

Abstract
We describe the development and testing of ab initio derived, AMBER ff03 compatible charge parameters for a large library of 147 noncanonical amino acids including β- and N-methylated amino acids for use in applications such as protein structure prediction and de novo protein design. The charge parameter derivation was performed using the RESP fitting approach. Studies were performed assessing the suitability of the derived charge parameters in discriminating the activity/inactivity between 63 analogs of the complement inhibitor Compstatin on the basis of previously published experimental IC50 data and a screening procedure involving short simulations and binding free energy calculations. We found that both the approximate binding affinity (K*) and the binding free energy calculated through MM-GBSA are capable of discriminating between active and inactive Compstatin analogs, with MM-GBSA performing significantly better. Key interactions between the most potent Compstatin analog that contains a noncanonical amino acid are presented and compared to the most potent analog containing only natural amino acids and native Compstatin. We make the derived parameters and an associated web interface that is capable of performing modifications on proteins using Forcefield_NCAA and outputting AMBER-ready topology and parameter files freely available for academic use at http://selene.princeton.edu/FFNCAA. The forcefield allows one to incorporate these customized amino acids into design applications with control over size, van der Waals, and electrostatic interactions.
Keywords: noncanonical amino acids, unnatural amino acids, AMBER partial charges, complement, inhibitors, Compstatin, molecular dynamics
A timely goal in drug discovery is to have the ability to design new analogs that will stimulate or inhibit a receptor involved in a particular disease process. Several approaches spanning different molecular-weight scales exist to do this. Namely the discovery of small molecules, peptides, peptidomimetics, and high molecular weight antibody therapeutics are all means to create new drugs to address a variety of disease targets. The discovery of such molecules is challenging, with pharmaceutical companies spending billions of dollars each year on the research, development, and optimization of the affinity and bioavailability properties. Problems with pharmacokinetics and bioavailability were estimated to be the cause of 40% of failures in clinical trials,1 which is troublesome considering the cost of getting a drug to market is approaching $1 billion.2
Protein design is increasingly becoming a means to address some of the challenges faced by small molecules. Over 200 peptides, proteins, or antibodies have been marketed as of 2010,3 and it has been predicted that by 2020 we will see a larger number of peptides as drugs.4 Protein/peptide design faces its own difficulties though. These include passively permeating the cell membrane, being soluble at biologically relevant concentrations and pH values, and being subjected to proteolytic cleavage, which quickly reduces the half-life. In fact, unmodified peptides cannot circulate in the bloodstream for longer than a few minutes due to proteolytic cleavage,5 which hinders any potential therapeutic application of the most specific and highest affinity binders that are designed against a target.
Synthetic biology can potentially address several of the limitations of traditional peptide design through the introduction of post-translational modifications (PTMs) and unnatural amino acids. These noncanonical amino acids (NCAAs) are chemical and biological derivatives of the 20 canonical amino acids and upon their introduction can improve both pharmacokinetic and pharmacodynamic properties of a peptide–drug candidate while maintaining some of the core of the side-chain scaffold to preserve key interactions. Additionally, backbone N-methylated and d-amino acids are a viable approach to block proteolytic cleavage and improve metabolic stability.6 From a protein design standpoint, they offer the ability to diversify the sequence space remarkably. A 10 amino acid peptide has a sequence space of 2010 = 1.024 × 1013. Considering the 20 amino acids and the over 400 PTMs that exist7,8 in nature, the sequence space of that same 10-amino acid peptide becomes 42010 = 1.71 × 1026, and that only accounts for the l-amino acids. Thus, introduction of modified amino acids as design choices substantially expands the complexity of reaching the globally optimal design solution as was pointed out in a recent review.9
It is not well understood how NCAAs affect protein structure. Unnatural amino acids have been shown to have utility in rational design applications, and thus, binding data exists of peptide analogues containing unnatural amino acids and corresponding performance metrics including KD and IC50/EC50s. Modified amino acids can change the local electrostatic and conformational environment of a protein and may cause a variety of downstream biological responses.10 Although incorporation of NCAAs has been shown to change the affinity and inhibitory/stimulatory potency of a peptide, it is currently difficult to pursue rationalizations into the key interactions contributing to that increase due to lack of forcefields to model them.
Experimentally, one can incorporate noncanonical amino acids by peptide synthesis,11 through bio-orthogonal chemistry,12,13 or genetically.14−16 Evolution so far has been unable to address NCAA design naturally, as the machinery to incorporate NCAAs does not exist in most organisms. Even though most organisms have not evolved to incorporate NCAAs, this does not imply that they are not beneficial; it means that there are reasons for why they were not incorporated. These reasons include the toxicity of some of the building blocks and precursors to the noncanonical amino acid, the lack of a metabolic pathway in an organism to create the building block, the inability of the NCAA to be incorporated to permeate the cytoplasm,16 the lack of a modified aminoacyl tRNA synthetase that can accept the modified amino acid, the lack of an expanded genetic code in the organism, or the lack of a tRNA to decode a modified genetic code. There have been examples where each of these reasons has been successfully engineered.16,17 Despite organisms having limited ability to naturally incorporate NCAAs, we have derived means to synthetically introduce them.
Complicating the matter is that it is prohibitively expensive to screen a large library of noncanonical amino acid designs. Even the cost of incorporating a few modified amino acids is often an order of magnitude higher than the same scaffold sequence with only natural amino acids. The extreme cost is attributed to raw materials, synthesis, and purification costs, which contribute their own unique difficulties and require attention. These difficulties can be overcome with attention to each modification’s characteristics but do not make the screening of large libraries of modified amino acids on peptides or proteins tractable. Nonetheless, there exist many examples where noncanonical amino acids were experimentally incorporated into therapeutic peptides targeting different disease and functional processes, as shown in Table 1. A novel approach that can computationally screen potential analogues and has agreement with experimental data would offer a competitive advantage to its possessor.
Table 1. Examples of Non-canonical Amino Acids Incorporated into Therapeutic Peptide Agonists and Antagonists Targeting Various Diseases.
| non-canonical amino acid | therapeutic peptide | diseases targeted | source |
|---|---|---|---|
| biphenylalanine 2′-et-4′-ome-biphenylalanine 2-napthylalanine | truncated variant of GLP1-peptide | diabetes | Mapelli et al.18 |
| d-amino acids | Rosetta designed peptides | Alzheimer’s | Sievers et al.19 |
| 2-indanylglycine | oxytocin variants | inhibition of uterine motor activity | Bakos et al.20 |
| 2-napthylalanine | T140 variants | CXCR4/HIV-1 | Tamamura et al.21 |
| O-methyltyrosine | carbetocin | prevention of uterine atony, induction, and control of postpartum bleeding or hemorrhage | Vlieghe et al.3 |
| O-ethyltyrosine | atosiban | delaying the birth in case of premature birth | Vlieghe et al.3 |
| 1-napthylalanine | angiotensin I variants | hypertension | Kokubu et al.22 |
| 1-methyltryptophan, 5-methyltryptophan | Compstatin variants | stroke, heart attack, Alzheimer’s, asthma, rheumatoid arthritis, systemic lupus | Mallik et al.23 |
| cyclohexylalanine | |||
| phosphotyrosine | |||
| aminoisobutyric acid | |||
| N-methylated amino acids | Qu et al.24 |
Several groups have created methods to design proteins and peptides containing natural amino acids computationally that have been experimentally validated. RosettaDesign25 has been applied to design a number of different peptides to bind to targets,26−28 as well as design enzymes for diverse applications.29−33 Citizen-scientists have applied algorithms available in the Rosetta suite of tools to design a new enzyme for higher Diels-Alderase activity using an online multiplayer game.34 An iterative optimization approach has been created and applied to designing chimeric variants of dihidrofolate reductase35 and to switch the cofactor preference of an enzyme.36 Until recently, methods for computational design only addressed the natural amino acids. Kuhlman and co-workers37 have recently extended the Rosetta25 suite of tools to be able to handle several noncanonical amino acids, constructed an extended rotamer library, and used the parameters with Rosetta to perform design to derivatize peptides based on calpastatin to calpain-1. Zagrovic and co-workers have created parameters38 and tools39 for a library of modified amino acids compatible with the GROMACS simulation engine.
The Floudas group had notable success designing peptides with natural amino acids that have been validated experimentally on 6 protein targets, producing inhibitors and agonists of proteins that are linked to different diseases.40−49,53 These designs include entry inhibitors of HIV gp41,42 agonists and antagonists of the Complement component C3a receptor,45 inhibitors of Complement component C3c,40,41,43,47,49,53 and the redesign of human β-defensin-2.44 We recently developed Forcefield_PTM,50 a set of AMBER parameters for 32 frequently occurring post-translational modifications. Here, we present new forcefield charge parameters and a web interface to allow for the introduction of 147 noncanonical amino acids into peptides and proteins. The optimized charge parameters are validated on their ability to discriminate between active and inactive analogs of Compstatin for the inhibition of complement component C3c with approximate binding affinity and binding free energy calculations. Subsequently, the new forcefield parameters are used to understand the precise interactions in the most potent Compstatin analog containing a noncanonical amino acid compared with its unmodified counterpart and the original native sequence. Thus, with this new parameter set, we can understand how unnatural amino acids affect binding and other structural properties through molecular simulations.
Results and Discussion
New Forcefield Charge Parameters for 147 Non-Canonical Amino Acids
Partial charges were calculated for every atom in the library of 147 noncanonical amino acids listed in Table 2 in accordance with the ff03 methodology.51 The new parameters for each NCAA are presented in the Supporting Information section “Forcefield Parameters for Each Non-canonical Amino Acid Modification Grouped by Scaffold Residue” and are freely available for download and direct import into AMBER at http://selene.princeton.edu/FFNCAA. Conventions for atom and three-letter code naming were done mainly in line with corresponding CIF files contained in the PDB when contained there so the parameters can be used directly with corresponding input PDB files. Images of each NCAA are also provided in the Supporting Information with all atoms explicitly labeled.
Table 2. Table of Modified Amino Acids for Which Charge Parameters Are Presented in This Work Grouped by Scaffold Amino Acida.
| alanine | phenylalanine | asparagine | aspartic acid | cysteine |
| α-aminoisobutyric acid (AIB) | (R)-α-methyl-phenylalanine (MPH) | N4-methyl-asparagine (MEN) | N-methylaspartic acid (NMD) | (R)-l-α-methylcysteine (MCY) |
| 2-aminobutyric acid (ABA) | 2-ethyl-4-O-methyl-biphenylalanine (TEF) | (2s,4s)-2,5-diamino-4-hydroxy-5-oxopentanoic acid (GHG) | 2-amino-propanedioic acid (FGL) | cysteine acetamide (YCM) |
| adamanthane (ADA) | 3-methyl-biphenylalanine (TMB) | glutamine hydroxamate (HGA) | 3-methyl-aspartic acid (2AS) | N-methylcysteine (NMC) |
| 2-aminoheptanoic acid (AHP) | 3-O-methyl-biphenylalanine (TOM) | N-methyl-asparagine (NMN) | 2-amino-6-oxopimelic acid (26P) | carboxymethylated cysteine (CCS) |
| 3-cyclopentylalanine (CP3) | 2-ethyl-biphenylalanine (EBP) | β-asparagine (NBA) | β-aspartic acid (DBA) | benzylcysteine (BCS) |
| diethylalanine (DLE) | 2-methyl-4-O-methyl-biphenylalanine (MFO) | tryptophan | glycine | s-(2-hydroxyethyl)-cysteine (OCY) |
| R(+)-α-Allylalanine (AAL) | 2-methyl-biphenylalanine (MBP) | 5-methyltryptophan (MTR) | 2-indanyl-glycine (IGL) | s-acetonylcysteine (CSA) |
| (R)-α-ethyl alanine (REA) | biphenylalanine (BFA) | 1-methyltryptophan (OMW) | vinylglycine (LVG) | β-cysteine (CBA) |
| (S)-α-ethyl alanine (SEA) | 2-methylphenylalanine (MH2) | N-methyltryptophan (NMW) | phenylglycine (004) | methionine |
| cyclohexylalanine (ALC) | 3-methylphenylalanine (APD) | 2-hydroxytryptophan (TRO) | 4-hydroxyphenylglycine (D4P) | hydroxyl-methionine (ME0) |
| 1-napthylalanine (ALN) | 4-methylphenylalanine (4PH) | 4-amino-tryptophan (4IN) | (2s)-amino(3,5-dihydroxyphenyl)-ethanoic acid (3FG) | ethionine (ESC) |
| 2-napthylalanine (NAL) | 4-tert-butyl-phenylalanine (TP4) | 6-methyltryptophan (TR6) | N-methylglycine (NMG) | N-methyl-methionine (MME) |
| 5-hydroxy-1-napthalene (NO1) | 4-amino-phenylalanine (HOX) | 5-methoxytryptophan (MT5) | 2-allyl-glycine (2AG) | β-methionine (MBA) |
| 6-hydroxy-2-naphthalene (NO2) | 4-methoxy-phenylalanine (0A1) | β-hydroxy-tryptophane (HTR) | β-glycine (GBA) | leucine |
| 3-(9-anthryl)-alanine (ANT) | m-amidinophenyl-3-alanine (APM) | 5-hydroxytryptophan (HRP) | valine | t-butyl-leucine (BUG) |
| 3-(2-pyridyl)-alanine (PY2) | 4-carbamimidoyl-phenylalanine (0BN) | β-tryptophan (WBA) | (R)-(+)-α-methylvaline (MVL) | norleucine (NLE) |
| 3-(3-pyridyl)-alanine (PY3) | 4-hydroxymethyl-phenylalanine (4HP) | tyrosine | norvaline (NVA) | 5-oxo-norleucine (ONL) |
| 3-(4-pyridyl)-alanine (PY4) | 3-ethyl-phenylalanine (DMP) | O-methyltyrosine (OMY) | N-methyl-valine (MVA) | N-methyl-leucine (MLE) |
| 3-(2-quinolyl)-alanine (Q32) | 3,4-dimethylphenylalanine (D34) | O-ethyltyrosine (OEY) | β-valine (VBA) | homoleucine (HLE) |
| 3-(3-quinolyl)-alanine (Q33) | phenylserine (BB8) | O-allyltyrosine (OAY) | threonine | (R)-α-methylleucine (RML) |
| 3-(4-quinolyl)-alanine (Q34) | homophenylalanine (HPE) | N-methyltyrosine (NMY) | N-methylthreonine (NMT) | β-hydroxyleucine (HLU) |
| 3-(5-quinolyl)-alanine (Q35) | 3,3-diphenylalanine (DIF) | O-tyrosine (OTY) | o-methyl-threonine (OLT) | hydroxynorvaline (VAH) |
| 3-(6-quinolyl)-alanine (Q36) | kynurenine (KYN) | 3-amino-tyrosine (TY2) | β-threonine (TBA) | β-leucine (LBA) |
| 3-(8-hydroxyquinolin-3-yl)-alanine (HQA) | N-methyl-phenylalanine (MEA) | 3-amino-6-hydroxy-tyrosine (TYQ) | lysine | serine |
| N-methylalanine (NMA) | β-phenylalanine (FBA) | (β-R)-β-hydroxy-tyrosine (OMX) | (R)-α-methylornithine (RMO) | homoserine (HSE) |
| 1-pyrenylalanine (PAL) | isoleucine | β-tyrosine (YBA) | (S)-α-methylornithine (SMO) | 2-amino-5-hydroxypentanoic acid (LDO) |
| (R)-2-(2′-propenyl)-alanine (PRP) | N-methylisoleucine (NMI) | glutamic acid | 2,3-diaminopropanoic acid (DPP) | 6-hydroxy-norleucine (AA4) |
| (R)-2-(4′-pentenyl)-alanine (PEN) | allo-isoleucine (IIL) | N-methylglutamic acid (NME) | diaminobutyric acid (DAB) | N-methyl-serine (NMS) |
| (R)-2-(7′-octenyl)-alanine (OCT) | 3-methyl-alloisoleucine (I2M) | (3r)-3-methyl-glutamic acid (LME) | (2s)-2,8-diaminooctanoic acid (HHK) | β-serine (SBA) |
| β-alanine (AAB) | β-isoleucine (IBA) | (3s)-3-methyl-glutamic acid (MEG) | N-methyl-lysine (NMK) | histidine |
| glutamine | arginine | 2s,4r-4-methylglutamate (SYM) | β-lysine (KBA) | N-methylhistidine (NMH) |
| N-methylglutamine (NMQ) | N-methylarginine (NMR) | 5-o-methyl-glutamic acid (GME) | ||
| N5-methyl-glutamine (MEQ) | ||||
| 3-methyl glutamine (LMQ) | ||||
| β-glutamine (QBA) |
Corresponding three-letter codes are listed in parentheses following each amino acid.
With the determination of these parameters, we next present the results of our efforts to test the parameters on experimental binding data and subsequently to understand the key interactions involved in the most potent Compstatin analog in Table 3 containing the unnatural amino acid 1-methyltryptophan relative to its scaffold sequence.
Table 3. Data Set Used for Testing the Optimized Charges Introduced in Forcefield_NCAAa.
| analog | source | sequence | SeqID | IC50 (μM) |
|---|---|---|---|---|
| 1 | pharmacophore | Ac-ICV(PTR)QDWGAHRCI-NH2 | 28 | 9.60 |
| 2 | pharmacophore | Ac-RCVVQDWGHHRCT-NH2 | 17 | 8.00 |
| 3 | pharmacophore | Ac-LCVVQDWGWHRCG-NH2 | 15 | 5.40 |
| 4 | pharmacophore | Ac-ICVWQDWGWHRCT-NH2 | 24 | 3.10 |
| 5 | pharmacophore | Ac-ICVVNDWGHHRCT-NH2 | 3 | 4.20 |
| 6 | structurekinetic | Ac-ICV(OMY)QDWGAHRCT-NH2 | 5 | 1.30 |
| 7 | pharmacophore | Ac-MCVHQDWGGHRCF-NH2 | 16 | 85.20 |
| 8 | pharmacophore | Ac-ICVWQDWGHHRCT-NH2 | 2 | 2.20 |
| 9 | structurekinetic | Ac-ICV(MTR)QDWGAHRCT-NH2 | 3 | 0.87 |
| 10 | novel analogues | Ac-ICVYQDWGAHRC(NMT)-NH2 | 12 | 1.90 |
| 11 | pharmacophore | Ac-ICV(OMW)QDWGAHRCT-NH2 | 1 | 0.21 |
| 12 | pharmacophore | Ac-ICVSQDWGHHRCT-NH2 | 20 | 50.90 |
| 13 | pharmacophore | Ac-ICVVQDWGHHSCT-NH2 | 10 | 25.00 |
| 14 | pharmacophore | Ac-ICVVQDWGHHRCI-NH2 | 13 | 3.20 |
| 15 | structurekinetic | Ac-ICVWQDWG(AIB)HRCT-NH2 | 12 | 1.50 |
| 16 | pharmacophore | Ac-ICVWQDWGAHRCT | 25 | 2.00 |
| 17 | pharmacophore | Ac-ICVVNDWGHHACT-NH2 | 11 | 60.00 |
| 18 | novel analogues | Ac-ICVYQDWGAHR(NMC)T-NH2 | 11 | 154.00 |
| 19 | structurekinetic | Ac-ICV(PAL)QDWGAHRCT-NH2 | 9 | 1.20 |
| 20 | pharmacophore | Ac-ICV(ALC)QDWGAHRCT | 27 | 53.60 |
| 21 | pharmacophore | Ac-ICVHQDWGHHRCT-NH2 | 21 | 10.50 |
| 22 | pharmacophore | Ac-ICVVQDWGAHACT-NH2 | 12 | 9.90 |
| 23 | structurekinetic | Ac-ICVWQDWGAHRCT-NH2 | 0 | 1.20 |
| 24 | pharmacophore | Ac-ICVWQD(OMW)GAHRCT-NH2 | 4 | 1000.00 |
| 25 | pharmacophore | Ac-ICLVQDWGHHRCT-NH2 | 8 | 10.00 |
| 26 | pharmacophore | Ac-ICVYQDWGAHRCT-NH2 | 23 | 3.80 |
| 27 | structurekinetic | Ac-ICVYQDWGAHRCT-NH2 | 4 | 2.40 |
| 28 | pharmacophore | Ac-ICVWQDWG(AIB)HRCT-NH2 | 29 | 1.50 |
| 29 | structurekinetic | Ac-ICVVQDWGHHRCT-NH2 | 15 | 4.50 |
| 30 | pharmacophore | Ac-ICVAQDWGAHRCI-NH2 | 7 | 12.00 |
| 31 | pharmacophore | Ac-ICLVNDWGHHRCT-NH2 | 9 | 8.30 |
| 32 | novel analogues | Ac-ICVYQD(NMW)GAHRCT-NH2 | 6 | 25.00 |
| 33 | pharmacophore | Ac-ICV(ALN)QDWGAHRCT | 31 | 1.80 |
| 34 | pharmacophore | Ac-ICVTQDWGHHRCT-NH2 | 19 | 68.30 |
| 35 | novel analogues | Ac-ICVYQ(NMD)WGAHRCT-NH2 | 5 | 44.00 |
| 36 | novel analogues | Ac-ICVYQDW(NMG)AHRCT-NH2 | 7 | 584.47 |
| 37 | novel analogues | Ac-ICVY(NMQ)DWGAHRCT-NH2 | 4 | 33.00 |
| 38 | novel analogues | Ac-ICVYQDWGAHRCT-NH2 | 0 | 2.40 |
| 39 | pharmacophore | Ac-LCVWQDWGRHQCF-NH2 | 14 | 131.00 |
| 40 | pharmacophore | Ac-ICVFQDWGHHRCT-NH2 | 22 | 10.20 |
| 41 | novel analogues | Ac-ICVYQDWGAH(NMR)CT-NH2 | 10 | 32.00 |
| 42 | novel analogues | Ac-ICVYQDWGA(NMH)RCT-NH2 | 9 | 94.00 |
| 43 | structurekinetic | Ac-ICV(OMW)QDWGPHRCT-NH2 | 14 | 0.54 |
| 44 | pharmacophore | Ac-DCVVQDWGHHRCT-NH2 | 18 | 22.00 |
| 45 | structurekinetic | Ac-ICV(OEY)QDWGAHRCT-NH2 | 6 | 1.30 |
| 46 | novel analogues | Ac-ICVYQDWG(NMA)HRCT-NH2 | 8 | 1000.00 |
| 47 | pharmacophore | CVVQDWGHHRC-NH2 | del1 | 33.00 |
| 48 | pharmacophore | CVVQDWGHC-NH2 | del9 | 600.00 |
| 49 | novel analogues | Ac–I(NMC)VYQDWGAHRCT-NH2 | 1 | 7.50 |
| 50 | novel analogues | Ac-ICV(NMY)QDWGAHRCT-NH2 | 3 | 1000.00 |
| 51 | pharmacophore | Ac-ICVVGDWGHHRCT-NH2 | 6 | 567.00 |
| 52 | pharmacophore | CVVQDWGHHRCT-NH2 | del0 | 25.00 |
| 53 | pharmacophore | ICVVQDWGHHRCT | 0 | 12.00 |
| 54 | pharmacophore | IAVVQDWGHHRAT | 5 (Linear) | 600.00 |
| 55 | pharmacophore | CVVQDWC-NH2 | del8 | 600.00 |
| 56 | pharmacophore | CAVQDWGHHRC | del10 | 1200.00 |
| 57 | pharmacophore | CWGHHRCT-NH2 | del4 | 600.00 |
| 58 | pharmacophore | CVVQDWAHHRC | del11 | 1200.00 |
| 59 | pharmacophore | CVQDWGHHRCT-NH2 | del7 | 600.00 |
| 60 | pharmacophore | CQDWGHHRCT-NH2 | del6 | 600.00 |
| 61 | pharmacophore | CDWGHHRCT-NH2 | del5 | 600.00 |
| 62 | pharmacophore | CHHRCT-NH2 | del2 | 600.00 |
| 63 | pharmacophore | CGHHRCT-NH2 | del3 | 600.00 |
The noncanonical amino acids studied are phosphotyrosine (PTR), O-methyltyrosine (OMY), N-methylthreonine (NMT), 5-methyltryptophan (MTR), 1-methyltryptophan (OMW), α-aminoisobutyric acid (AIB), N-methylcysteine (NMC), 1-pyrenylalanine (PAL), cyclohexylalanine (ALC), N-methyltryptophan (NMW), 1-naphthylalanine (ALN), N-methylaspartic acid (NMD), N-methylglycine (NMG), N-methylglutamine (NMQ), N-methyl-arginine (NMR), N-methylhistidine (NMH), O-ethyltyrosine (OMY), N-methylalanine (NMA), N-methyltyrosine (NMY). Several of these non-canonical amino acids were substituted in different positions on the Compstatin sequence. ACE and NH2 correspond to the N-terminal and C-terminal blocking groups acetyl and amide to keep the termini neutrally charged.
Predictive Ability of Approximate Binding Affinity in Discriminating Active and Inactive Analogs of Compstatin
Independent molecular dynamics simulations for all 63 Compstatin variants reported in Table 3 were carried out in complex with C3c and in isolation. An independent simulation of the protein C3c without the peptide bound was also carried out. The approximate binding affinity was calculated as in eq 7 (see Methods). The analogs were rank ordered by K* from highest to lowest. Then, an ROC curve was constructed to assess the predictive ability of the approximate binding affinity metric to discriminate between active and inactive analogs. Two cutoffs were chosen for defining whether an analog was active or inactive: 20 μM and 200 μM.
Parts A and B of Figure 1 present the results of the predictive ability of this metric for this system using the forcefield parameters derived. The approximate binding affinity metric can reasonably discriminate between active and inactive analogs with areas under the ROC curve of 0.691 and 0.721 corresponding to a cutoff for active analogs with IC50 < 20 μM and <200 μM, respectively. Only 1 false positive was observed in the top 10 ranked analogs by K*, and 6 false positives by the 20 μM IC50 cutoff were observed in the top 20. This result is encouraging, since K* has previously been used as the final discriminating metric in our de novo protein design framework42,43,46 using only natural amino acids. This suggests that K* can be used to reasonably discriminate between active/inactive analogs for new designs of Compstatin with the AMBER energy function and molecular dynamics simulations.
Figure 1.

Receiver operating characteristic (ROC) curves constructed from rank-ordered lists of Compstatin variants’ binding metrics. ROC curve for rank ordered list by K* corresponding to an active IC50 cutoff of <20 μM (A) and 200 μM (B). ROC curve for rank ordered list by ΔGBind,Solv°GBSA to an active IC50 cutoff of <20 M (C) and 200 μM (D).
Predictive Ability of Binding Free Energy Calculations in Discriminating Active and Inactive Analogs of Compstatin
An independent molecular dynamics simulation of the complex with each of the 63 variants of Compstatin and C3c was carried out. Snapshots from the simulations were used to perform MM-GBSA calculations to evaluate the binding free energies of each analog. Parts C and D of Figure 1 present the results of the ability for MM-GBSA to discriminate between active and inactive analogs. Binding free energy calculations performed using the forcefield parameters and MM-GBSA yielded the greatest discriminatory ability with areas under the ROC curve of 0.808 and 0.936 corresponding to a cutoff for active analogs with IC50 < 20 μM and <200 μM, respectively. This suggests that using the forcefield parameters with MM-GBSA can accurately discriminate between active and inactive analogs of Compstatin. In fact, in the top 10 sequences’ calculated ΔGBind,Solv°GBSA, there was only 1 false positive for either of the IC50 cutoffs (in the seventh position), and 6 false positives in the top 20 sequences. Removing the sequences corresponding to deletions of Compstatin in order to compare only equal sequence-length analogs yields an area under the curve of 0.702 and 0.869 for a cutoff IC50 of <20 μM and <200 μM, respectively.
We next asked whether there exists a correlation observed between experimental IC50 and calculated binding free energy as IC50 values were found to be strongly correlated (R2 = 0.887) with KD values in the work of Magotti and co-workers.52 IC50 data was used over KD due to the much larger number of experimental data points available for this system, important for achieving statistical significance in the results. Ideally, a larger number of KD values would be available to perform similar assessments. We observed a weak correlation between IC50 and binding free energy in Figure 2. The existence of a correlation is in agreement with data presented by Magotti.52
Figure 2.
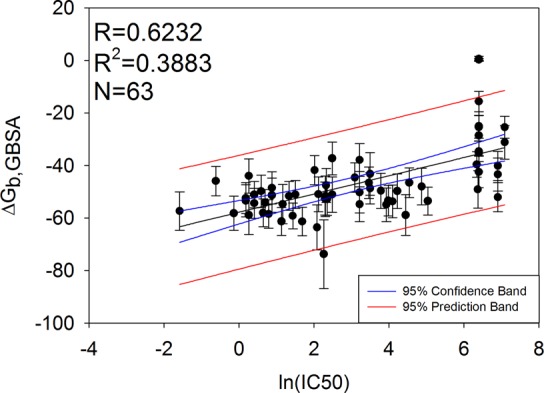
Correlation between IC50 and calculated binding free energies using MM-GBSA for 63 Compstatin analogs. The blue bands correspond to the 95% confidence interval for the regression line. The red bands correspond to the 95% confidence interval for a new value to lie in the prediction band. Error bars are ±1 standard deviation from the mean binding free energy calculated.
It is interesting that the binding free energy metric performed better than the approximate binding affinity (K*), since the approximate binding affinity of each analog compared to the template sequence has been used as the final discriminator of which analogs to send for experimental testing in our de novo design framework,46 and has been successfully applied to designing inhibitors of HIV entry42 and inhibitors of Complement activation43 previously. This suggests that we should use the calculated binding free energy if possible instead of the approximate binding affinity when designing new Compstatin variants. Despite these results, the approximate binding affinity calculation will still be useful to assess the docking of complexes when the binding mode is unknown. Additional testing with different sets of protein/peptide complexes beyond C3c/Compstatin should be evaluated in the future to provide further evidence supporting the use of these binding evaluation metrics. Similarly, future testing with different systems having more experimental KD data can be performed.
Discriminating Differences in Interactions in Compstatin Analogs with Natural and Modified Amino Acids
Due to the new forcefield parameters derived in this work, we have the capability to discriminate interfacial interactions involving noncanonical amino acids at atomistic detail. The most potent analog of Compstatin in Table 3 contains the noncanonical amino acid 1-methyltryptophan in position 4. We aimed to gain insight as to why this substitution is more potent than the analog W4A9 and native Compstatin based on strengths of residue–residue interactions. Therefore, for multiple independent simulation trajectories of analogs W4(OMW)A9, W4A9, and native Compstatin (Sequence 11: Ac-ICV(OMW)QDWGAHRCT-NH2, Sequence 23: Ac-ICVWQDWGAHRCT-NH2, and Sequence 53: ICVVQDWGHHRCT in Table 3), we decomposed the polar and nonpolar interaction free energy contributions and present the results of the average interaction free energies as two-dimensional density maps in Figure 3. Here, we focus on differences in interactions due to 1-methyltryptophan relative to tryptophan or valine present in other Compstatin sequences. A full detailed analysis of all pairwise interactions is presented in Supporting Information section “Use of FF_NCAA to Discriminate Specific Contributing Interactions to Antagonistic Activity for Unnaturally Modified Analog Compared to Native and Variant E1 Compstatin.”
Figure 3.

(A) Nonpolar and (B) polar interaction maps for analog W4(OMW)A9. (C) Nonpolar and (D) polar interaction maps for analog W4A9. (E) Nonpolar and (F) polar interaction maps for native Compstatin. The color bar represents the interaction free energy between the corresponding residue–residue pairs in kcal/mol. The color bar was scaled to be the same for the nonpolar and polar interaction free energy contributions so that the different analogues’ energetic contributions can be directly compared.
The substitutions of Val4 to Trp4 and from Trp4 to W4(OMW) result in an increase of the overall nonpolar interactions between residue 4 and C3c residues Gly345, Met346, Asn390, Thr391, His392, Pro393, and Arg456; the increase is more pronounced in the 390–393 C3c residue moiety, as was previously proposed through molecular modeling by Magotti and co-workers.52 During the simulations of analog W4(OMW)A9, the methyl group of Trp4 is frequently found to lay upon the Cys2-Cys12 disulfide bridge, and this leads to increased intramolecular nonpolar interactions between Trp4 and Cys2-Cys12. Similar behavior was observed in recently published MD computational studies53 investigating two novel analogs, R1W4(OMW)A9 and R-1S0W4(OMW)A9, which also contain 1-methyltryptophan in position 4 and using the CHARMM forcefield.54 W4(OMW) forms increased intra- and intermolecular interactions compared with the less potent analogs containing Trp4 and Val4. In all systems, the backbone N in Val/Trp4 is hydrogen bonded to C3c Gly345 O, and the backbone O in Val/Trp4 is hydrogen bonded to the charged amide group of C3c Arg456; the former hydrogen bond-related polar interaction is stronger in the parent Compstatin, whereas the latter is stronger in W4(OMW)A9. The full set of intermolecular interactions formed between the Compstatin analogs and C3c residues in Figure 3 are in agreement with the X-ray structure of W4A9 in complex with C3c55 and with all previous MD simulation studies in explicit water solvent49,56,57 using the CHARMM forcefield. Despite the specific differences between the analogs, overall, the strengths of the residue pairwise interaction free energies of the three systems were similar.
FF_NCAA Web Interface
To disseminate the ability to use FF_NCAA to the broader academic community, we have also created a web interface http://selene.princeton.edu/FFNCAA, as shown in Figure 4. The interface allows one to upload a PDB structure to be modified by single or multiple noncanonical amino acids and/or simultaneously mutated. Additionally, the interface allows a user to download the forcefield parameters calculated and derived for FF_NCAA, as well as read instructions for use with AMBER directly.
Figure 4.

Web interface for the dissemination of Forcefield_NCAA. The web interface has static links to download and use Forcefield_NCAA in AMBER locally, as well as an interactive interface to make noncanonical amino acid substitutions and mutations to an input PDB structure. Screenshot taken April 25, 2013.
After user submission, the interface performs the requested modifications and minimizes the structure to remove any clashes that have been formed by introducing the noncanonical amino acid to the nearest local minimum. This step utilizes the parameters from FF_NCAA for the noncanonical amino acids coupled with the parameters in ff03.51 After completion, the user will receive an e-mail indicating the structure’s successful modification with a unique link to download their results. The user can visualize the modified and input structure using a Jmol applet integrated into the web interface. Additionally, the interface provides links to relevant information tabulated about the structures including the TMScore and58 and RMSD between the structures, and the molecular-mechanics calculated energy of the structure. In addition, the topology and parameter files are generated for AMBER and are provided so that one can directly use them as input for further molecular dynamics simulation on the user’s local computing systems. Disulfide bridges in the input structure are automatically detected and introduced by the web interface. The web interface will be useful to researchers aiming to interactively make site-specific noncanonical amino acid substitutions on a protein structure/complex and to perform subsequent binding calculations in AMBER locally.
Methods
Procedure for Calculation and Derivation of Forcefield Parameters in Forcefield_NCAA
Quantum calculations to derive partial charges compatible with AMBER ff03 were performed consistent with the procedure of Duan and co-workers51 for the 20 natural amino acids. The ff03 methodology was chosen due to the noncanonical amino acids having multiple R-groups attached to the Cα or substitutions such as methylation on the backbone nitrogen. Therefore, the consensus fixed-backbone charges in other AMBER fixed-charge forcefields would not be applicable.50 The choices for NCAAs chosen to be parametrized were based on those found in the literature to enhance the binding affinity of a peptide to a receptor protein, α,α-disubstituted modified amino acids, N-methylated amino acids, and β-amino acids. The procedure used is shown in Figure 5 and is adapted from the procedure we developed to derive parameters in Forcefield_PTM50 for parametrizing frequently occurring post-translational modifications.
Figure 5.

Automated framework for AMBER partial charge parametrization for noncanonical, α,α-disubstituted, β- and N-methylated amino acids. Adapted with permission from ref (50). Copyright 2013, American Chemical Society.
In the first step, structures of each unnatural amino acid dipeptide were built using the MarvinSketch program.59 Dipeptides are constructed to mimic the peptide backbone with a core amino acid with a preceding and subsequent amino acid. An acetyl group precedes the amino acid to be parametrized, with an N-methyl group following it in the form ACE-X-NME, where X is the amino acid to be parametrized. l-Amino acids were constructed, unless otherwise noted by designations of (R) or (S) for the disubstituted amino acids. In the second step, the distance geometry (distgeom) module in TINKER 5.160 is used to construct α-helix (ϕ = −60, ψ = −40) and β-strand (ϕ = −120, ψ = −140) conformers. In Step 3, through TINKER, utilizing the AMBER ff9461 parameter set, and using restraints on the main-chain torsion angles, each conformer was subjected to 25 simulated annealing calculations (using the anneal routine with default parameters). Ti and Tf were 1000 K and 0 K in the annealing. 2000 steps of cooling are done in each simulation, employing a linear cooling protocol and a 1 fs time step. This procedure was performed to find suitable feasible points for subsequent detailed optimization at the quantum mechanical level. For both the helix and strand conformer, the lowest energy structure for each was minimized to the nearest local minima using the optimize routine, with a RMS gradient cutoff of 0.01. Gaussian09(62) was used to optimize (Step 5) each conformer at the HF/6-31G** level of theory, with restraints on the ϕ and ψ angles to preserve the backbone secondary structure. Single point energy calculations are next performed on the dipeptide structures using the density functional theory method and the B3LYP exchange and correlation functionals63−65 and the cc-pVTZ66 basis set. An ether-like organic solvent environment (ε = 4) was mimicked by applying the IEFPCM implicit solvent model,67,68 as suggested by Duan.51 The electrostatic potential (ESP) was calculated at a set of gridpoints defining the molecular surface in the solvent-accessible region around each optimized conformation at 1.4, 1.6, 1.8, and 2.0 times the vdw radii using the program DMS at a density of 0.5, yielding 2.5–2.8 points/Å2.69 The ESP values calculated from each conformation were next used as input to a two-stage RESP fit of the partial atomic charges using Antechamber in AmberTools 1.4.70 In the RESP model, atoms are reasonably approximated as spherical points having fixed charges rather than as nuclei with shared electrons. Bond, angle, and dihedral force constants were perceived by matching atom types contained in the General Amber Forcefield (GAFF)71 using Antechamber.70 GAFF torsion parameters, when applied to post-translational modifications, were shown previously to be highly correlated with and reproduce the locations of local maxima/minima on the quantum mechanically calculated potential energy surface of key torsion angles, although their amplitudes needed refinement.50 Subsequent refinement of key bonded parameters may be warranted but are not developed in this work due to the extreme computational cost involved and since the parameters developed herein are intended to be used mainly in a design context. This procedure was completed for a total of 147 diverse noncanonical amino acids spanning all amino acids except Proline. No new atom types were needed in the matching of force constants and equilibrium values. Further, in 99 of the 147 molecules parametrized, no parameters were utilized from GAFF since AMBER already had defined the parameters for all of the atom types for those noncanonical amino acids. The method described is similar to that described by Khoury et al.50 with the changes being able to handle α,α-disubstituted and backbone modified amino acids. The parameters for noncanonical amino acids were next tested using binding calculations.
Testing the Parameters Using Approximate Binding Affinity and Binding Free Energy Calculations of 63 Compstatin Analogs
Selection of System for Testing and Extraction of Data from the Literature
We aimed to create charge parameters for noncanonical amino acids that would be compatible with the existing AMBER ff03 forcefield parameters for natural amino acids. We focused on assessing whether the derived parameters can discriminate between active/inactive analogs of an inhibitor, since we expect the charge parameters derived to be used mainly in a protein design context. This approach is viable since there are sets of binding data available in the literature where others have experimentally incorporated noncanonical amino acids for various therapeutic applications.
The availability of (i) an experimentally solved structure for Compstatin variant E1 (the ligand) bound to human Complement component C3c55 (the receptor) and (ii) an abundance of experimental binding and IC50 data available, and the observation that (iii) the ligand peptide is relatively rigid because of a disulfide bond that cyclizes it which limits the entropic contribution to the binding free energy, led us to focus our testing efforts on the Complement/Compstatin system. There are three pathways in the Complement system: the classical pathway, the lectin pathway, and the alternative pathway.72 All three pathways converge on a single step where the key protein, C3 binds to C3 convertase and causes it to become cleaved into C3a and C3b (which contains C3c), which allows downstream events to occur leading to the membrane attack complex that can cleave cells. Compstatin binds to C3 and the C3 convertase, which blocks them from coming together and becoming cleaved.73 Inhibiting their ability to bind together and cleave inhibits the activation of C3 and disrupts the formation of the membrane attack complex downstream. This is important as its improper activation has been linked to over 10 autoimmune, inflammation, and neurological disorders.24,74−77
Figure 6 shows a schematic diagram of Complement component C3c with the coordinates of the ligand peptide Compstatin variant E1 (Ac-ICVWQDWGAHRCT-NH2) bound. Since the region that Compstatin binds to C3c is confined and localized in one site, utilizing the entire C3c:Compstatin complex is not necessary. In addition, because of unresolved/missing residues in the crystal structure, and because of its large size (643 amino acids in the solved PDB structure), using the full structure of C3c only serves to complicate subsequent calculation complexities, extend simulation times, and potentially add unnecessary noise.49,56,57 Therefore, residues F335 (macroglobulin domain 4,MG4) to D535 (MG5) were extracted from PDB: 2QKI and used in all subsequent calculations. Previous simulations to study this complex have shown that using these regions were sufficient for modeling the binding interface.49,57
Figure 6.
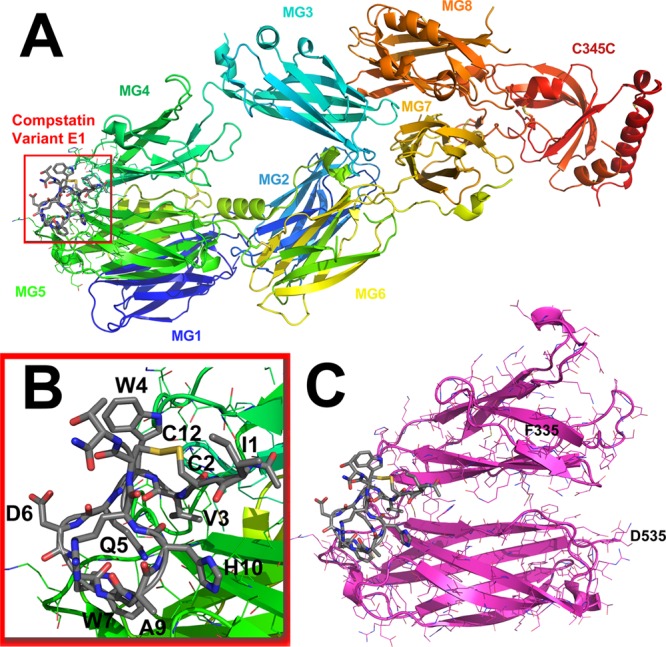
(A) Crystal structure of Compstatin Variant E1 bound to Complement component C3c (PDB: 2QKI). Each macroglobulin domain (MG) is denoted by color. (B) The region where modified amino acids are to be substituted in is shown in the inset. This is the interface that is being designed when making natural or noncanonical amino acid substitutions. A disulfide bridge cyclizes Compstatin between residues 2 and 12. (C) This is the full region all calculations will be performed on, beginning with F335 from MG4 to D535 on MG5 as labeled. The images are visualized in PyMOL.78
All experimental binding data (IC50’s) on Compstatin variants were tabulated from Magotti et al.,52 Chiu et al.,79 and Qu et al.24 These data sets are denoted by structurekinetic, pharmacophore, and novel analogues, respectively, and are shown in Table 3. The data set included 47 analogues with IC50 < 200 μM, and 16 analogs with IC50 > 200 μM, as well as 31 analogues with IC50 < 20 μM and 32 analogs with IC50 > 20 μM. In terms of diversity, the data set included 23 noncanonical amino acid modifications, of which 11 were backbone N-methylations, 28 were natural amino acid substitutions, and 12 were deletions. There was no training done specific to this system in the derivation of the parameters for the NCAAs used. All subsequent results are based only on physical interactions derived from the physics governing the AMBER forcefield.
Screening Compstatin Analogs with Short Molecular Dynamics Simulations and Binding Calculations
The starting coordinates of variant E1 of Compstatin bound to Complement component C3c were taken from PDB: 2QKI. Each variant was constructed using the program tleap by stripping off the side-chains and substituting them with the new amino acid, natural or modified. Deletions were constructed by deleting the amino acids on the peptide chain. During the initial minimization the atoms moved to adjust for the deletions. The cysteine residues in the Compstatin variants were connected to form disulfide bridges in all simulations using the bond command in tleap. Acetyl and N-methyl blocking groups were added if they were present in the experimental analog.
The molecular dynamics and screening procedure used was done identically for each variant with AMBER11.80 Partial charges compatible with AMBER ff0351 derived in this work were used for all noncanonical amino acids. All simulations were performed with the generalized Born implicit solvent model of Onufriev81,82 with a 16 Å nonbonded interaction cutoff. The surface energy term was activated and a salt concentration of 0.1 mol/L was used to account for charge screening. Structures were minimized with 600 steps of steepest descent followed by 400 steps of conjugate gradient minimization. The structures were next heated by rescaling the velocities in 6 stages with increments of 50 K from 0 K to 300 K over 30 ps and a 0.5 fs time step to heat the structures. The collision frequency γ was 5 ps–1. Shake constraints were used to constrain all bonds between heavy atoms and hydrogens and reduce the number of degrees of freedom. Next, each C3c/Compstatin variant underwent a short 0.5 ns production simulation using a 1 fs time step at 300 K. Short (<1 ns) MD simulations with the ff03 charge model have previously been shown to give better performance in ranking binding energies than longer simulations83 and gave the best overall ranking results in an assessment of 5 AMBER forcefields (ff99, ff99SB, ff99SB-ILDN, ff03, and ff12SB) and 46 small molecules targeting 5 protein receptors.84 No restraints were employed in any step of any simulation to strictly assess the suitability of the parameters to discriminate between active/inactive analogs of Compstatin.
Approximate Binding Affinity Calculations
Three independent simulations as described above were performed for the complex, the protein, and the peptide, respectively. One simulation of the protein was utilized to assess the contributions of the protein and to remove any variability from its contribution. Sixty three independent simulations of each complex and 63 independent simulations of each peptide were performed in isolation using the procedure described above. The derivation of the Approximate Binding Affinity (K*) from statistical mechanics is described in the following.
The equilibrium of complex formation PL from a protein P and ligand L is defined as
| 1 |
Assuming a dilute mixture, one can assume ideal behavior. The Helmholtz free energy A can be related to the total partition function, Q in eq 2. Q is in turn a function of the individual partition functions qi, where i is PL, P, L. This assumes independence of subsystems and that the particles are indistinguishable.
| 2 |
The chemical potential μ of species i is the partial derivative of the Helmholtz Free Energy A with respect to Ni, denoted in eq 3.
| 3 |
Equality of chemical potentials at equilibrium is denoted in eq 4.
| 4 |
Next, substituting eq 3 into eq 4 and rearranging yields.
| 5 |
qi is a product of the translational (qt), rotational (qr), and vibrational (qv) partition functions. Since qt is only a function of the coordinates x, y, and z, it integrates out to a volume V, leaving qi = V * qi′(T). qi(T) is the rotational and vibrational partition functions, which is what is approximated by evaluation of Eintra during the snapshots sampled over the course of a molecular dynamics trajectory. This is shown in eq 6.
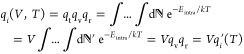 |
6 |
Next, the volume term can be taken out of each partition function and therefore the ratio of the partition functions equals the ratio of concentrations (since Ni/Vi is a concentration). K* = KA when the intrapartition functions (qr and qv) are exactly calculated.
| 7 |
Currently, it is not possible to compute exact partition functions for a complex molecular species due to the inability to integrate an exact energy function over the molecule’s entire conformational space.85 Therefore, we approximate the partition functions as the sampled space of a molecular dynamics trajectory scored with the AMBER potential energy function.
This derivation is based on an initial derivation by Lilien et al.85 using rotamerically based ensembles with modifications resulting from a personal communication between Dr. Meghan Bellows Peterson and Professor Pablo G. Debenedetti. Using the ratios of the partition function of the complex, peptide, and protein, the Approximate Binding Affinity, K* was calculated and placed in a rank ordered list from highest to lowest, which would correspond to the largest to smallest predicted association affinity. The Jacobi logarithm was used to handle numerical overflows that can be caused by summing exponential terms.86
Binding Free Energy Calculations
Binding free energies were calculated using the states produced over the time course of each of the 63 molecular dynamics simulations of the complex. These were calculated to test the forcefield parameters and also to compare its predictive ability to the K*. In the approximate binding affinity, independent simulations of the peptide, protein, and complex are performed. In the binding free energy calculation, only one simulation of the complex is required. The binding free energy is calculated as in eq 8, utilizing the thermodynamic cycle denoted in Figure 7.
| 8 |
Tamamis et al., using the CHARMM54 suite of tools, showed MM-GBSA was able to show the difference in binding Compstatin in complex with human versus rat C3c56 and mouse C3c.57 Tamamis et al. further showed MM-GBSA was helpful in discriminating potential analogs for experimental testing.49 Based on these previous findings, it is clear that MM-GBSA can be used to study the Compstatin/C3c system. Therefore, in this study, binding free energies were calculated using the MM-GBSA module87 in AMBER11 complemented by the charge parameters for the noncanonical amino acids introduced in this work.
Figure 7.

Thermodynamic cycle used to calculate the binding free energies. Ideally one can calculate the binding free energy for the association of [A] + [B] ⇌ [AB] directly in solvent. This calculation is expensive and contains much noise due to the contribution of the solvent. Therefore, a different approach was used exploiting a thermodynamic cycle that can calculate the same difference by utilizing the solvation free energies of the protein (receptor), peptide (ligand), and complex, with the binding energy calculated in vacuo.87
For this system, we assume that the entropic contribution due to the binding free energy is small since the ligand’s structure is relatively rigid due to the disulfide bridges (the maximum Cα RMSD between analogs W4(OMW)A9, W4A9, and native Compstatin was 1.38 at the end of the simulation). The entropic contribution, which can be calculated through a normal-mode analysis, can have a large uncertainty.80 Given that we expect the Compstatin analogs to have similar entropies due to their cyclic nature and being bound to the same binding pocket of C3c, and because of the expected large error, the entropic contribution in the calculation was ignored. This “one-trajectory” approximation where we assume similar structure in the bound and unbound state is reasonable and has been applied elsewhere49,53 and also has been compared to the results of a “three-trajectory” approximation for this system.56 Using the “one-trajectory” approximation, all of the bonded terms (bonds, angles, torsions) cancel out in the evaluation of the binding free energies, leaving only nonbonded terms, of which the electrostatic component is a significant contributor.
Construction of Receiver Operating Characteristic Curves
After the approximate binding affinity and binding free energies were calculated for each variant of Compstatin following the procedure described above, the values were rank ordered from most favorable to least favorable. For K*, this was largest to smallest, and for ΔGBind,Solv°, this was smallest to largest. Receiver operating characteristic (ROC) curves were constructed based on the rank-ordered lists of the analogs. Two cutoff values of IC50 values defining active and inactive were chosen. The first cutoff was 20 M, which corresponded to an even split between active and inactive in the data set. That is, analogs with IC50 values < 20 μM were considered active. Similarly, a cutoff of 200 μM was chosen to perform the analysis with a less stringent cutoff of active/inactive. These values as cutoffs were determined based on the fact that the most active Compstatin analog in Table 3 is Ac-ICV(OMW)QDWGAHRCT-NH2,88 which has an IC50 of 0.21 μM. The axes on the ROC curve correspoμnd to the True Positive Rate (Sensitivity) vs the False Positive Rate (1-Specificity). The ROC curve constructed aimed to find how many of the most favorable analogs by calculated approximate binding affinity or binding free energy were experimentally active when rank-ordered. In a virtual screen, this is often one of the first steps in design: to assess whether a particular ligand will bind to its target favorably, and ranking the affinity of a ligand to the targeted receptor. Sensitivity and specificity are defined as.
| 9 |
where TP, FP, TN, and FN stand for true/false positives/negatives, respectively.
Additional Simulations for Analysis of Interaction Free Energies of Compstatin Derivatives and C3 Residue Pairs
Three biologically relevant complexes containing derivatives of Compstatin (Sequence 11: Ac-ICV(OMW)QDWGAHRCT-NH2, Sequence 23: Ac-ICVWQDWGAHRCT-NH2, and Sequence 53: ICVVQDWGHHRCT in Table 3) were assessed for their interactions through multiple longer-time molecular dynamics simulations. Four independent trajectories were performed for each complex. Each of the four trajectories for each complex were appended together for subsequent analysis. The starting complex structures were constructed and minimized as described previously deriving from PDB: 2QKI. The complexes were heated stepwise from 0 to 300K over 30 ps using restraints on all backbone atoms with a force constant of 10 kcal/(mol·Å2). The complexes were carefully equilibrated in 3 stages; each stage was run for 200 ps. In the first stage, all atoms were restrained with a force constant of 5.0 kcal/(mol·Å2) for 200 ps. In the second stage, all backbone atoms outside of the binding pocket were constrained (residues 335–343, 350–387, 394–453, 463–487, 493–535), and all atoms in the binding pocket were constrained with a force constant of 5.0 kcal/(mol·Å2). In the final stage, all backbone atoms outside of the binding pocket were constrained and all atoms in the binding pocket were constrained with a force constant of 1.5 kcal/(mol·Å2). After equilibration, four independent 10 ns production simulations were performed for each complex. These simulations were used to produce maps of residue–residue polar and nonpolar interaction free energies to identify key energetic interactions important for binding through the simulation trajectories.
The interaction free energies between two residues (R and R′) were computed by the relation:
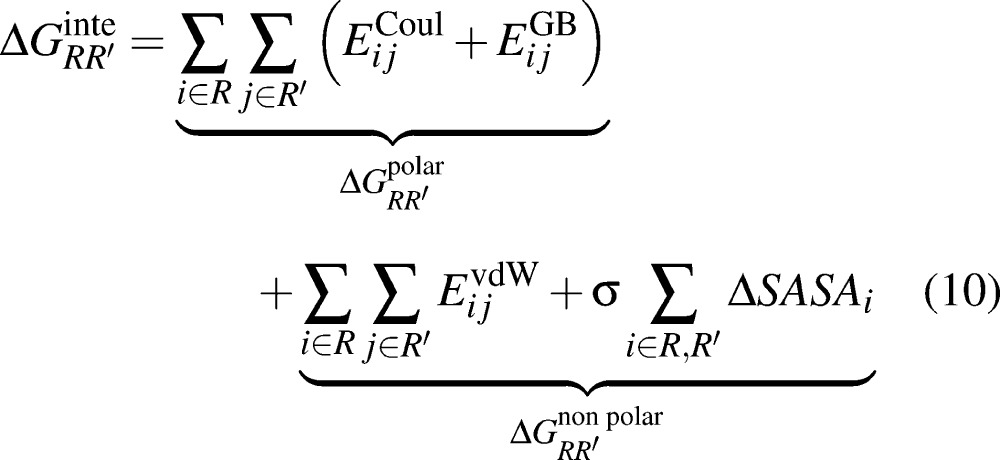 |
10 |
for each of the 3 Compstatin variants evaluated. The first and second group of terms on the right-hand side of eq 10 describe polar and nonpolar interactions between R and R′. In the calculations, R corresponds to a residue on Compstatin and R′ corresponds to a residue on C3c. The details of the calculation have been presented previously.49,53,56,57 For each complex, 4000 frames over the 40 ns total simulation time were evaluated for their average interaction free energies using a spacing of 10 ps per frame.
Acknowledgments
C.A.F. acknowledges support from the National Institutes of Health (R01GM052032) and the National Science Foundation. G.A.K. is grateful for support by a National Science Foundation Graduate Research Fellowship under grant No. DGE-1148900. The authors gratefully acknowledge that the calculations reported in this paper were performed at the TIGRESS high performance computing center at Princeton University which is supported by the Princeton Institute for Computational Science and Engineering (PICSciE) and the Princeton University Office of Information Technology. The authors are grateful to Eric First for expert help with making the webtool. We are grateful to Dr. Meghan Bellows Peterson and Professor Pablo G. Debenedetti for helpful discussions.
Glossary
Abbreviations
- μM
micromolar concentration
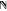
all degrees of freedom
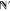
all the degrees of freedom minus the translational degrees of freedom
- Eintra
the intramolecular energy of a system interacting with itself
- Q
the total partition function
- qi′
the partition function over the internal degrees of freedom
- B
the set of bound configurations
- F
the set of free protein configurations
- L
the set of free ligand peptide configurations
- K*
the approximate binding affinity
- KA
the binding equilibrium constant for the association reaction P + L ⇌ PL
Supporting Information Available
(1) Table containing the raw calculated binding affinities of different Compstatin variants. (2) Instructions for importing new parameters into AMBER. (3) Explanation of the contents of the forcefield parameter file. (4) Images and atom namings of each parametrized noncanonical amino acid grouped by scaffold residue. (5) Forcefield parameters for each noncanonical amino acid grouped by scaffold residue. (6) The full pairwise residue–residue analysis of interaction energy contributions for 3 important analogs of Compstatin. (7) Zip file containing the Forcefield parameters in AMBER formats directly importable into AMBER. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
G.A.K. and C.A.F. conceived the project. G.A.K. and J.S. contributed to the source code enabling the calculation of the parameters. G.A.K. and A.V. calculated and compiled the partial charges. J.S., C.A.K., G.A.K. contributed to the construction of the ROC curves. G.A.K. performed the simulations and constructed the ROC curves. G.A.K., J.S., C.A.K., and C.A.F. analyzed the ROC curves. P.T. and G.A.K. constructed the interaction maps and analyzed the simulation results. G.A.K. created the web interface. G.A.K., P.T., and C.A.F. wrote the manuscript. All authors have read and approved the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Wang J.; Hou T. (2011) Recent advances on aqueous solubility prediction. Comb. Chem. High Throughput Screening 14, 328–338. [DOI] [PubMed] [Google Scholar]
- Andrade C. H.; Pasqualoto K. F. M.; Ferreira E. I.; Hopfinger A. J. (2009) Rational design and 3D-pharmacophore mapping of 5′-thiourea-substituted α-thymidine analogues as mycobacterial TMPK inhibitors. J. Chem. Inf. Model. 49, 1070–1078. [DOI] [PubMed] [Google Scholar]
- Vlieghe P.; Lisowski V.; Martinez J.; Khrestchatisky M. (2010) Synthetic therapeutic peptides: Science and market. Drug Discovery Today 15, 40–56. [DOI] [PubMed] [Google Scholar]
- Craik D. J.; Fairlie D. P.; Liras S.; Price D. (2013) The future of peptide-based drugs. Chem. Biol. Drug Des. 81, 136–147. [DOI] [PubMed] [Google Scholar]
- Adessi C.; Soto C. (2002) Converting a peptide into a drug: Strategies to improve stability and bioavailability. Curr. Med. Chem. 9, 963–978. [DOI] [PubMed] [Google Scholar]
- Miller S. M.; Simon R. J.; Ng S.; Zuckermann R. N.; Kerr J. M.; Moos W. H. (1995) Comparison of the proteolytic susceptibilities of homologous l-amino acid, d-amino acid, and N-substituted glycine peptide and peptoid oligomers. Drug Dev. Res. 35, 20–32. [Google Scholar]
- Khoury G. A.; Baliban R. C.; Floudas C. A. (2011) Proteome-wide post-translational modification statistics: Frequency analysis and curation of the SWISS-PROT database. Sci. Rep. 1, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A.; Apweiler R. (2000) The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury G. A.; Smadbeck J.; Kieslich C. A.; Floudas C. A. (2013) Protein folding and de novo protein design for biotechnological applications. Trends Biotechnol. 10.1016/j.tibtech.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C. T. (2006) Posttranslational Modification of Proteins: Expanding Nature’s Inventory; Roberts and Co. Publishers, Englewood, CO, pp xxi, 490. [Google Scholar]
- Merrifield R. B. (1963) Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 85, 2149–2154. [Google Scholar]
- Chalker J. M.; Davis B. G. (2010) Chemical mutagenesis: selective post-expression interconversion of protein amino acid residues. Curr. Opin. Chem. Biol. 14, 781–789. [DOI] [PubMed] [Google Scholar]
- Chalker J. M.; Bernardes G. J. L.; Davis B. G. (2011) A ″tag-and-modify″ approach to site-selective protein modification. Acc. Chem. Res. 44, 730–741. [DOI] [PubMed] [Google Scholar]
- Johnson J. A.; Lu Y. Y.; Van Deventer J. A.; Tirrell D. A. (2010) Residue-specific incorporation of non-canonical amino acids into proteins: Recent developments and applications. Curr. Opin. Chem. Biol. 14, 774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H.; Wang K.; Davis L.; Garcia-Alai M.; Chin J. W. (2010) Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature 464, 441–444. [DOI] [PubMed] [Google Scholar]
- Wang L.; Xie J.; Schultz P. G. (2006) Expanding the genetic code. Annu. Rev. Biophys. Biomol. Struct. 35, 225–249. [DOI] [PubMed] [Google Scholar]
- Watanabe T.; Muranaka N.; Hohsaka T. (2008) Four-base codon-mediated saturation mutagenesis in a cell-free translation system. J. Biosci. Bioeng. 105, 211–215. [DOI] [PubMed] [Google Scholar]
- Mapelli C.; et al. (2009) Eleven amino acid glucagon-like peptide-1 receptor agonists with antidiabetic activity. J. Med. Chem. 52, 7788–7799. [DOI] [PubMed] [Google Scholar]
- Sievers S. A.; Karanicolas J.; Chang H. W.; Zhao A.; Jiang L.; Zirafi O.; Stevens J. T.; Munch J.; Baker D.; Eisenberg D. (2011) Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature 475, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakos K.; Havass J.; Fülöp F.; Gera L.; Stewart J. M.; Falkay G.; Tóth G. K. (2001) Synthesis and receptor binding of oxytocin analogs containing conformationally restricted amino acids. Lett. Pept. Sci. 8, 35–40. [Google Scholar]
- Tamamura H.; Hiramatsu K.; Kusano a. S.; Terakubo S.; Yamamoto N.; Trent J. O.; Wang Z.; Peiper S. C.; Nakashima H.; Otaka A.; Fujii N. (2003) Synthesis of potent CXCR4 inhibitors possessing low cytotoxicity and improved biostability based on T140 derivatives. Org. Biomol. Chem. 1, 3656–3662. [DOI] [PubMed] [Google Scholar]
- Kokubu T.; Hiwada K.; Murakami E.; Imamura Y.; Matsueda R.; Yabe Y.; Koike H.; Iijima Y. (1985) Highly potent and specific inhibitors of human renin. Hypertension 7, I8–11. [DOI] [PubMed] [Google Scholar]
- Mallik B.; Katragadda M.; Spruce L. A.; Carafides C.; Tsokos C. G.; Morikis D.; Lambris J. D. (2005) Design and NMR characterization of active analogues of compstatin containing non-natural amino acids. J. Med. Chem. 48, 274–286. [DOI] [PubMed] [Google Scholar]
- Qu H.; Magotti P.; Ricklin D.; Wu E. L.; Kourtzelis I.; Wu Y.-Q.; Kaznessis Y. N.; Lambris J. D. (2011) Novel analogues of the therapeutic complement inhibitor compstatin with significantly improved affinity and potency. Mol. Immunol. 48, 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver-Fay A.; et al. (2011) Rosetta3 an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia B. E.; et al. (2010) Computational design of epitope-scaffolds allows induction of antibodies specific for a poorly immunogenic HIV vaccine epitope. Structure 18, 1116–1126. [DOI] [PubMed] [Google Scholar]
- Fleishman S. J.; Whitehead T. A.; Ekiert D. C.; Dreyfus C.; Corn J. E.; Strauch E.-M.; Wilson I. A.; Baker D. (2011) Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science 332, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead T. A.; Chevalier A.; Song Y.; Dreyfus C.; Fleishman S. J.; De Mattos C.; Myers C. A.; Kamisetty H.; Blair P.; Wilson I. A.; Baker D. (2012) Optimization of affinity, specificity, and function of designed influenza inhibitors using deep sequencing. Nat. Biotechnol. 30, 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L.; Althoff E. A.; Clemente F. R.; Doyle L.; Rothlisberger D.; Zanghellini A.; Gallaher J. L.; Betker J. L.; Tanaka F.; Barbas I.; Carlos F.; Hilvert D.; Houk K. N.; Stoddard B. L.; Baker D. (2008) De novo computational design of retro-aldol enzymes. Science 319, 1387–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter F.; et al. (2012) Computational design of catalytic dyads and oxyanion holes for ester hydrolysis. J. Am. Chem. Soc. 134, 16197–16206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Althoff E. A.; Wang L.; Jiang L.; Giger L.; Lassila J. K.; Wang Z.; Smith M.; Hari S.; Kast P.; Herschlag D.; Hilvert D.; Baker D. (2012) Robust design and optimization of retroaldol enzymes. Protein Sci. 21, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel J. B.; Zanghellini A.; Lovick H. M.; Kiss G.; Lambert A. R.; St.Clair J. L.; Gallaher J. L.; Hilvert D.; Gelb M. H.; Stoddard B. L.; Houk K. N.; Michael F. E.; Baker D. (2010) Computational design of an enzyme catalyst for a stereoselective bimolecular Diels–Alder reaction. Science 329, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothlisberger D.; Khersonsky O.; Wollacott A. M.; Jiang L.; DeChancie J.; Betker J.; Gallaher J. L.; Althoff E. A.; Zanghellini A.; Dym O.; Albeck S.; Houk K. N.; Tawfik D. S.; Baker D. (2008) Kemp elimination catalysts by computational enzyme design. Nature 453, 190–195. [DOI] [PubMed] [Google Scholar]
- Eiben C. B.; Siegel J. B.; Bale J. B.; Cooper S.; Khatib F.; Shen B. W.; Players F.; Stoddard B. L.; Popovic Z.; Baker D. (2012) Increased Diels-Alderase activity through backbone remodeling guided by Foldit players. Nat. Biotechnol. 30, 190–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraf M. C.; Moore G. L.; Goodey N. M.; Cao V. Y.; Benkovic S. J.; Maranas C. D. (2006) IPRO: an iterative computational protein library redesign and optimization procedure. Biophys. J. 90, 4167–4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury G. A.; Fazelinia H.; Chin J. W.; Pantazes R. J.; Cirino P. C.; Maranas C. D. (2009) Computational design of Candida boidinii xylose reductase for altered cofactor specificity. Protein Sci. 18, 2125–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renfrew P. D.; Choi E. J.; Bonneau R.; Kuhlman B. (2012) Incorporation of noncanonical amino acids into Rosetta and use in computational protein–peptide interface design. PLoS One 7, e32637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov D.; Margreitter C.; Grandits M.; Oostenbrink C.; Zagrovic B. (2013) A systematic framework for molecular dynamics simulations of protein post-translational modifications. PLoS Comput. Biol. 9, e1003154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margreitter C.; Petrov D.; Zagrovic B. (2013) Vienna-PTM web server: A toolkit for MD simulations of protein post-translational modifications. Nucleic Acids Res. 41, W422–W426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klepeis J. L.; Floudas C. A.; Morikis D.; Tsokos C. G.; Argyropoulos E.; Spruce L.; Lambris J. D. (2003) Integrated computational and experimental approach for lead optimization and design of compstatin variants with improved activity. J. Am. Chem. Soc. 125, 8422–8423. [DOI] [PubMed] [Google Scholar]
- Klepeis J. L.; Floudas C. A.; Morikis D.; Tsokos C. G.; Lambris J. D. (2004) Design of peptide analogues with improved activity using a novel de novo protein design approach. Ind. Eng. Chem. Res. 43, 3817–3826. [Google Scholar]
- Bellows M. L.; Taylor M. S.; Cole P. A.; Shen L.; Siliciano R. F.; Fung H. K.; Floudas C. A. (2010) Discovery of entry inhibitors for HIV-1 via a new de novo protein design framework. Biophys. J. 99, 3445–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellows M. L.; Fung H. K.; Taylor M. S.; Floudas C. A.; Lopez de Victoria A.; Morikis D. (2010) New compstatin variants through two de novo protein design frameworks. Biophys. J. 98, 2337–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung H. K.; Floudas C. A.; Taylor M. S.; Zhang L.; Morikis D. (2008) Toward full-sequence de novo protein design with flexible templates for human β-defensin-2. Biophys. J. 94, 584–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellows-Peterson M. L.; Fung H. K.; Floudas C. A.; Kieslich C. A.; Zhang L.; Morikis D.; Wareham K. J.; Monk P. N.; Hawksworth O. A.; Woodruff T. M. (2012) De novo peptide design with C3a receptor agonist and antagonist activities: Theoretical predictions and experimental validation. J. Med. Chem. 55, 4159–4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smadbeck J.; Peterson M. B.; Khoury G. A.; Taylor M. S.; Floudas C. A. (2013) Protein WISDOM: A workbench for in silico de novo design of biomolecules. J. Visualized Exp. e50476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Victoria A.; Gorham R. D.; Bellows-Peterson M. L.; Ling J.; Lo D. D.; Floudas C. A.; Morikis D. (2011) A new generation of potent complement inhibitors of the compstatin family. Chem. Biol. Drug Des. 77, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellows M. L.; Floudas C. A. (2010) Computational methods for de novo protein design and its applications to the human immunodeficiency virus 1, purine nucleoside phosphorylase, ubiquitin specific protease 7, and histone demethylases. Curr. Drug Targets 11, 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamis P.; López de Victoria A.; Gorham R. D.; Bellows-Peterson M. L.; Pierou P.; Floudas C. A.; Morikis D.; Archontis G. (2012) Molecular dynamics in drug design: New generations of compstatin analogs. Chem. Biol. Drug Des. 79, 703–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury G. A.; Thompson J. P.; Smadbeck J.; Kieslich C. A.; Floudas C. A. (2013) Forcefield_PTM: Ab initio charge and AMBER forcefield parameters for frequently occurring post-translational modifications. J. Chem. Theory Comput. 9, 5653–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y.; Wu C.; Chowdhury S.; Lee M. C.; Xiong G.; Zhang W.; Yang R.; Cieplak P.; Luo R.; Lee T.; Caldwell J.; Wang J.; Kollman P. (2003) A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 24, 1999–2012. [DOI] [PubMed] [Google Scholar]
- Magotti P.; Ricklin D.; Qu H.; Wu Y.-Q.; Kaznessis Y. N.; Lambris J. D. (2009) Structure–kinetic relationship analysis of the therapeutic complement inhibitor compstatin. J. Mol. Recognit. 22, 495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorham R. D. Jr; Forest D. L.; Tamamis P.; López de Victoria A.; Kraszni M.; Kieslich C. A.; Banna C. D.; Bellows-Peterson M. L.; Larive C. K.; Floudas C. A.; Archontis G.; Johnson L. V.; Morikis D. (2013) Novel compstatin family peptides inhibit complement activation by drusen-like deposits in human retinal pigmented epithelial cell cultures. Exp. Eye Res. 116C, 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R.; et al. (2009) CHARMM: The biomolecular simulation program. J. Comput. Chem. 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen B. J. C.; Halff E. F.; Lambris J. D.; Gros P. (2007) Structure of compstatin in complex with complement component C3c reveals a new mechanism of complement inhibition. J. Biol. Chem. 282, 29241–29247. [DOI] [PubMed] [Google Scholar]
- Tamamis P.; Morikis D.; Floudas C. A.; Archontis G. (2010) Species specificity of the complement inhibitor compstatin investigated by all-atom molecular dynamics simulations. Proteins: Struct., Funct., Bioinf. 78, 2655–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamis P.; Pierou P.; Mytidou C.; Floudas C. A.; Morikis D.; Archontis G. (2011) Design of a modified mouse protein with ligand binding properties of its human analog by molecular dynamics simulations: The case of C3 inhibition by compstatin. Proteins: Struct., Funct., Bioinf. 79, 3166–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Skolnick J. (2005) TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MarvinSketch, version 6.0.3; (2010) ChemAxon, Budapest, Hungary. [Google Scholar]
- TINKER, version 5.0; (2010) Washington University, St. Louis, MO. [Google Scholar]
- Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Merz K. M.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. (1995) A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117, 5179–5197. [Google Scholar]
- Frisch M. J.et al. (2004) Gaussian 03, Revision C.02; Gaussian Inc., Wallingford, CT.
- Lee C.; Yang W.; Parr R. G. (1988) Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785. [DOI] [PubMed] [Google Scholar]
- Miehlich B.; Savin A.; Stoll H.; Preuss H. (1989) Results obtained with the correlation energy density functionals of Becke and Lee, Yang, and Parr. Chem. Phys. Lett. 157, 200–206. [Google Scholar]
- Becke A. D. (1993) Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652. [Google Scholar]
- Kendall R. A.; Thom H. Dunning J.; Harrison R. J. (1992) Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 96, 6796–6806. [Google Scholar]
- Mennucci B.; Cammi R.; Tomasi J. (1999) Analytical free energy second derivatives with respect to nuclear coordinates: Complete formulation for electrostatic continuum solvation models. J. Chem. Phys. 110, 6858–6870. [Google Scholar]
- Tomasi J.; Mennucci B.; Cances E. (1999) The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct.: THEOCHEM 464, 211–226. [Google Scholar]
- Richards F. M. (1977) Areas, Volumes, Packing, and Protein Structure. Annu. Rev. Biophys. Bioeng. 6, 151–176. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wang W.; Kollman P. A.; Case D. A. (2006) Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 25, 247–260. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. (2004) Development and testing of a general Amber force field. J. Comput. Chem. 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- Janeway C. A., Travers P., Walport M., and Shlomchik M. J. (2001) Immunobiology; Garland Science, New York. [Google Scholar]
- Sahu A.; Morikis D.; Lambris J. D. (2003) Compstatin, a peptide inhibitor of complement, exhibits species-specific binding to complement component C3. Mol. Immunol. 39, 557–566. [DOI] [PubMed] [Google Scholar]
- Kovacs G. G.; Gasque P.; Ströbel T.; Lindeck-Pozza E.; Strohschneider M.; Ironside J. W.; Budka H.; Guentchev M. (2004) Complement activation in human prion disease. Neurobiol. Dis. 15, 21–28. [DOI] [PubMed] [Google Scholar]
- Java A.; Atkinson J.; Salmon J. (2013) Defective complement inhibitory function predisposes to renal disease. Annu. Rev. Med. 64, 307–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklin D.; Lambris J. D. (2013) Complement in immune and inflammatory disorders: Therapeutic interventions. J. Immunol. 190, 3839–3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rensen S. S.; Slaats Y.; Driessen A.; Peutz-Kootstra C. J.; Nijhuis J.; Steffensen R.; Greve J. W.; Buurman W. A. (2009) Activation of the complement system in human nonalcoholic fatty liver disease. Hepatology 50, 1809–1817. [DOI] [PubMed] [Google Scholar]
- PyMOL, version 1.3; Delano Scientific LLC, 2008. [Google Scholar]
- Chiu T.-L.; Mulakala C.; Lambris J. D.; Kaznessis Y. N. (2008) Development of a new pharmacophore model that discriminates active compstatin analogs. Chem. Biol. Drug Des. 72, 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.et al. (2010) Amber 11, University of California, San Francisco.
- Onufriev A.; Bashford D.; Case D. A. (2000) Modification of the generalized Born Model suitable for macromolecules. J. Phys. Chem. B 104, 3712–3720. [Google Scholar]
- Onufriev A.; Bashford D.; Case D. A. (2004) Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins: Struct., Funct., Bioinf. 55, 383–394. [DOI] [PubMed] [Google Scholar]
- Hou T.; Wang J.; Li Y.; Wang W. (2011) Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 51, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L.; Sun H.; Li Y.; Wang J.; Hou T. (2013) Assessing the performance of MM/PBSA and MM/GBSA methods. 3. The impact of force fields and ligand charge models. J. Phys. Chem. B 117, 8408–8421. [DOI] [PubMed] [Google Scholar]
- Lilien R. H.; Stevens B. W.; Anderson A. C.; Donald B. R. (2005) A novel ensemble-based scoring and search algorithm for protein redesign and its application to modify the substrate specificity of the gramicidin synthetase a phenylalanine adenylation enzyme. J. Comput. Biol. 12, 740–761. [DOI] [PubMed] [Google Scholar]
- Lidl R., and Neiderreiter H. (1983) Finite fields. In Encyclopedia of Mathematics and its Applications, Addision-Wesley, Reading, MA. [Google Scholar]
- Miller B. R. III; McGee T. D. Jr; Swails J. M.; Homeyer N.; Gohlke H.; Roitberg A. E. (2012) MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 8, 3314–3321. [DOI] [PubMed] [Google Scholar]
- Katragadda M.; Magotti P.; Sfyroera G.; Lambris J. D. (2006) Hydrophobic effect and hydrogen bonds account for the improved activity of a complement inhibitor, compstatin. J. Med. Chem. 49, 4616–4622. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.