

Abstract
The development of readily tunable and regioselective C–H functionalization reactions that operate solely through catalyst control remains a challenge in modern organic synthesis. Herein, we report that simple silver catalysts supported by common nitrogenated ligands can be used to tune a nitrene transfer reaction between two different types of C–H bonds. The results reported herein represent the first example of ligand-controlled and site-selective silver-promoted C–H amination.
Achieving chemo- and site-selective C–H bond functionalizations in complex molecules remains a challenging problem in synthetic organic chemistry. In many cases, the inherent steric and electronic features of a given substrate dictate the reaction outcome.1 While directing groups are often employed to override inherent reactivity preferences in a molecule and can be very powerful for the functionalization of a specific C–H bond,2,3 these approaches are not ideal if the aim is to achieve tunable and site-selective transformations in the presence of multiple possible reactive sites. Our group is interested in developing inexpensive, catalyst-controlled methods for late-stage C–H oxidation chemistries, with the ultimate goal of reshaping current synthetic strategies to complex molecules. Here, we describe our progress in promoting tunable silver-catalyzed nitrene transfer reactions where the selectivity of the C–H amination event is controlled solely by the identity of the ligand.
The development of catalysts capable of tunable aminations of C–H bonds in different steric and electronic environments via nitrene transfer has been focused mainly on the use of powerful dinuclear Rh(II) complexes, especially Rh2(esp)2 and related compounds.4,5 Such catalysts can show excellent selectivity in the presence of multiple reactive sites, but the transformations are primarily substrate-controlled; changing the nature of the ligand does not permit tunable C–H functionalization.6 Changing from Rh to another metal, such as Fe, can be employed to switch the selectivity of the nitrene transfer, but again, the ability to choose between two electronically or sterically similar C–H bonds by tuning the ligand is not yet possible.4c,7
Our group has recently reported that a simple change in the AgOTf/ligand ratio enables highly chemoselective aminations of C=C and C–H bonds (Scheme 1, top).8 The dynamic behavior of these silver complexes, coupled with steric effects, was primarily responsible for the ability to tune the reactivity between C=C and C–H groups. However, another striking feature of silver-catalyzed nitrene transfer, as compared to Rh- and Fe-based platforms, is the breadth of different ligands that are capable of promoting the amination event.9 We were curious if the changes in the coordination geometries of silver complexes enforced by different types of ligands translated into tunable, site-selective amination between C–H bonds in different chemical environments (Scheme 1, bottom).10
Scheme 1. Tunable Ag-Catalyzed Nitrene Transfers.
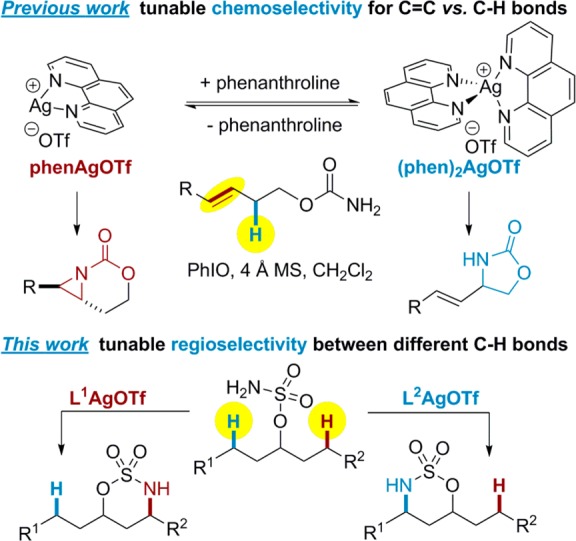
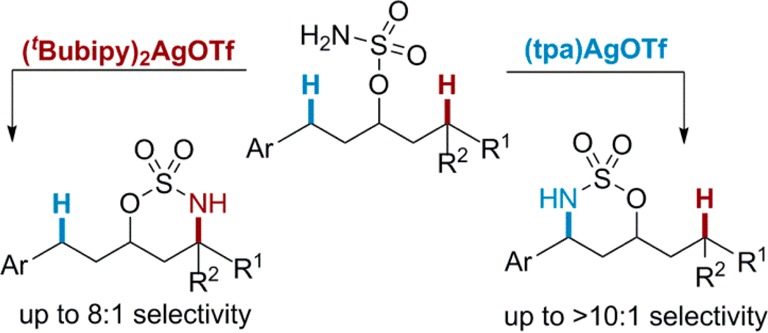
Sulfamate 1 (Table 1) was used to compare the reactivity between a benzylic C–H bond (Bn) and an electron-rich 3° C–H bond (T). As expected, the amination of 1 with dinuclear Rh(II) catalysts (Table 1, entries 1–4) favors reaction at T to yield 2b, even when the steric bulk of the catalyst is increased (entry 4).4c As previously shown, activation of Bn was favored using Ru- and Fe-based catalysts (entries 5–6).7 In contrast, the modularity and breadth of ligands that could be employed with AgOTf showed that catalyst control of the nitrene transfer could be achieved. The substituted 2,2′-bipyridine (tBubipy, 3, entry 8) preferred activation of T, as did ligands 4 and 5 (entries 9–10). Phenanthroline-derived ligands 6–8 (entries 11–13), 2,2′,2″-terpyridines 10–11 (entries 15–16), and Tp ligand 12 (entry 17)9f−9j all showed reduced selectivity in favor of 2b. Interestingly, a silver catalyst supported by tris(2-pyridylmethyl)amine (tpa, 9, entry 14) preferred activation of Bn, indicating that Ag-catalyzed C–H amination is tunable through the ligand.
Table 1. Ligand-Controlled, Silver-Catalyzed C–H Amination.

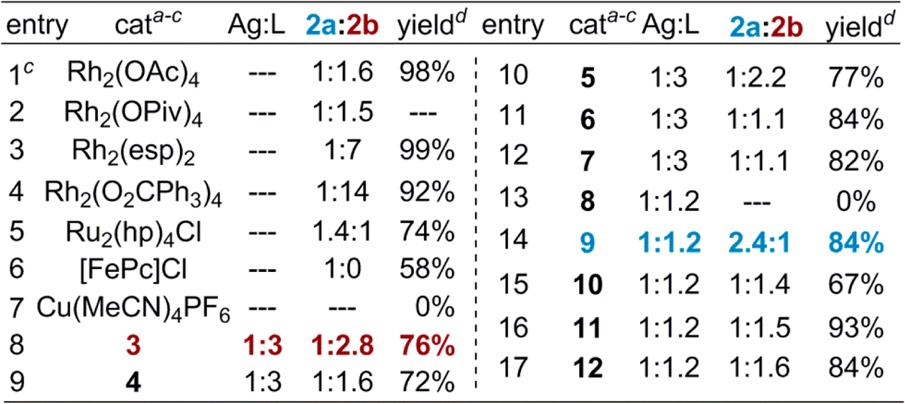
C–H insertion: 10 mol % AgOTf, 10–30 mol % ligand, 3.5 equiv of PhIO, 4 A MS, 0.05 M CH2Cl2.
Rh cat.: 2 mol %, 1.1 equiv of PhI(OAc)2, 2.3 equiv of MgO, CH2Cl2.
See Supporting Information for the conditions for entries 5–7.
Total NMR yield. Average of 2 runs,
mesitylene internal standard.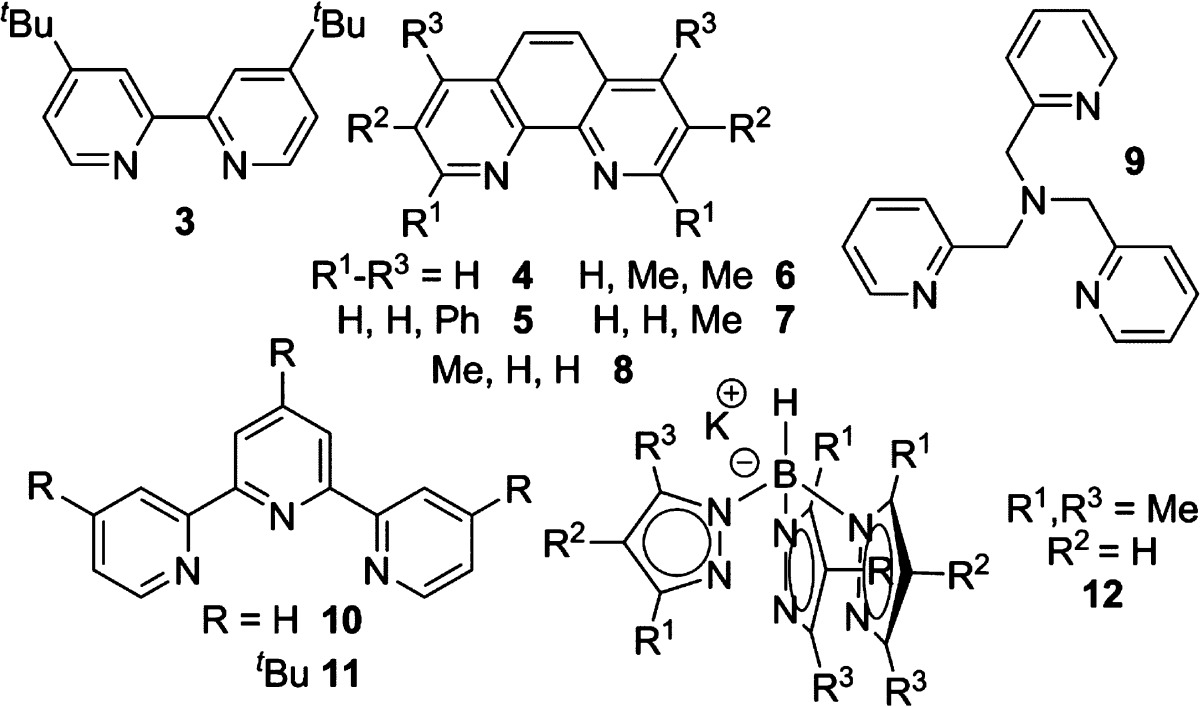
Based on these initial results, tBubipy 3 (Condition A) and the tpa ligand 8 (Condition B) were chosen to compare the reactivity between a Bn and various T bonds. Substrates in Table 2 were designed to elucidate the relationship between the steric environment around T and the site selectivity. The moderate preference for insertion into T over Bn in 1 using (tBubipy)2AgOTf parallels the reactivity of Rh(II) catalysts in intramolecular aminations.4c Moving from the isopropyl group of 1 to a cyclopropyl group in 13 gave exclusive Bn activation with both catalysts; however, the yield was superior using (tpa)AgOTf. The high bond dissociation energy (BDE) of the cyclopropyl C–H in 13 (∼106 kcal/mol) compared to Bn (∼89 kcal/mol) may preclude the formation of 13b.11 The two catalysts responded differently to increasing the size of the alkyl group. For (tBubipy)2AgOTf, the selectivity for amination at T vs Bn improved as the size of the alkyl group was increased from the cyclopropyl of 13 to the cyclohexyl of 17 (17b:17a = 8:1). This indicates that moderate steric bulk does not hinder C–H insertion; however, further steric congestion in 18 lowered the T/Bn selectivity to 2.6:1, similar to the ratio observed for 1. In contrast, (tpa)AgOTf reveals selective functionalization of Bn over T for all substrates in Table 2. When T was contained in a cyclic alkane, (tpa)AgOTf appeared to favor Bn to a greater extent as the size of the ring increased, with the exception of 16, where lower selectivity was observed. This could be due to the lower BDE of the cyclopentyl C–H bond of 16 (∼96 kcal/mol)11 compared to the other substrates. Substrates 15 and 18, containing acyclic alkyl groups, showed higher selectivity for the benzylic position as the steric bulk increased. In general, it appears that steric effects influence the selectivity of (tpa)AgOTf to a greater extent than (tBubipy)2AgOTf, which tends to promote reaction at the most electron-rich C–H bond.
Table 2. Investigation of Steric Effects on Selectivity.
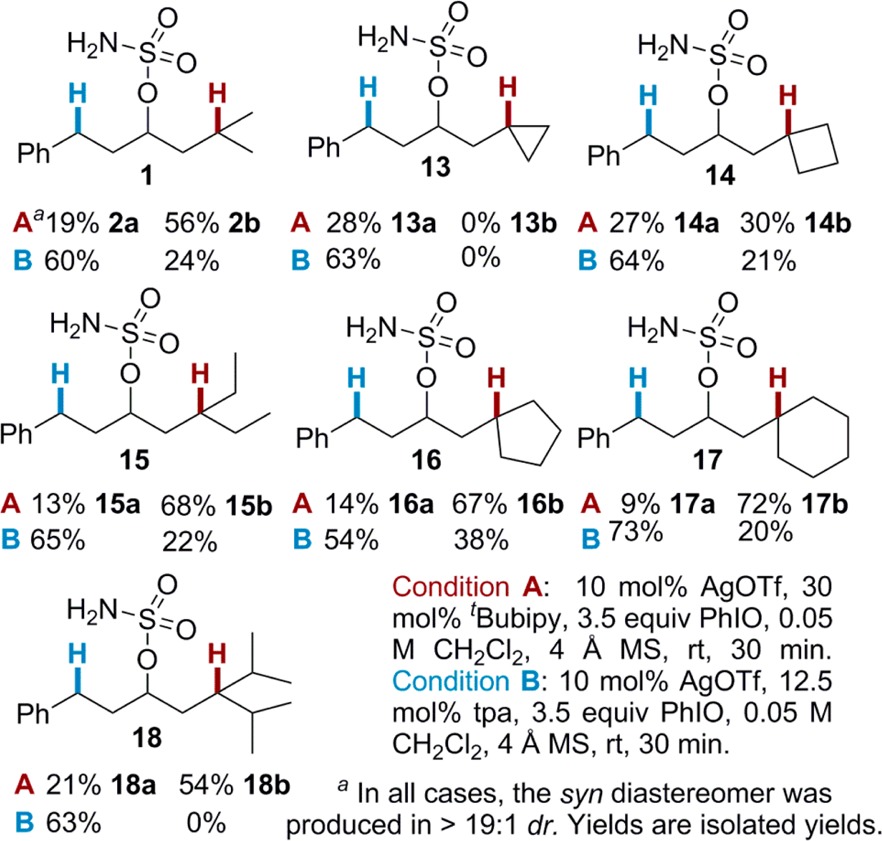
The reactivity of the isopropyl 3° C–H bond T was further compared to other types of C–H bonds (Table 3). Reaction of 19 with (tpa)AgOTf preferred the allylic C–H bond, while that of 20 with (tBubipy)2AgOTf favored T over the propargylic C–H bond. The higher BDE of the propargylic C–H of 20, as compared to the allylic C–H of 19, might be responsible for the decreased selectivity with (tpa)AgOTf. As expected, amination of 21 gave 21b as the exclusive product, as 2° C–H bonds are generally less reactive than 3° C–H’s. Amination of 21 with (tpa)AgOTf again gave exclusive insertion at the 3 °C–H bond, but with an improved yield compared to (tBubipy)2AgOTf. Finally, the amination of 22 shows selectivity between two similar 3° C–H bonds is achievable. This intriguing result demonstrates that even slight differences in sterics and electronics can yield site selectivity with silver catalysis.
Table 3. Further Study of Site Selectivity.
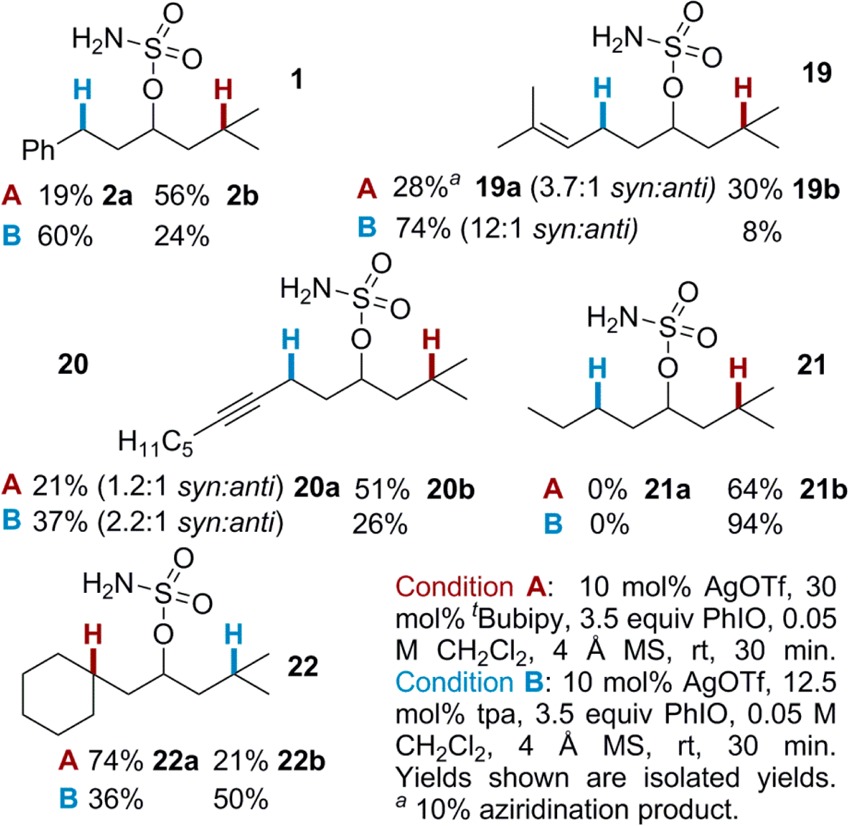
Further exploration of the impact of substrate electronics on the selectivity of the nitrene transfer was carried out (Table 4). In 23, the additional Me group increases the electron density of the 3° Bn C–H, resulting in lower selectivity using (tBubipy)2AgOTf. In contrast, the steric congestion at Bn in 23 decreased the preference of (tpa)AgOTf for benzylic amination. Moving the Me group from the benzylic carbon of 23 to the ortho position in 24 restores selectivity with (tpa)AgOTf, further supporting the observation that steric effects influence the behavior of (tpa)AgOTf to a greater extent than (tBubipy)2AgOTf, which tends to be more sensitive to the electron density at the C–H bond.
Table 4. Exploration of Electronic Effects.
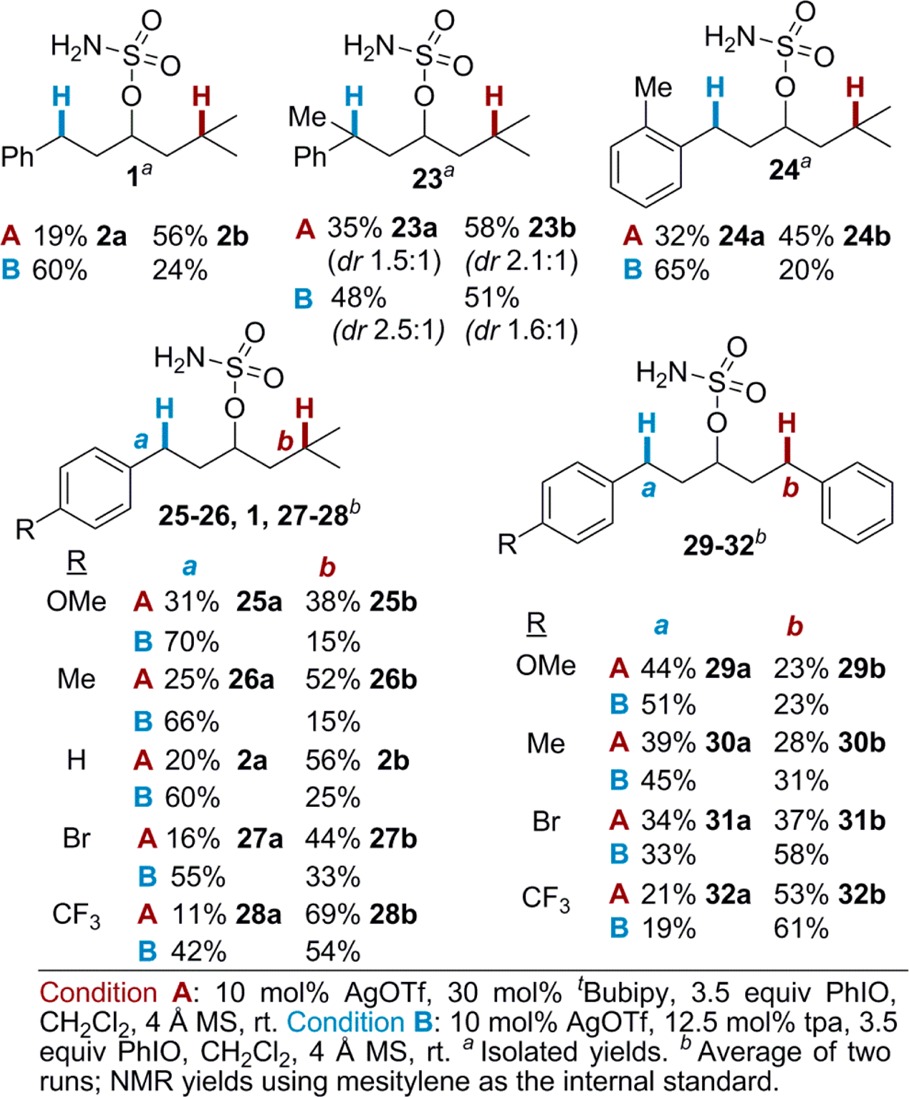
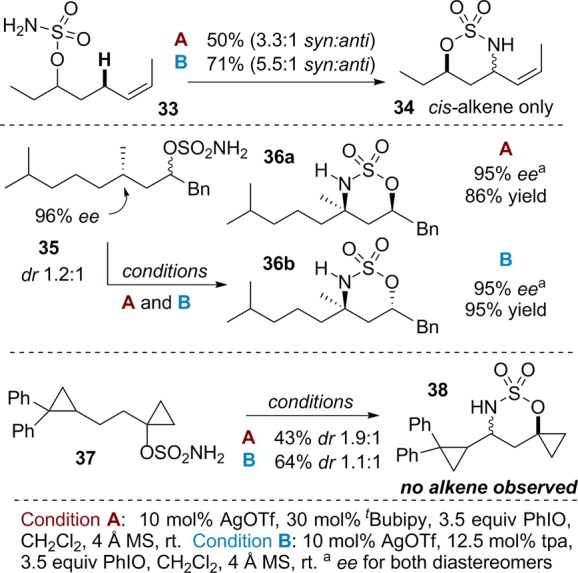
The sensitivity of (tBubipy)2AgOTf for activation of the most electron-rich C–H bond was further supported by substrates 25–28, where amination of T increased as the electron density of Bn was decreased. Interestingly, the same trend held true for (tpa)AgOTf, indicating that the two catalysts react similarly to changes in the electronics of the substrate. Insight into the nature of the transition state was obtained through Hammett correlations using the diaryls 29–32. Because selectivity occurs during the product-determining step, the ratio of the isomeric products offers a direct measure of kAr/kPh and does not require knowledge of the rate-determining step.4a,4c,7b Hammett plots of both catalysts support the buildup of positive charge at the carbon undergoing reaction (see the Supporting Information for details).8
Further exploration of the mechanistic pathway of silver-promoted nitrene transfer was carried out (Scheme 2). No isomerization of the double bond of 33 was noted using either catalyst. A stereochemical probe 35 resulted in no loss in ee in the product 36a or 36b with either catalyst. Finally, employing the radical clock 37 yielded no alkene products with either (tBubipy)2AgOTf or (tpa)AgOTf. All three of these experiments argue against the presence of long-lived radical species in the reaction pathway.
Scheme 2. Mechanistic Studies of Nitrene Transfer.
Our initial empirical results for catalyst-controlled tunable amination suggest design principles for developing more selective catalysts. The sensitivity of the bipy-supported catalyst to the electron density of the C–H bond prompted us to briefly explore the impact of the ligand electronics on the site selectivity of nitrene transfer. We were pleased to find that installing an electron-donating p-OMe group on the bipy ligand increased the selectivity for 2b:2a from 1.6:1 in the unsubstituted ligand 39 to 4.0:1 in 41, providing a clear path for the design of more selective catalysts. In contrast, the tpa-supported catalysts were not as sensitive to ligand electronics (Table 5, compare catalysts 9, 42, and 43). Interestingly, installing a Me group ortho to the pyridines of 44 reversed the selectivity to again favor T, while replacing a pyridine arm with a benzyl in 45 also eroded selectivity, suggesting future studies of the tpa-based ligand should focus on the steric features of the catalyst and the oxidant. Indeed, replacing PhIO with PhI(OPiv)2 increased both the yield and the selectivity. The dynamic behavior of tpa-derived catalysts is influenced by the substitution of the tpa scaffold, and studies to determine how this design feature impacts selectivity are currently underway.
Table 5. Insight into Future Catalyst Design.

10 mol % catalyst, 3.5 equiv of PhIO, 4 A MS, CH2Cl2.
NMR yields, mesitylene internal standard.
One of the pyridines of the ligand was replaced with a benzyl group.
The oxidant was PhI(OPiv)2 instead of PhIO.
In conclusion, we have developed two silver catalyst systems capable of promoting tunable, regioselective amination of C–H bonds in different chemical environments. Silver catalysts supported by tBubipy appear to prefer amination of the most electron-rich C–H bond, while silver supported by a tpa ligand is more sensitive to the steric environment around the C–H bond, as well as the bond dissociation energy. Preliminary mechanistic work indicates that if radical intermediates are formed in either pathway, they undergo a rapid rebound, as no loss of stereochemical information is noted.8 Computational studies are currently underway to obtain a more detailed mechanistic picture of both reaction pathways and gain a better understanding of how the ligand identity affects the electronic and steric nature of the purported silver nitrene intermediate. Another exciting ongoing investigation is utilizing these initial results to parametrize features of both the silver nitrenes and the substrates to develop predictive models for site selectivity in complex substrates.
Acknowledgments
This research was supported by start-up funds provided by the University of Wisconsin. J.M.S. is a Sloan Research Fellow. The Varian INOVA 500 was obtained with NSF Award No. CHE-9629688. The Varian UNITY 500 was obtained with NSF Award Nos. CHE-8813550 and CHE-9629688, as well as NIH Award No. 1-S10-RR04981-01. The Varian Nanoprobe was obtained with NIH Award No. 1-S10-RR04981-01.
Acknowledgments
The authors thank Ryan Van Hoveln for helpful discussions, James Jirak for substrate synthesis, Charles Fry and Heike Hofstetter for NMR assistance, and Martha Vestling for assistance with mass spectrometry.
Supporting Information Available
Experimental procedure and characterization is provided for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† J.M.A., A.M.P., and R.J.S. contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For selected references on selective C–H functionalizations, see:; a Roizen J. L.; Harvey M. E.; Du Bois J. Acc. Chem. Res. 2012, 45, 911. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lescot C.; Darses B.; Collet F.; Retailleau P.; Dauban P. J. Org. Chem. 2012, 77, 7232. [DOI] [PubMed] [Google Scholar]; c Newhouse T.; Baran P. S. Angew. Chem., Int. Ed. 2011, 50, 3362. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Collet F.; Lescot C.; Liang C.; Dauban P. Dalton Trans. 2010, 39, 10401. [DOI] [PubMed] [Google Scholar]; e Bess E. N.; DeLuca R. J.; Tindall D. J.; Oderinde M. S.; Roizen J. L.; Du Bois J.; Sigman M. S. J. Am. Chem. Soc. 2014, 136, 5783. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Chen M. S.; White M. C. Science 2007, 318, 783. [DOI] [PubMed] [Google Scholar]; g Chen M. S.; White M. C. Science 2010, 327, 566. [DOI] [PubMed] [Google Scholar]; h White M. C. Science 2012, 335, 807. [DOI] [PubMed] [Google Scholar]; i Bagchi V.; Paraskevopoulou P.; Das P.; Chi L.; Wang Q.; Choudhury A.; Mathieson J. S.; Cronin L.; Pardue D. B.; Cundari T. R.; Mitrikas G.; Sanakis Y.; Stavropoulos P. J. Am. Chem. Soc. 2014, 136, 11362. [DOI] [PubMed] [Google Scholar]
- For selected examples of directing groups used in conjunction with Ir catalysts, see:; a Kim H. J.; Ajitha M. J.; Ryu J.; Kim J.; Lee Y.; Jung Y.; Chang S. J. Am. Chem. Soc. 2014, 136, 1132. [DOI] [PubMed] [Google Scholar]; b Kang T.; Kim Y.; Lee D.; Wang Z.; Chang S. J. Am. Chem. Soc. 2014, 136, 4141. [DOI] [PubMed] [Google Scholar]; c Simmons E. M.; Hartwig J. F. Nature 2012, 483, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]; For examples involving Pd, see:; d Shao J.; Chen W.; Giulianotti M. A.; Houghten R. A.; Yu Y. Org. Lett. 2012, 14, 5452. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Musaev D. G.; Kaledin A.; Shi B.; Yu J. J. Am. Chem. Soc. 2012, 134, 1690. [DOI] [PubMed] [Google Scholar]; f Desai L. V.; Stowers K. J.; Sanford M. S. J. Am. Chem. Soc. 2008, 130, 13285. [DOI] [PMC free article] [PubMed] [Google Scholar]; For examples involving Ru, see:; g Allu S.; Swarmy K. C. K. J. Org. Chem. 2014, 79, 3963. [DOI] [PubMed] [Google Scholar]
- For selected reviews on C–H functionalization strategies, see:; a Godula K.; Sames D. Science 2006, 312, 67. [DOI] [PubMed] [Google Scholar]; b Neufeldt S. R.; Sanford M. S. Acc. Chem. Res. 2012, 45, 936. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bruckl T.; Baxter R. D.; Ishihara Y.; Baran P. S. Acc. Chem. Res. 2012, 45, 826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Espino C. G.; Wehn P. M.; Chow J.; Du Bois J. J. Am. Chem. Soc. 2001, 123, 6935. [Google Scholar]; b Collet F.; Dodd R. H.; Dauban P. Chem. Commun. 2009, 5061. [DOI] [PubMed] [Google Scholar]; c Fiori K. W.; Espino C. G.; Brodsky B. H.; Du Bois J. Tetrahedron 2009, 65, 3042. [Google Scholar]; d Norder A.; Warren S. A.; Herdtweck E.; Huber S. M.; Bach T. J. Am. Chem. Soc. 2012, 134, 13524. [DOI] [PubMed] [Google Scholar]
- For other examples of Rh-catalyzed C–H amination, see:; a Lescot C.; Darses B.; Collet C.; Retailleau P.; Dauban P. J. Org. Chem. 2012, 77, 7232. [DOI] [PubMed] [Google Scholar]; b Cochet T.; Bellosta V.; Roche D.; Ortholand J.; Greiner A.; Cossy J. Chem. Commun. 2012, 48, 10745. [DOI] [PubMed] [Google Scholar]; c Grohmann C.; Wang H.; Glorius F. Org. Lett. 2012, 14, 656. [DOI] [PubMed] [Google Scholar]; d Ng K.; Zhou Z.; Yu W. Org. Lett. 2012, 14, 272. [DOI] [PubMed] [Google Scholar]; e Lebel H.; Spitz C.; Leogane O.; Trudel C.; Parmentier M. Org. Lett. 2011, 13, 5460. [DOI] [PubMed] [Google Scholar]
- a Nguyen Q.; Sun K.; Driver T. G. J. Am. Chem. Soc. 2012, 134, 7262. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tang C.; Yuan Y.; Cui Y.; Jiao N. Eur. J. Org. Chem. 2013, 7480. [Google Scholar]; c Beltran A.; Lescot C.; Díaz-Reqeujo M.; Pérez P. J.; Dauban P. Tetrahedron 2013, 69, 4488. [Google Scholar]; d Ryu J.; Shin K.; Park S. H.; Kim Y.; Chang S. Angew. Chem., Int. Ed. 2012, 51, 9904. [DOI] [PubMed] [Google Scholar]; e Lebel H.; Trudel C.; Spitz C. Chem. Commun. 2012, 48, 7799. [DOI] [PubMed] [Google Scholar]; f Hayes C. J.; Beavis P. W.; Humphries L. A. Chem. Commun. 2006, 4501. [DOI] [PubMed] [Google Scholar]; g Kornecki K. P.; Berry J. F. Eur. J. Inorg. Chem. 2012, 3, 562. [Google Scholar]
- For recent selected examples, see:; a Collet F.; Lescot C.; Dauban P. Chem. Soc. Rev. 2011, 40, 1926. [DOI] [PubMed] [Google Scholar]; b Harvey M. E.; Musaev D. G.; Du Bois J. J. Am. Chem. Soc. 2011, 133, 17207. [DOI] [PubMed] [Google Scholar]; c Paradine S. M.; White M. C. J. Am. Chem. Soc. 2012, 134, 2036. [DOI] [PubMed] [Google Scholar]
- Rigoli J. W.; Weatherly C. D.; Alderson J. M.; Vo B. T.; Schomaker J. M. J. Am. Chem. Soc. 2013, 135, 17238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected examples of aminations mediated by silver catalysis, see:; a Cui Y.; He C. J. Am. Chem. Soc. 2003, 125, 16202. [DOI] [PubMed] [Google Scholar]; b Cui Y.; He C. Angew. Chem., Int. Ed. 2004, 43, 4210. [DOI] [PubMed] [Google Scholar]; c Li Z.; Capretto D. A.; Rahaman R. H.; He C. Angew. Chem., Int. Ed. 2007, 46, 5184. [DOI] [PubMed] [Google Scholar]; d Silver in Organic Chemistry; Harmata M., Ed.; John Wiley & Sons: Hoboken, NJ, 2010. [Google Scholar]; e Llaveria J.; Beltran A.; Diaz-Requejo M. M.; Matheu M. I.; Castillon S.; Pérez P. J. Angew. Chem., Int. Ed. 2010, 49, 7092. [DOI] [PubMed] [Google Scholar]; f Fructos M. R.; Trofimenko S.; Diaz-Requejo M. M.; Pérez P. J. J. Am. Chem. Soc. 2006, 128, 11784. [DOI] [PubMed] [Google Scholar]; g Braga A. A. C.; Maseras F.; Urbano J.; Caballero A.; Diaz-Requejo M. M.; Pérez P. J. Organometallics 2006, 25, 5292. [Google Scholar]; h Maestre L.; Sameera W. M. C.; Dia-Requejo M. M.; Maseras F.; Pérez P. J. J. Am. Chem. Soc. 2013, 135, 1338. [DOI] [PubMed] [Google Scholar]; i Arenas I.; Fuentes M.Á.; Álvarez E.; Díaz Y.; Caballero A.; Castillón S.; Pérez P. J. Inorg. Chem. 2014, 53, 3991. [DOI] [PubMed] [Google Scholar]; j Gómez-Emeterio B. P.; Urbano J.; Díaz-Requejo M. M.; Pérez P. J. Organometallics 2008, 27, 4126. [Google Scholar]
- For selected examples illustrating the various coordination geometries that can be adopted by silver complexes, see:; a Hung-Low F.; Renz A.; Klausmeyer K. K. Polyhedron 2009, 28, 407. [Google Scholar]; b Hung-Low F.; Renz A.; Klausmeyer K. K. J. Chem. Cryst. 2011, 41, 1174. [Google Scholar]; c Du J.-L.; Hu T.-L.; Zhang S.-M.; Zeng Y.-F.; Bu X.-H. CrystEngComm 2008, 10, 1866. [Google Scholar]; d Hung-Low F.; Renz A.; Klausmeyer K. K. J. Chem. Cryst. 2009, 39, 438. [Google Scholar]; e Levason W.; Spicer M. D. Coord. Chem. Rev. 1987, 76, 45. [Google Scholar]
- a Yao Y.-R.Handbook of Bond Dissocation Energies in Organic Compounds; CRC Press: Boca Raton, FL, 2003. [Google Scholar]; b CRC Handbook of Chemistry and Physics, 85th ed.; Lide D. R., Ed.; CRC Press: Boca Raton, FL, 2004. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.