. [DOI] [PubMed] [Google Scholar]; g Wender P. A.; Gamber G. G.; Scanio M. J. C. Angew. Chem., Int. Ed. 2001, 40, 3895. [PubMed] [Google Scholar]; h Wender P. A.; Williams T. J. Angew. Chem., Int. Ed. 2002, 41, 4550. [DOI] [PubMed] [Google Scholar]; i Wender P. A.; Love J. A.; Williams T. J. Synlett 2003, 1295. [Google Scholar]; j Wegner H. A.; de Meijere A.; Wender P. A. J. Am. Chem. Soc. 2005, 127, 6530. [DOI] [PubMed] [Google Scholar]; k Wender P. A.; Haustedt L. O.; Lim J.; Love J. A.; Williams T. J.; Yoon J.-Y. J. Am. Chem. Soc. 2006, 128, 6302. [DOI] [PubMed] [Google Scholar]; l Lee S. I.; Park Y.; Park J. H.; Jung G.; Choi S. Y.; Chung Y. K.; Lee B. Y. J. Org. Chem. 2006, 71, 91. [DOI] [PubMed] [Google Scholar]; m Saito A.; Ono T.; Hanzawa Y. J. Org. Chem. 2006, 71, 6437. [DOI] [PubMed] [Google Scholar]; n Gomez F. J.; Kamber N. E.; Deschamps N. M.; Cole A. P.; Wender P. A.; Waymouth R. M. Organometallics 2007, 26, 4541. [Google Scholar]; o Shintani R.; Nakatsu H.; Takatsu K.; Hayashi T. Chem.—Eur. J. 2009, 15, 8692. [DOI] [PubMed] [Google Scholar]; p Liu P.; Sirois L. E.; Cheong P. H. Y.; Yu Z. X.; Hartung I. V.; Rieck H.; Wender P. A.; Houk K. N. J. Am. Chem. Soc. 2010, 132, 10127. [DOI] [PubMed] [Google Scholar]; q Wender P. A.; Stemmler R. T.; Sirois L. E. J. Am. Chem. Soc. 2010, 132, 2532. [DOI] [PubMed] [Google Scholar]; r Wender P. A.; Sirois L. E.; Stemmler R. T.; Williams T. J. Org. Lett. 2010, 12, 1604. [DOI] [PubMed] [Google Scholar]; s Wender P. A.; Lesser A. B.; Sirois L. E. Org. Synth. 2011, 88, 109. [Google Scholar]; t Wender P. A.; Lesser A. B.; Sirois L. E. Angew. Chem., Int. Ed. 2012, 124, 2790. [DOI] [PubMed] [Google Scholar]
- For a ruthenium catalyst, see:; a Trost B. M.; Toste F. D.; Shen H. J. Am. Chem. Soc. 2000, 122, 2379. [Google Scholar]; b Trost B. M.; Shen H. C. Org. Lett. 2000, 2, 2523. [DOI] [PubMed] [Google Scholar]; c Trost B. M.; Toste F. D. Angew. Chem., Int. Ed. 2001, 40, 1114. [DOI] [PubMed] [Google Scholar]; d Trost B. M.; Shen H. C. Angew. Chem., Int. Ed. 2001, 40, 2313. [DOI] [PubMed] [Google Scholar]; e Trost B. M.; Shen H. C.; Schulz T. Org. Lett. 2003, 5, 4149. [DOI] [PubMed] [Google Scholar]; f Trost B. M.; Shen H. C.; Horne D. B.; Toste E. D.; Steinmetz B. G.; Koradin C. Chem.—Eur. J. 2005, 11, 2577. [DOI] [PubMed] [Google Scholar]; For a nickel catalyst, see:; g Zuo G.; Louie J. J. Am. Chem. Soc. 2005, 127, 5798. [DOI] [PubMed] [Google Scholar]; For an iron catalyst, see:; h Fürstner A.; Majima K.; Martin R.; Krause H.; Kattnig E.; Goddard R.; Lehmann C. W. J. Am. Chem. Soc. 2008, 130, 1992. [DOI] [PubMed] [Google Scholar]
- For representative total syntheses based on Rh(I)-catalyzed (5 + 2) and (5 + 2 + 1) cycloadditions, see:; a Wender P. A.; Fuji M.; Husfeld C. O.; Love J. A. Org. Lett. 1999, 1, 137. [Google Scholar]; b Wender P. A.; Zhang L. Org. Lett. 2000, 2, 2323. [DOI] [PubMed] [Google Scholar]; c Ashfeld B. L.; Martin S. F. Org. Lett. 2005, 7, 4535. [DOI] [PubMed] [Google Scholar]; d Ashfeld B. L.; Martin S. F. Tetrahedron 2006, 62, 10497. [Google Scholar]; e Fan X.; Zhuo L.-G.; Tu Y.-Q.; Yu Z.-X. Tetrahedron 2009, 65, 4709. [Google Scholar]; f Yuan C.; Jiao L.; Yu Z.-X. Tetrahedron Lett. 2010, 51, 5674. [Google Scholar]; g Liang Y.; Jiang X.; Yu Z.-X. Chem. Commun. 2011, 47, 6659. [DOI] [PubMed] [Google Scholar]; h Liang Y.; Jiang X.; Fu X.-F.; Ye S.; Wang T.; Yuan J.; Wang Y.; Yu Z.-X. Chem.—Asian. J. 2012, 7, 593. [DOI] [PubMed] [Google Scholar]; For related total syntheses based on Ru(II)-catalyzed (5 + 2) cycloadditions, see:; i Trost B. M.; Waser J.; Meyer A. J. Am. Chem. Soc. 2007, 129, 14556. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Trost B. M.; Waser J.; Meyer A. J. Am. Chem. Soc. 2008, 130, 16424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For reviews, see:; a Kitagaki S.; Inagaki F.; Mukai C. J. Synth. Org. Chem., Jpn. 2009, 67, 618. [Google Scholar]; b Ma S. Aldrichimica Acta 2007, 40, 91. [Google Scholar]; c Croatt M. P.; Wender P. A. Eur. J. Org. Chem. 2010, 19. [Google Scholar]; For selected examples of Rh(I)-catalyzed intramolecular cycloadditions with allenes, see:; d Wender P. A.; Correa A. G.; Sato Y.; Sun R. J. Am. Chem. Soc. 2000, 122, 7815. [Google Scholar]; e Inagaki F.; Sugikubo K.; Miyashita Y.; Mukai C. Angew. Chem., Int. Ed. 2010, 49, 2206. [DOI] [PubMed] [Google Scholar]; f Inagaki F.; Sugikubo K.; Oura Y.; Mukai C. Chem.—Eur. J. 2011, 17, 9062. [DOI] [PubMed] [Google Scholar]; g Oonishi Y.; Hosotani A.; Sato Y. J. Am. Chem. Soc. 2011, 133, 10386. [DOI] [PubMed] [Google Scholar]; h Mukai C.; Ohta Y.; Oura Y.; Kawaguchi Y.; Inagaki F. J. Am. Chem. Soc. 2012, 134, 19580. [DOI] [PubMed] [Google Scholar]; i Oonishi Y.; Hosotani A.; Sato Y. Angew. Chem., Int. Ed. 2012, 51, 11548. [DOI] [PubMed] [Google Scholar]; j Sugikubo K.; Omachi F.; Miyanaga Y.; Inagaki F.; Matsumoto C.; Mukai C. Angew. Chem., Int. Ed. 2013, 52, 11369. [DOI] [PubMed] [Google Scholar]; k Oonishi Y.; Kitano Y.; Sato Y. Tetrahedron 2013, 69, 7713. [Google Scholar]; l Oonishi Y.; Yokoe T.; Hosotani A.; Sato Y. Angew. Chem., Int. Ed. 2014, 53, 1135. [DOI] [PubMed] [Google Scholar]
- For other related Rh-catalyzed reactions with allenes or cycloadditions, see:; a Wang Y.; Wang J.; Su J.; Huang F.; Jiao L.; Liang Y.; Yang D.; Zhang S.; Wender P. A.; Yu Z.-X. J. Am. Chem. Soc. 2007, 129, 10060. [DOI] [PubMed] [Google Scholar]; b Jiao L.; Yuan C.; Yu Z.-X. J. Am. Chem. Soc. 2008, 130, 4421. [DOI] [PubMed] [Google Scholar]; c Evans P. A.; Inglesby P. A. J. Am. Chem. Soc. 2008, 130, 12838. [DOI] [PubMed] [Google Scholar]; d Ma S. Acc. Chem. Res. 2009, 42, 1679. [DOI] [PubMed] [Google Scholar]; e Jiao L.; Lin M.; Zhuo L.-G.; Yu Z.-X. Org. Lett. 2010, 12, 2528. [DOI] [PubMed] [Google Scholar]; f Jiao L.; Lin M.; Yu Z.-X. Chem. Commun. 2010, 46, 1059. [DOI] [PubMed] [Google Scholar]; g Brusoe A. T.; Alexanian E. J. Angew. Chem., Int. Ed. 2011, 123, 6726. [DOI] [PubMed] [Google Scholar]; h Zhang X.; Fu C.; Ma S. Org. Lett. 2011, 13, 1920. [DOI] [PubMed] [Google Scholar]; i Lin M.; Li F.; Jiao L.; Yu Z.-X. J. Am. Chem. Soc. 2011, 133, 1690. [DOI] [PubMed] [Google Scholar]; j Evans P. A.; Inglesby P. A. J. Am. Chem. Soc. 2012, 134, 3635. [DOI] [PubMed] [Google Scholar]; k Zeng R.; Fu C.; Ma S. J. Am. Chem. Soc. 2012, 134, 9597. [DOI] [PubMed] [Google Scholar]; l Zeng R.; Wu S.; Fu C.; Ma S. J. Am. Chem. Soc. 2013, 135, 18284. [DOI] [PubMed] [Google Scholar]; m Yu S.; Ma S. Angew. Chem., Int. Ed. 2012, 51, 3074. [DOI] [PubMed] [Google Scholar]; n Jiao L.; Yu Z.-X. J. Org. Chem. 2013, 78, 6842. [DOI] [PubMed] [Google Scholar]; o Evans P. A.; Inglesby P. A.; Kilbride K. Org. Lett. 2013, 15, 1798. [DOI] [PubMed] [Google Scholar]; p Wender P. A.; Fournogerakis D. N.; Jeffreys M. S.; Quiroz R. V.; Inagaki F.; Pfaffenbach M. Nat. Chem. 2014, 6, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]; q Inglesby P. A.; Bacsa J.; Negru D. E.; Evans P. A. Angew. Chem., Int. Ed. 2014, 53, 3952. [DOI] [PubMed] [Google Scholar]
- Wender P. A.; Jenkins T. E.; Suzuki S. J. Am. Chem. Soc. 1995, 117, 1843. [Google Scholar]
- Wender P. A.; Glorius F.; Husfeld C. O.; Langkopf E.; Love J. A. J. Am. Chem. Soc. 1999, 121, 5348. [Google Scholar]
- Wender P. A.; Inagaki F.; Pfaffenbach M.; Stevens M. C. Org. Lett. 2014, 16, 2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Brummond K. M.; Chen H.; Fisher K. D.; Kerekes A. D.; Rickards B.; Sill P. C.; Geib S. J. Org. Lett. 2002, 4, 1931. [DOI] [PubMed] [Google Scholar]; b Brummond K. M.; Sill P. C.; Rickards B.; Geib S. J. Tetrahedron Lett. 2002, 43, 3735. [Google Scholar]; c Brummond K. M.; Mitasev B. Org. Lett. 2004, 6, 2245. [DOI] [PubMed] [Google Scholar]; d Bayden A. S.; Brummond K. M.; Jordan K. D. Organometallics 2006, 25, 5204. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Brummond K. M.; Chen D. Org. Lett. 2008, 10, 705. [DOI] [PubMed] [Google Scholar]; f Brummond K. M.; Davis M. M.; Huang C. J. Org. Chem. 2009, 74, 8314. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Grillet G.; Huang C.; Brummond K. M. Org. Lett. 2011, 13, 6304. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Grillet F.; Brummond K. M. J. Org. Chem. 2013, 78, 3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mukai C.; Nomura I.; Yamanishi K.; Hanaoka M. Org. Lett. 2002, 4, 1755. [DOI] [PubMed] [Google Scholar]; b Mukai C.; Nomura I.; Kitagaki S. J. Org. Chem. 2003, 68, 1376. [DOI] [PubMed] [Google Scholar]; c Mukai C.; Inagaki F.; Yoshida T.; Kitagaki S. Tetrahedron 2004, 45, 4117. [Google Scholar]; d Mukai C.; Hirose T.; Teramoto S.; Kitagaki S. Tetrahedron 2005, 61, 10983. [Google Scholar]; e Mukai C.; Inagaki F.; Yoshida T.; Yoshitani K.; Hara Y.; Kitagaki S. J. Org. Chem. 2005, 70, 7159. [DOI] [PubMed] [Google Scholar]; f Inagaki F.; Mukai C. Org. Lett. 2006, 8, 1217. [DOI] [PubMed] [Google Scholar]; g Inagaki F.; Kawamura T.; Mukai C. Tetrahedron 2007, 63, 5154. [Google Scholar]; h Hirose T.; Miyakoshi N.; Mukai C. J. Org. Chem. 2008, 73, 1061. [DOI] [PubMed] [Google Scholar]; i Inagaki F.; Narita S.; Hasegawa T.; Kitagaki S.; Mukai C. Angew. Chem., Int. Ed. 2009, 48, 2007. [DOI] [PubMed] [Google Scholar]; j Inagaki F.; Kitagaki S.; Mukai C. Synlett 2011, 594. [Google Scholar]; k Inagaki F.; Itoh N.; Hayashi Y.; Matsui Y.; Mukai C. Beilstein J. Org. Chem. 2011, 7, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Hayashi Y.; Ogawa K.; Inagaki F.; Mukai C. Org. Biomol. Chem. 2012, 10, 4747. [DOI] [PubMed] [Google Scholar]; m Shafawati M. T. S.; Inagaki F.; Kawamura T.; Mukai C. Tetrahedron 2013, 69, 1509. [Google Scholar]; n Iwata T.; Inagaki F.; Mukai C. Angew. Chem., Int. Ed. 2013, 52, 11138. [DOI] [PubMed] [Google Scholar]; o Mukai C.; Takahashi Y.; Ogawa K.; Hayashi Y.; Inagaki F. Chem. Pharm. Bull. 2014, 62, 84. [DOI] [PubMed] [Google Scholar]
- Wender P. A.; Croatt M. P.; Deschamps N. M. Angew. Chem., Int. Ed. 2006, 45, 2459. [DOI] [PubMed] [Google Scholar]
- For a review on allenic Pauson–Khand reactions, see:Alcaide B.; Almendros P. Eur. J. Org. Chem. 2004, 3377. [Google Scholar]
- Murakami M.; Ubukata M.; Itami K.; Ito Y. Angew. Chem., Int. Ed. 1998, 37, 2248. [DOI] [PubMed] [Google Scholar]
- Murakami M.; Itami K.; Ito Y. Angew. Chem., Int. Ed. 1996, 34, 2691. [Google Scholar]
- Wender P. A.; Deschamps N. M.; Sun R. Can. J. Chem. 2005, 83, 838. [Google Scholar]
- Wender P. A.; Deschamps N. M.; Sun R. Angew. Chem., Int. Ed. 2006, 45, 3957. [DOI] [PubMed] [Google Scholar]
- Murakami M.; Itami K.; Ubukata M.; Tsuji I.; Ito Y. J. Org. Chem. 1998, 63, 4. [DOI] [PubMed] [Google Scholar]
- a Brummond K. M.; Chen H.; Mitasev B.; Casarez A. D. Org. Lett. 2004, 6, 2161. [DOI] [PubMed] [Google Scholar]; b Brummond K. M.; Chen D.; Davis M. M. J. Org. Chem. 2008, 73, 5064. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Aragoncillo C. Chem. Soc. Rev. 2010, 39, 783.and references therein. [DOI] [PubMed] [Google Scholar]
- For Johnson’s work, see:; a Skraba S. L.; Johnson R. P. J. Org. Chem. 2012, 77, 11096. [DOI] [PubMed] [Google Scholar]; For other related works, see:; b Siebert M. R.; Osbourn J. M.; Brummond K. M.; Tantillo D. J. J. Am. Chem. Soc. 2010, 132, 11952. [DOI] [PubMed] [Google Scholar]; c Soriano E.; Fernández I. Chem. Soc. Rev. 2014, 43, 3041. [DOI] [PubMed] [Google Scholar]
- Ingrosso G.; Immirzi A.; Porri L. J. Organomet. Chem. 1973, 60, C35. [Google Scholar]
- Toyoshima T.; Miura T.; Murakami M. Angew. Chem., Int. Ed. 2011, 50, 10436. [DOI] [PubMed] [Google Scholar]
- Huang G.; Kalek M.; Himo F. J. Am. Chem. Soc. 2013, 135, 7647. [DOI] [PubMed] [Google Scholar]
- a Brusoe A. T.; Alexanian E. J. Angew. Chem., Int. Ed. 2011, 50, 6596. [DOI] [PubMed] [Google Scholar]; b Brusoe A. T.; Edwankar R. V.; Alexanian E. J. Org. Lett. 2012, 14, 6096. [DOI] [PubMed] [Google Scholar]
- a Lu P.; Ma S. Org. Lett. 2007, 9, 2095. [DOI] [PubMed] [Google Scholar]; b Ma S.; Lu P.; Lu L.; Hou H.; Wei J.; He Q.; Gu Z.; Jiang X.; Jin X. Angew. Chem., Int. Ed. 2005, 44, 5275. [DOI] [PubMed] [Google Scholar]; c Jiang X.; Cheng X.; Ma S. Angew. Chem., Int. Ed. 2006, 45, 8009. [DOI] [PubMed] [Google Scholar]; d Ma S.; Lu L. Chem.—Asian J. 2007, 2, 199. [DOI] [PubMed] [Google Scholar]; e Chen G.; Jiang X.; Fu C.; Ma S. Chem. Lett. 2010, 39, 78. [Google Scholar]; f Lu P.; Ma S. Chin. J. Chem. 2010, 28, 1600. [Google Scholar]; g Aubert C.; Fensterbank L.; Garcia P.; Malacria M.; Simonneau A. Chem. Rev. 2011, 111, 1954. [DOI] [PubMed] [Google Scholar]
- Yu Z.-X.; Wender P. A.; Houk K. N. J. Am. Chem. Soc. 2004, 126, 9154. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Keith T.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dap-prich S.; Daniels A. D.; Farkas O.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2013.
- Zhao Y.; Truhlar D. G. Theor. Chem. Acc. 2008, 120, 215. [Google Scholar]
- a Barone V.; Cossi M. J. Phys. Chem. A 1998, 102, 1995. [Google Scholar]; b Cossi M.; Rega N.; Scalmani G.; Barone V. J. Comput. Chem. 2003, 24, 669. [DOI] [PubMed] [Google Scholar]
- For computational studies on rhodium-catalyzed (5 + 2) cycloadditions, see:; a Liu P.; Cheong P. H.-Y.; Yu Z.-X.; Wender P. A.; Houk K. N. Angew. Chem., Int. Ed. 2008, 47, 3939. [DOI] [PubMed] [Google Scholar]; b Yu Z.-X.; Cheong P. H.-Y.; Liu P.; Legault C. Y.; Wender P. A.; Houk K. N. J. Am. Chem. Soc. 2008, 130, 2378. [DOI] [PubMed] [Google Scholar]; c Xu X.; Liu P.; Lesser A.; Sirois L. E.; Wender P. A.; Houk K. N. J. Am. Chem. Soc. 2012, 134, 11012. [DOI] [PubMed] [Google Scholar]; For computational studies on other metal-catalyzed (5 + 2) cycloadditions, see:; d Hong X.; Liu P.; Houk K. N. J. Am. Chem. Soc. 2013, 135, 1456. [DOI] [PubMed] [Google Scholar]; e Hong X.; Trost B. M.; Houk K. N. J. Am. Chem. Soc. 2013, 135, 6588. [DOI] [PubMed] [Google Scholar]
- For other related DFT studies on Rh(I)-catalyzed reactions, see:; a Baik M.-H.; Baum E. W.; Burland M. C.; Evans P. A. J. Am. Chem. Soc. 2005, 127, 1602. [DOI] [PubMed] [Google Scholar]; b Chung L. W.; Wiest O.; Wu Y.-D. J. Org. Chem. 2008, 73, 2649. [DOI] [PubMed] [Google Scholar]; c Pitcock W. H.; Lord R. L.; Baik M.-H. J. Am. Chem. Soc. 2008, 130, 5821. [DOI] [PubMed] [Google Scholar]; d Wang H.; Sawyer J. R.; Evans P. A.; Baik M.-H. Angew. Chem., Int. Ed. 2008, 47, 342. [DOI] [PubMed] [Google Scholar]; e Yang Y.-F.; Shi T.; Zhang X.-H.; Tang Z.-X.; Wen Z.-Y.; Quan J.-M.; Wu Y.-D. Org. Biomol. Chem. 2011, 9, 5845. [DOI] [PubMed] [Google Scholar]; f Baik M.-H.; Mazumder S.; Ricci P.; Sawyer J. R.; Song Y.-G.; Wang H.; Evans P. A. J. Am. Chem. Soc. 2011, 133, 7621. [DOI] [PubMed] [Google Scholar]; g Jiao L.; Lin M.; Yu Z.-X. J. Am. Chem. Soc. 2011, 133, 447. [DOI] [PubMed] [Google Scholar]; h Lin M.; Kang G.-Y.; Guo Y.-A.; Yu Z.-X. J. Am. Chem. Soc. 2012, 134, 398. [DOI] [PubMed] [Google Scholar]; i Mazumder S.; Shang D.; Negru D.; Baik M.-H.; Evans P. A. J. Am. Chem. Soc. 2012, 134, 20569. [DOI] [PubMed] [Google Scholar]; j Li Z.; Boyarskikh V.; Hansen J. H.; Autschbach J.; Musaev D. G.; Davies H. M. L. J. Am. Chem. Soc. 2012, 134, 15497. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Yu H.; Lu Q.; Dang Z.; Fu Y. Chem.—Asian J. 2013, 8, 2262. [DOI] [PubMed] [Google Scholar]; l Zhang Q.; Yu H.-Z.; Li Y.-T.; Liu L.; Huang Y.; Fu Y. Dalton Trans. 2013, 42, 4175. [DOI] [PubMed] [Google Scholar]; m Xu X.; Liu P.; Shu X.; Tang W.; Houk K. N. J. Am. Chem. Soc. 2013, 135, 9271. [DOI] [PMC free article] [PubMed] [Google Scholar]; n Sun H.; Wang C.; Yang Y. −F.; Chen P.; Wu Y.-D.; Zhang X.; Huang Y. J. Org. Chem. 2014, 10.1021/jo500807d. [DOI] [PubMed] [Google Scholar]
- The cyclopropane cleavage is very facile with a 1.3 kcal/mol barrier relative to intermediate 15.
- Both the insertion transition states leading to Z- and E-cycloadducts were studied computationally, and only TS18 is shown in Figure 1. Experimentally, a mixture of Z- and E-cycloadducts is found.
- We studied the C–C reductive elimination from 27, the barrier is 24.7 kcal/mol, which is significantly higher than
the barrier of the observed (5 + 2) cycloaddition with allene-yne 10 under the same conditions (15.0 kcal/mol).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.