

Abstract
Prokaryotic regulatory proteins respond to diverse signals and represent a rich resource for building synthetic sensors and circuits. The TetR family contains >105 members that use a simple mechanism to respond to stimuli and bind distinct DNA operators. We present a platform that enables the transfer of these regulators to mammalian cells, which is demonstrated using human embryonic kidney (HEK293) and Chinese hamster ovary (CHO) cells. The repressors are modified to include nuclear localization signals (NLS) and responsive promoters are built by incorporating multiple operators. Activators are also constructed by modifying the protein to include a VP16 domain. Together, this approach yields 15 new regulators that demonstrate 19- to 551-fold induction and retain both the low levels of crosstalk in DNA binding specificity observed between the parent regulators in Escherichia coli, as well as their dynamic range of activity. By taking advantage of the DAPG small molecule sensing mediated by the PhlF repressor, we introduce a new inducible system with 50-fold induction and a threshold of 0.9 μM DAPG, which is comparable to the classic Dox-induced TetR system. A set of NOT gates is constructed from the new repressors and their response function quantified. Finally, the Dox- and DAPG- inducible systems and two new activators are used to build a synthetic enhancer (fuzzy AND gate), requiring the coordination of 5 transcription factors organized into two layers. This work introduces a generic approach for the development of mammalian genetic sensors and circuits to populate a toolbox that can be applied to diverse applications from biomanufacturing to living therapeutics.
Keywords: mammalian synthetic biology; systems biology; eukaryote; inducible system; 2,4-diacetylphloroglucinol (DAPG)
Realizing the potential of engineering mammalian cells requires the predictable construction of synthetic sensors and circuits. In the clinic, cell-based therapies could function to integrate physiological markers, migrate to a disease location, and execute a multistep treatment.1−4 Cells involved in the manufacturing of biologics, such as the workhorse Chinese hamster ovary (CHO) cell line, could be engineered to respond to inducers that stage a multistep production process.5−7 Other applications include the programmable spatial organization needed for artificial organs and regenerative medicine, responsive living prosthetics (e.g., sensing blood glucose and controlling insulin production), and high-throughput drug screens based on readouts of cell state.1,8,9 These advanced applications require circuitry that encodes signal processing and control algorithms, the implementation of which requires more regulatory parts than are currently available.2,4 While the number of such parts for prokaryotes has exploded,10 there is currently a lag in building the analogous toolboxes for mammalian circuit design.11
In prokaryotes, TetR transcriptional repressors constitute one of the most abundant and plastic family of regulators.12,13 These repressors consist of a single protein that contains both small molecule sensing and DNA-binding domains. Over 200 000 TetR homologues have been sequenced that are representative of a wide range of sensing and DNA binding specificities. To date, sensing domains have been characterized that respond to >60 ligands, including antibiotics, metabolites, hormones, cell–cell signaling molecules, and metals.13 In the absence of its ligand, TetR forms a dimer that strongly binds to the TetR operator sequence (tetO). In the presence of ligand, the dimer is disrupted, TetR dissociates from the DNA, and gene expression is activated. The DNA-binding domains of different repressors bind unique 17–30 bp sequences, and it has been shown that these are highly orthogonal, with few off-target interactions between noncognate operators.14 Thus, this family provides a rich resource for mining ligand- and DNA-binding domains to build synthetic sensors and circuits.
The most common method for inducing mammalian gene expression is based on the TetR repressor, whose ligand is the antibiotic tetracycline.15,16 In mammalian cells, TetR can be used as both a repressor, or converted into an activator by fusing it to the transactivation domain from virion protein 16 of the Herpes simplex virus (VP16),17 which recruits RNA polymerase (RNAP). Multiple copies of tetO are placed upstream of the minimal CMV promoter (referred to as a Tetracycline Response Element or TRE), and in this fashion, reporter expression is activated when TetR is bound to the TRE, and becomes inactivated upon the addition of doxycycline (this system is referred to as “Tet-Off”). A “Tet-On” system has also been developed, whereby reporter expression is activated upon addition of doxycycline; this behavior is mediated by the reverse TetR transcription factor (rtTA).18 In both cases, the VP16 domain recruits RNA polymerase (RNAP) when TetR is bound to a synthetic, TRE-containing promoter. These switches typically have low basal expression, exhibit a large dynamic range (from 10 to several thousand-fold induction), and have been shown to function in a wide range of tissue culture systems, including embryonic stem cells,19,20 CHO,21 HEK,22 HeLa,23 and MCF-724 cells, as well as in living animals.25
Homologues of TetR have been used to build synthetic gene switches for various applications,2,26 and switches responding to other antibiotics, including erythromycin (MphR)27 and pristinamycin (Pip)28 have also been constructed. To expand upon the available inducible systems, sensors that respond to other small molecules (cumate, CymR29) have been developed, including some that can be delivered to cells in gas form (acetaldehyde, AlcR;30 6-hydroxy-nicotine, HdnoR31). Quorum sensing systems involved in cell–cell communication have been ported from Streptomyces (ScbR and SpbR),32Agrobacterium (TraR),32,33 and Vibrio fischeri (LuxR).34 These sensors have largely been developed for research purposes or in the context of a bioreactor. For clinical uses in patients, switches have been built that respond to nontoxic molecules, including amino acids (arginine, ArgR;35 tryptophan, TrpR36), food additives and metabolites (vanillic acid, VanR;37 phlorectin, TtgR38), and vitamins (biotin,39 BirA40) . Beyond cell cultures, many of these switches have been demonstrated to function in living animals, including mice.27 In one compelling application, a uric acid (HucR) sensing circuit was constructed as part of a feedback mechanism to maintain blood urate homeostasis, the disruption of which can lead to gout.41 Furthermore, a sensor that reacts to the inactivation of antituberculosis compounds (EthR),42 which serves as an application for drug discovery, has also been constructed.
Of the many synthetic mammalian circuits that have been built using TetR and its homologues,43 several of the resulting genetic switches and cascades based on these regulators exhibit ultrasensitivity and bistability.44−47 To build more sophisticated functions, logic operations such as inverters and 2-input Boolean gates have been layered together to generate feedforward circuits,48 half adders (and subtractors),49 2-input decoders,50 and a cell type classifier.51 Dynamic circuits have also been constructed, including time delays and oscillators.40,52−54 Furthermore, channels for cell–cell communication have also been developed where the sender signal (which consists of a metabolic pathway) produces the signaling molecule and the receiver acts as the signal sensor.40,55,56 To date, as many as 3 TetR homologues have been incorporated into a single mammalian circuit (tTA, PIP-KRAB, and E-KRAB57,58), and in one case, up to 3 repressors (TtgR, TetR, and ScbR) were combined into a single protein.59 However, the construction of circuits that can perform more sophisticated signal processing operations will require a larger set of transcription factors that are orthogonal to one another, or in other words, that do not cross react with one another’s DNA operators.
TetR and its homologues are not the only transcriptional regulators commonly used to construct genetic circuits, and several classes of transcription factors have modular DNA-binding domains that allow them to be programmed to target a specific nucleotide sequence.59 This can be based on a combination of residues that bind to specific base pairs, as is the case for zinc finger proteins (ZFPs)60 and transcription-activator-like effectors (TALEs).61 Similarly, the CRISPRi technique is based on the targeting of a catalytically inactive Cas9 protein to a specific DNA sequence through the use of a guide RNA.62,63 All of these systems can be moved into mammalian cells and retooled to function as activators or repressors by fusing VP16- or KRAB-like peptides, respectively,64−72 or by relying on steric hindrance of Cas9 alone.67 However, it remains a challenge to add sensing capability to these DNA-binding domains. A generalizable approach (based on two-hybrid systems) has been to utilize two proteins whose dimerization is induced by a stimulus; such an approach has been used to build ZFPs and TALEs that respond to small molecules (e.g., rapamycin, hydroxytamoxifen, or RU486),73,74 hypoxia,75 and light.76,77 The advantage of the TetR family is that a compact single protein has both the capability to sense a wide range of stimuli and transduce this to a DNA-binding event. Further, TetR and its homologues bind to small operator sequences with high specificity, which is desirable for promoter design but also comes at the cost of the inability to target them to arbitrary sequences.
Here, we present a systematic approach to retool a group of TetR-family repressors to operate as repressors and activators in mammalian cells. In previous work, we applied a part mining approach to build a set of 20 TetR homologues and characterized their orthogonality in Escherichia coli. Borrowing a strategy based off of designs used to convert TALEs into potent mammalian transcription factors,78 we move 8 new TetR homologues (AmtR, BM3R1, ButR, IcaR, LmrA, McbR, PhlF, and QacR) into human embryonic kidney (HEK293) cells, retooled as 15 new activators and repressors. Remarkably, these transcription factors retain both the orthogonality and fold-change observed in prokaryotic cells.14 Ligand sensing is also preserved, and we use this to build a new inducible system, which we characterize in both HEK293 and CHO cells. We also measure their response functions as gates to aide in the construction of larger circuits. Collectively, this work demonstrates that prokaryotic part mining is an effective strategy for expanding the regulatory parts available for mammalian cell engineering.
Results and Discussion
Functional Characterization of Retooled TetR Homologues in HEK293 Cells
In previous work, we used DNA synthesis to build a library of 73 TetR homologues,12 of which we built responsive promoters for 20 in E. coli. The crosstalk within this subset was quantified by measuring the activity of 400 combinations of repressors and promoters. From these data, we selected a subset of 8 that are highly orthogonal to move into mammalian cells. The mammalian regulators were built using the complete protein sequence for each TetR homologue, where the corresponding gene was codon optimized for expression in mammalian cells and resynthesized (Methods). Both activator (Figure 1a) and repressor (Figure 1b) versions were generated. Activators (TFA) were built by adding a destabilization domain,79 a Nuclear Export signal (NES), a VP16 activation domain,80 and a Nuclear Localization signal (NLS). Due to high levels of activation observed in the absence of inducer, destabilization domains were added to the activator design. Only an NLS was added to build repressors (TFR), which therefore rely on steric hindrance to achieve repression. The genes encoding TFA and TFR were placed under the control of the human elongation factor 1α promoter (hEF1a)81 and inserted into the pZDonor 1-GTW-2 plasmid82 (Supporting Information Figures 1 and 2).
Figure 1.

Design and characterization of synthetic transcription factors. (a) Expression of TFA is controlled by the constitutive hEF1a promoter. Operator sequences are shown as boxes. pTFA controls expression of the YFP output, which is activated by its cognate transcription factor. (b) The control system for TFR is similar to part a except that Gal4-VP16 is constitutively expressed from a third plasmid. pTFR controls expression of the YFP output, which is activated by Gal4-VP16 and repressed by its cognate transcription factor. (c) A detailed positional view of the activated (pLmrAA, top) and repressed (pLmrAR, bottom) LmrA promoters is illustrated. The pLmrAA promoter contains a minimal CMV promoter core with six upstream operators. The pLmrAR promoter consists of a minimal CMV promoter that is surrounded by two LmrA operators and five upstream Gal4 operators. The corresponding transcriptional start site (TSS) and TATA box are illustrated. (d) The function of the activators are shown and compared to the TetR activator (TetRA). The fold-activation was calculated by comparing the average fluorescence in the presence of a plasmid encoding the activator (P-constitutive TFA) with that obtained from the reporter plasmid (P-pTFA reporter) in the absence of the P-constitutive TFA plasmid. Cells were grown for 48 h post-transfection and assayed using flow cytometry (Methods). Representative histograms are shown in Supporting Information Figure 5. Microscopic images of cells transfected with the reporter only (−, top panel) or the cotransfected reporter and activator (+, bottom panel) are shown. BFP transfection controls are shown in Supporting Information Figure 6. (e) The function of the repressors are shown and compared to the TetR repressor (TetRR). Fold-repression is calculated by comparing the average fluorescence in the presence and absence of the plasmid containing the repressor (P-constitutive TFR). Microscopic images of cells transfected with the reporter and Gal4-VP16 (−, top panel) or the reporter, Gal4-VP16, and the repressor (+, bottom panel) are shown. Fluorescence histograms generated from the FITC-A geometric mean and BFP transfection control images are shown in Supporting Information Figures 7 and 8, respectively. In both parts d and e, the error bars were calculated based on the standard deviation of three independent experiments performed on different days. Cells are visualized using a YFP filter at 10× magnification, and images were taken 48 h post-transfection. The scale bars correspond to 400 μm. Gray boxes indicate that a particular TetR homologue was converted into only an activator or repressor and the other version was either not built or is nonfunctional.
Synthetic promoters were built for each of the mammalian transcription factors (Figure 1c). To generate activatable promoters, six copies of the cognate operator (Table 1) were inserted upstream of a minimal CMV promoter.78,83 A more complex promoter architecture is required in order to generate the repressible promoters, as the promoter itself must be activated in the absence of repressor. This behavior was achieved by designing a Gal4-VP16 activatable promoter. Specifically, each repressible promoter was designed to contain 5 Gal4 binding sequences upstream of a minimal CMV promoter, where Gal4-VP16 is constitutively expressed.67,78 The resulting promoter is rendered repressible by the inclusion of operators on either side of the CMV promoter. To measure activity, the promoters were placed upstream of a yellow fluorescent protein (YFP) coding sequence (Supporting Information Figure 3).84 The transcription factors and reporters were maintained on separate pZDonor 1-GTW-2 plasmids.
Table 1. Transcription Factor Operators and Inducer Molecules.
| TF | operator sequence | inducer molecule |
|---|---|---|
| AmtR | TTCTATCGATCTATAGATAAT | Gln K protein112 |
| BM3R1 | CGGAATGAACGTTCATTCCG | pentobarbital113 |
| ButR | GTGTCACTTTGACAGCAGTGTCAC | unknown |
| IcaR | TTCACCTACCTTTCGTTAGGTTAGGTTGT | gentamicin114 |
| LmrA | GATAATAGACCAGTCACTATATTT | lincomycin115 |
| McbR | ATAGACTGGCCTGTCTA | l-methionine116 |
| PhlF | ATGATACGAAACGTACCGTATCGTTAAGGT | 2,4-diacetylphloroglucinol85 |
| QacR | TATAGACCGTGCGATCGGTCTATA | plant alkaloids117 |
| TetR | TCCCTATCAGTGATAGA | doxycycline118 |
The two-plasmid system containing the constitutively expressed transcription factor and the reporter were transiently transfected into HEK293 cells, as well as a single-plasmid transfection of the reporter alone. For the repressible system, a third plasmid was included from which Gal4-VP16 was expressed, and in all cases, a plasmid containing the constitutively expressed eBFP transfection control plasmid was included (Supporting Information Figure 4). Cells were then trypsinized 48 h post-transfection, and their fluorescence quantified using flow cytometry (Methods). The induction of the reporter in the presence and absence of the plasmid containing the constitutively expressed activator or repressor was then compared (Figure 1c and d, respectively, and Supporting Information Figures 5–8). Seven of the activators are highly functional and demonstrate an average of 225-fold activation (ranging from 33- to 416-fold). For comparison, an activator based on TetR is able to achieve 75-fold activation. In addition, six new repressors were obtained with an average of 172-fold repression (ranging from 18- to 551-fold). These levels of repression are comparable to the 50-fold repression that is achieved by TetRR. While most of the TetR homologues could be systematically converted into both repressors and activators, for some only a single variant was found to be both functional and robust (BM3R1R, ButRA, and IcaRA).
The division of transcription factors and reporters on separate plasmids facilitates the rapid measurement of crosstalk between noncognate pairs, and all combinations of reporters and transcription factors were cotransfected into HEK293 cells. The activators are largely orthogonal, with the exception of a few cross-reactions (Figure 2a and Supporting Information Figure 9). Notably, LmrAA activates pQacRA, and LmrAA and QacRA both activate pMcbRA. The repressors are also highly orthogonal, although there is some activity of LmrAR against pMcbRR and pQacRR (Figure 2b and Supporting Information Figure 10). Interestingly, the off-target interactions observed here are not present in the E. coli system.14 This may be due to changes in the expression level of the transcription factors, having multiple operators in the synthetic promoters, and/or the ability of VP16 to recruit the transcriptional machinery even when delivered to a promoter at low affinity.
Figure 2.
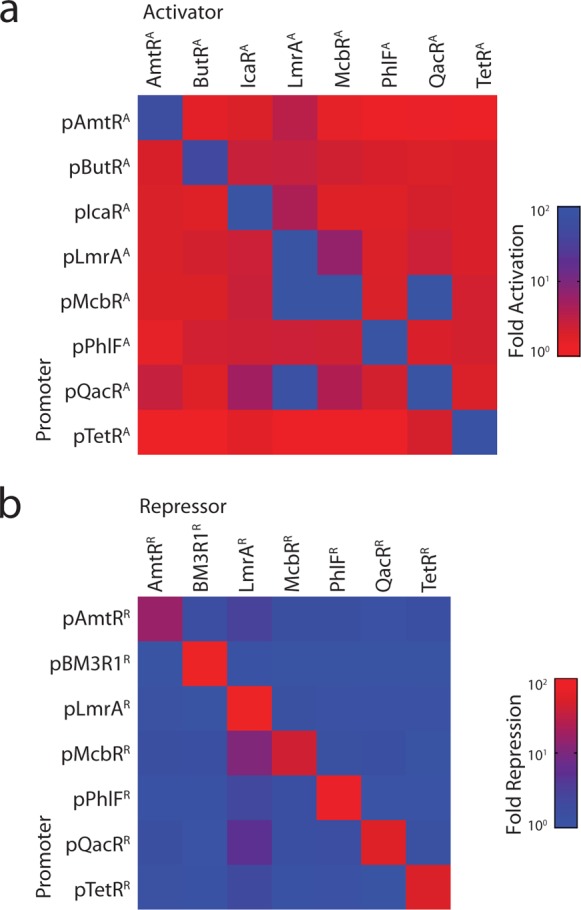
Orthogonality between synthetic transcription factors. (a) Crosstalk is shown between all combinations of activators and promoters. The fold-activation is calculated by dividing the average fluorescence of cells containing both the reporter and activator plasmids by the average fluorescence of cells only transfected with the reporter plasmid. Raw data underlying the matrix are shown in Supporting Information Figure 9, and data correspond to the average FITC-A geometric mean values from flow cytometry data collected from three independent transfections carried out on separate days. (b) Crosstalk is shown between all combinations of repressors and promoters. The fold-repression is calculated by dividing the average fluorescence of cells containing the reporter and Gal4-VP16 encoded plasmids by the fluorescence of cells transfected with plasmids encoding the reporter, Gal4-VP16, and cognate repressor. Raw data underlying the matrix are shown in Supporting Information Figure 10, and data correspond to the average FITC-A geometric mean values from flow cytometry data collected from three independent transfections carried out on separate days.
Construction of a DAPG-Inducible System
The TetR homologues that were selected for this study are associated with different classes of ligands, including metabolites, natural products, and plant alkaloids (Table 1). Similar to the doxycycline (Dox) induction of TetR in the Tet-On inducible system (Figure 3a),18 the PhlF repressor responds to 2,4-diacetylphloroglucinol (DAPG), which is a polyketide antibiotic produced by Pseudomonas fluorescens that has activity against plant pathogens (Figure 3b).85,86 DAPG has the potential to be a similarly useful inducible system, because it freely diffuses through eukaryotic membranes and can be purchased from chemical suppliers (Methods).
Figure 3.
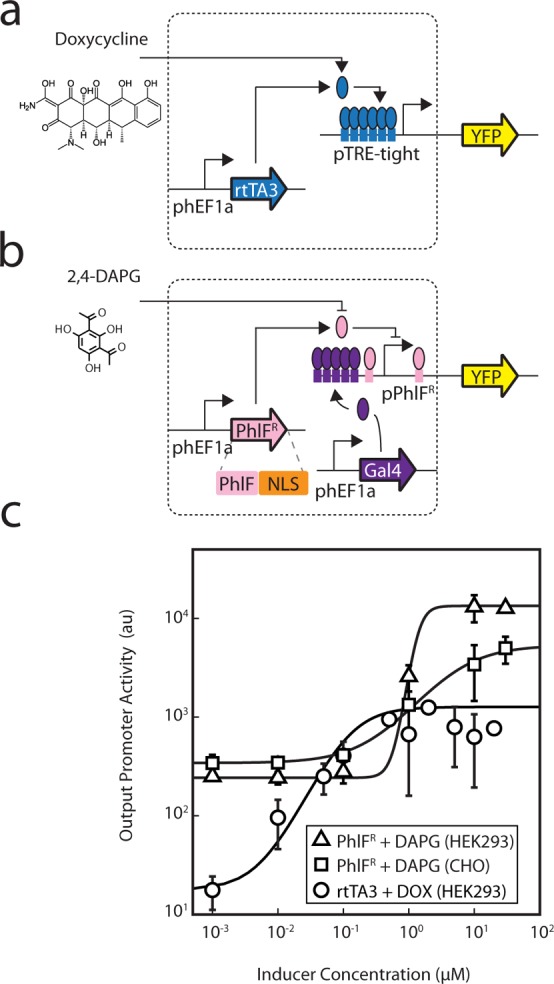
Characterization of the DAPG-inducible PhlFR system. (a) The structure of doxycycline and the Tet-On inducible system, comprised of the rtTA3 regulator, are shown.111 In this system, rtTA3 is constitutively expressed from the phEF1a constitutive promoter and activates expression of its cognate promoter which contains 6 copies of the TetR operator sequence situated upstream of the minimal CMV promoter (referred to as pTRE-tight). The rtTA3 regulator binds to and activates expression from the pTRE-tight promoter in the presence of doxycycline. (b) The structure of DAPG and the PhlF inducible system are shown. In this system, PhlFR is constitutively expressed from the phEF1a promoter. The pPhlFR output promoter is activated by Gal4-VP16, which is constitutively expressed by the phEF1a promoter. PhlFR binds to and represses expression from the pPhlFR promoter in the absence of DAPG. (c) Induction of the Dox- and DAPG- inducible systems are compared and were measured in both HEK293 (Dox and DAPG systems) and CHO cells (DAPG system only). YFP fluorescence was measured after induction at [0, 0.01, 0.1, 1, 10, and 30 μM DAPG] or [0, 0.01, 0.05, 0.1, 0.5, 1, 2, 5, 10, 20 μM Dox]. The lines were fit to a Hill equation (Methods), the parameters for which are shown in Supporting Information Table 1. The data shown correspond to the average of three experiments from different transfections performed on different days, and error bars correspond to the standard deviation. Representative cytometry histograms for the three inducible systems are shown in Supporting Information Figure 11.
The inducibility of PhlFR was tested by adding DAPG to transfected cells and measuring the response from the pPhlFR reporter (Figure 3c; Supporting Information Figure 11 and Table 1). For cells supplemented with DAPG at the time of transfection, a drastic decrease in transfection efficiency was observed. To alleviate this decrease in transfection efficiency, the inducer was instead added 6 h post transfection, and cells were incubated for 42 h (Methods). After induction, YFP expression was measured using flow cytometry. In HEK293 cells, the response yields a robust 54-fold induction with a notably ultrasensitive transition (n = 4.7 when fit to a Hill function), with a threshold (half-maximum) of 1 μM DAPG. This response is similar to what has been observed for the Tet-On inducible system, which has a similar dynamic range (70-fold) but a less cooperative transition (n = 1.4). However, greater leakiness is associated with the DAPG-inducible system (240 versus 18 au), and because of this, the response curve is shifted higher. We also tested the PhlFR system in CHO cells, due to their importance in the manufacturing of biologics. This yielded a strong response, albeit with a lower dynamic range (15-fold) and less cooperative behavior (n = 1.0). The threshold of the switch is nearly identical among the two cell lines (5 μM DAPG in CHO cells), and the leakiness is also greater.
These discrepancies between the Dox- and DAPG-inducible systems can likely be attributed to their variable mechanisms used to control expression. For instance, our PhlFR system is based on dual and opposing activities (activation by Gal4-VP16 and repression by PhlF). Such an architecture has been shown to result in ultrasensitivity.87 In contrast, the Tet-On system relies on a more direct mechanism, whereby Dox induces rtTA3 binding to the promoter and subsequent activation of gene expression. Because of the large dynamic range associated with varying their inducer concentrations, both systems can be used to examine input-output relationships.
Measurement of 1-Input Response Functions
The response function of a gate captures how the output changes as a function of the input; for transcriptional gates, promoter activity serves as both the input and output. Our new repressors (TFRs) were used to build NOT gates,88,89 (which can be further converted into NOR gates by placing several upstream promoters in series).14,90 To deliver an input to the gate, the TRE-tight promoter (inducible by Dox) was used to drive expression of each TFR (Figure 4a). The response function of this inducible system was measured separately in the same genetic context using a fluorescent reporter, where the output of each gate corresponds to the fluorescence of the TFR-responsive promoter.
Figure 4.

Gate and circuit response functions. (a) The Dox inducible system is used to characterize NOT gates. Symbols are as described in Figure 1. Expression of TFR is controlled by the TRE-tight promoter, which is activated by the TetR activator (rtTA3) in the presence of Dox. Expression of the rtTA3 gene is controlled by the constitutive hEF1a promoter. (b) The response of each NOT gate is shown: McbR (blue inverted triangles), PhlF (red squares), AmtR (green circles), BM3R1 (purple triangles), and LmrA (light blue diamonds). The expression of the fluorescent reporter from the output promoter (pTFR) with respect to the induction of the input promoter (pTRE-tight) via Dox is shown. The average and standard deviation are plotted from three replicates from transfections performed on different days. Cytometry distributions corresponding to the FITC-A geometric mean of the 0, 0.5, and 5 μM induction points are shown in Supporting Information Figure 12 and fit parameters for each curve are listed in Table 2. (c) Two inducible systems (Dox or DAPG via pPhlFR) are used to measure the response function of the buffer gates based on transcriptional activators (TFA). (d) The response functions of the buffer gates are shown. The Dox-inducible system is used to characterize the AmtRA gate (circles) and the DAPG-inducible system is used to characterize the QacRA gate (squares). The inset shows the response as a function of input promoter activity (pTRE-tight or pPhlFR), rather than inducer concentration (Methods). Cytometry distributions corresponding to data for several induction points are shown in Supporting Information Figure 13 and fit parameters for each curve are listed in Table 3. (e) A schematic of the circuit that behaves as an enhancer is shown. The pAmtRA-QacRA promoter contains three upstream operators for each TF. (f) Enhancer fold activation of the output promoter (pAmtRA-QacRA) is shown as a function of the two inducers, where inducer concentrations vary from 0 to 20 μM doxycyline and 0–30 μM DAPG. Activation is indicated in blue, and data correspond to average fluorescence values from three replicates collected on different days. Cytometry distributions and error bars are shown in Supporting Information Figures 15 and 16, respectively.
The response function for five repressors (McbRR, PhlFR, AmtRR, BM3R1R, and LmrAR) was determined (Figure 4b and Supporting Information Figure 12). The average fluorescence was calculated by taking the mean YFP fluorescence from three experiments for each data point in the response curve; from these values, background fluorescence was subtracted, and the resulting output fluorescence values were converted into units of output promoter activity (this is done by separately measuring the activity of the various input promoters as a function of inducer). These values were used to generate a response function for each gate, where data were fit to a hill equation:
| 1 |
where y is the activity of the output promoter, ymin is the minimum output, ymax is the maximum output, n is the Hill coefficient, and K is the threshold level of input where the output is half-maximal (Table 2). The output from the ON state (Dox = 1 nM) differs between each gate because it depends on the activity of the TFR-responsive promoter, which vary based on operator sequence. When maximally induced (Dox = 20 μM), all of the response functions converge on the same OFF state. The dynamic range is defined as the ON state divided by the OFF state, and this varies from 23- to 78-fold. All of the switches are noncooperative with a Hill coefficient approaching unity (n ≈ 1), which is expected because the promoters contain two noninteracting operators.
Table 2. NOT Gate Response Function Parameters.
| name | inducer | Ka | n | ymaxb | yminb | fold-changec |
|---|---|---|---|---|---|---|
| McbRR | Dox | 0.13 | 1.22 | 1.6 × 104 | 6.6 × 102 | 24 |
| PhlFR | Dox | 0.05 | 1.50 | 1.2 × 104 | 3.6 × 102 | 33 |
| AmtRR | Dox | 0.17 | 1.07 | 8.4 × 104 | 2.9 × 102 | 28 |
| BM3R1R | Dox | 0.09 | 1.07 | 3.6 × 104 | 1.7 × 102 | 21 |
| LmrAR | Dox | 0.12 | 1.46 | 9.3 × 104 | 1.2 × 102 | 77 |
The threshold at which the NOT gate is at the half-maximum output, in μM doxycycline.
The maximum and minimum levels of expression, in arbitrary units of YFP fluorescence.
The fold-change is calculated by dividing the maximum average fluorescence (20 μM Dox) by the fluorescence of cells containing no inducer.
Buffer gates were also built based on the activators, which turn ON in response to induction from their input promoter. The response functions of the activators were measured either using the Dox-inducible pTRE-tight promoter, as above, or the DAPG-inducible pPhlFR promoter from this study (Figure 4c). Using this approach, the response function of two activators (AmtRA and QacRA) was determined following the same approach used for the NOT gates (Figure 4d and Supporting Information Figure 13). The data for each switch were fit using the following hill equation:
| 2 |
where the variables correspond to those used in equation 1 (parameters listed in Table 3).
Table 3. Activator Response Function Parameters.
| name | inducer | Ka | n | ymaxb | yminb | fold-changec |
|---|---|---|---|---|---|---|
| AmtRA | Dox | 0.1 | 3.00 | 1.3 × 103 | 15 | 82 |
| QacRA | DAPG | 4.6 | 1.89 | 1.1 × 104 | 122 | 91 |
The threshold at which the buffer gate is at the half-maximum output, in μM doxycyline (for AmtRA) or μM DAPG (for QacRA).
The maximum and minimum levels of expression, in arbitrary units of YFP fluorescence.
The fold-change is calculated by dividing the maximum average fluorescence (20 μM Dox or 30 μM DAPG) by the fluorescence of cells containing no inducer.
When characterizing gates, it is useful to report the input and output promoters in the same units,14,91,92 which would allow the predictable connection of gates to form larger circuits (although this can be complicated by context effects93,94). Yet the main challenge in doing so is that gates are typically measured using inducible systems and reported in terms of the concentration of the chemical inducer. When characterizing prokaryotic gates, we have separately measured the response of promoter output of the inducible system, and this information is used to build a response function that has the same units for the inputs and outputs.95 Similarly, we could characterize the Dox- and DAPG-inducible systems and use this to renormalize the transfer functions of the NOT gates (Supporting Information Figure 15) and the switches (inset, Figure 4d). The hill coefficients for the inverters change after renormalization but are consistent with respect to one another. This variation can be attributed to the limited resolution in input promoter activity in our measurements that increases regression error. The characterized switches illustrated above act individually upon a promoter, yet composite promoters that respond to multiple transcription factors can also be constructed to provide tunable output control.
Signal Integration: Construction of an “Enhancer” Promoter That Responds to Two Activators
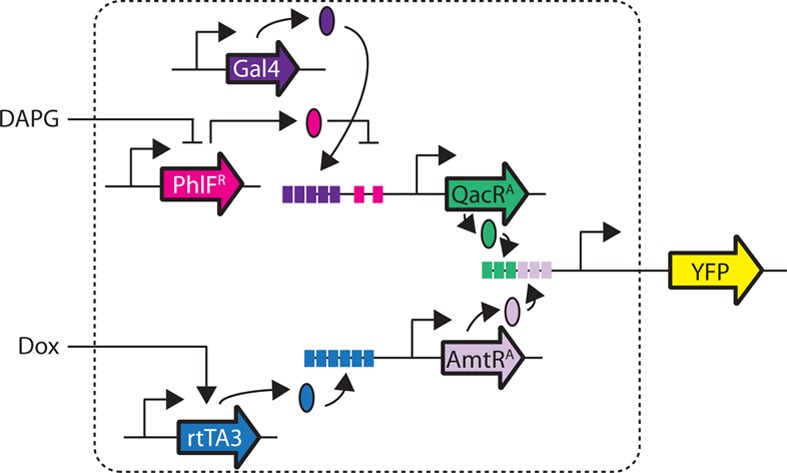
To generate a promoter capable of responding to combinations of input signals, operators for different transcription factors are typically combined into a single synthetic promoter. Similar approaches have been applied to build several classes of 2-input gates based on modified TetR homologues.58 These circuits consist of a single activator (e.g., ScbR modified with VP16) and up to two repressors (e.g., Pip modified with KRAB). For example, a NOT IF gate was built by constructing a promoter that contains 8 upstream ScbR operators, followed by 3 pir operators in between and a minimum promoter motif. The resulting promoter is ON only in the presence of ScbR and in the absence of Pip. Here, we sought to determine whether our promoter architecture could integrate multiple positive regulators to converge on a single output.
To construct a hybrid promoter that is responsive to multiple transcription factors, we modified our initial architecture used to build synthetic promoters containing six upstream operators.78 We postulated that this architecture could be altered to integrate signals from multiple TFs whose corresponding operators are present in different locations within the promoter. The full output of the promoter would not be achievable without induction of all of the TFs; thus, they would collectively enhance the activity of the promoter. The resulting circuit is not expected to function as an “AND gate” because each input increases activity toward the maximum. However, it does have features similar to fuzzy logic96 and analog adder circuitry.97
The integrating promoter was constructed by combining the operators for AmtRA (3 downstream) and QacRA (3 upstream, Figure 4e). Specifically, AmtRA expression is controlled by the Dox-inducible Tet-ON system, while QacRA expression is controlled by the DAPG-inducible PhlFR system (which also requires Gal4-VP16). Thus, the resulting circuit requires the control of 5 transcription factors carried on 7 distinct plasmids. All of the plasmids were cotransfected and the resulting YFP fluorescence measured using flow cytometry (Methods). The output was measured across varying concentrations of the two inducers (Dox and DAPG), and the resulting 25 data points were used to build a two-dimensional response function (Figure 4f and Supporting Information Figures 15 and 16). As expected, each inducible system is able to turn on the promoter independently, and the Dox-inducible system alone is able to induce the system 4.5-fold, while the DAPG-inducible system independently activates the system 8.5-fold. When both systems are maximally induced, the promoter is activated 19-fold. Thus, there is a near-perfect multiplicative effect between the induction of the two systems in isolation, compared to their collective impact on the promoter.
To gain insight into how transfection efficiency affects circuit performance, the fluorescence of the BFP-transfection control plasmid (a plasmid that constitutively expresses eBFP under the control of the hEF1a promoter) was used as a proxy for “copy number.” Since all plasmids are transfected in equal concentrations, it is expected that transfected cells contain the same relative amount of individual plasmids.98 Therefore, cells with a higher “copy number” will have higher levels of eBFP expression, and a larger quantity of each plasmid. To assess the effect of “copy number” on circuit performance, cells were separated into 360 logarithmically spaced bins based on their BFP fluorescence, and the maximally inducing and noninducing conditions were compared for each bin (Supporting Information Figure 17). The “fuzzy” AND gate is quite robust, as it exhibits a consistent fold activation over a wide range of “copy numbers”.
Expanding the Mammalian Parts Toolbox and Beyond
The “fuzzy” AND gate demonstrated here, as well as the increased number of both sensors and circuits illustrated throughout, significantly expands upon the tools available for use in mammalian cells. We also systematically verify that these components exhibit minimal crosstalk and robust levels of fold change similar to their bacterial predecessors.14 Furthermore, we demonstrate their functionality across a variety of cell types including HEK293 and CHO cells. Finally, we reveal that these components can be combined in a single cell to coordinately fine-tune the expression of an individual output.
To obtain variable and specific output levels, we utilized a hybrid promoter architecture whereby two distinct TFs converge on a single promoter, through the inclusion of multiple copies of each TFs operator sequence. In mammalian cells, variable output levels are typically achieved through adjusting the number of transcriptional enhancer elements.99 Enhancers integrate multiple signals in vivo, and act in cis to regulate transcriptional activity.100 Not only the spacing but also the content of cis-regulatory elements have been shown to have a dramatic effect on biological processes (such as development) in eukaryotes.101 While enhancer elements alone can lack discernible activity, in concert with other elements they typically evoke robust expression patterns upon associated genes.102 Recent efforts have been dedicated to identifying mammalian enhancer elements, where naturally occurring sequences were assessed in parallel to identify the essential elements of transcriptional networks.103,104
Although much work has been done to characterize the behavior and identity of naturally occurring enhancers, and to develop synthetic tools to control mammalian gene expression, issues persist in the implementation of such components toward broader applications. For example, the development of systems via transient transfection of tissue culture cells, and nonsite-specific integration make measurements difficult, and systems developed in this manner are not suited for clinical applications.3 Furthermore, it is known that enhancers exhibit negligible activity when transiently transfected but far more robust activity upon genomic integration.105−107 For these and other reasons, a safe harbor for genetic insertions should be developed, either through artificial chromosomes or designed integration sites.108 Based on these findings, future efforts should focus upon rigorously characterizing the behavior of these and other components upon genomic integration. Delineating the contribution of integration site and copy number should be at the forefront of these efforts, as well as the engineering of epigenetic tools to ensure active expression of integrated circuitry. Breakthroughs in these areas will aide in the implementation of the tools presented here toward real world applications that span from living therapeutics to the production of complex pharmaceuticals.
Methods
Cell Culture, Strains, and Media
E. coli strain DH10B [F–mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15ΔlacX74 recA1 endA1 araΔ139 Δ(ara, leu)7697 galU galK λ-rpsL (StrR) nupG] was used for cloning and to propagate DNA, except in the case where the propagated plasmids were used for Gateway cloning. In such cases, the ccdB Survival 2 T1R strain (Life Technologies, [F-mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15 ΔlacX74 recA1 araΔ139 Δ(ara-leu)7697 galU galK rpsL (StrR) endA1 nupG fhuA::IS2]) was used. The HEK293 (293FT) cell line was purchased from Invitrogen (product number R700-07), and CHO cells were obtained from ATCC (strain number CCL-61). HEK293 and CHO cells were cultured in high-glucose DMEM complete media (Dulbecco’s modified Eagle’s medium (DMEM), 4.5 g/L glucose, 0.045 units/mL of penicillin and 0.045 g/mL streptomycin and 10% FBS (Sigma)) at 37 °C, 100% humidity, and 5% CO2. Doxycycline was purchased from Clontech (product number 631311), and 2,4-diacetylphloroglucinol (DAPG) was purchased from Santa Cruz Biotechnology (product number 206518).
Mammalian Genetic Parts
Supporting Information Table 3 contains all of the part sequences used in this study. Plasmid maps are provided in Supporting Information Figures 1–4. Prokaryotic repressor coding sequences were optimized for production in mammalian cells using multiparameter gene optimization methods and synthesized by Geneart.109 The constitutive mammalian promoter (human elongation factor 1 alpha promoter, phEF1a) was from pLEIGW, a gift from Ihor R. Lemischka. The rtTA3 coding sequence and the pTRE-tight promoter (containing the CMV minimal promoter) were amplified from pTRIPZ (GE Healthcare, product number RHS4743). Constitutively expressed BFP (phEF1a-eBFP2) was used as a transfection control and was purchased from Addgene (plasmid 14891). The rb glob PA terminator was amplified from Addgene vector AAV-CAGGS-EGFP (plasmid 22212). In all cases, the reporter used corresponds to the Yellow Fluorescent Protein (eYFP),84 and the DD-tag was purchased from Clonetech (product number 632172).
Plasmids were constructed using a combination of GeneArt gene synthesis, Gateway cloning,110 and/or inverse PCR. Specifically, transcription factor coding sequences and their cognate promoters were synthesized into basic cloning vectors and were subcloned into expression or reporter vectors, respectively, via Gateway cloning. Hybrid promoters were constructed using inverse PCR to insert operator sequences upstream of the CMV minimal promoter within the reporter vector. In the case where inverse PCR was used to construct reporter vectors, whole plasmids were PCR amplified using Phusion DNA polymerase (NEB) along with multiple operator containing oligonucleotides. The resulting product was run on an agarose gel, extracted, and digested with DpnI. The blunted-ended, DpnI-digested product was phosphorylated (T4 Polynucleotide Kinase) and ligated (T4 DNA ligase) in a single reaction at room temperature, transformed into chemically competent DH10B cells, and plated on selective LB medium.
Transfection, Growth, and Processing of Cells
HEK293 FT and CHO cells were transfected using the Attractene transfection reagent (Qiagen) as described in the manual with several modifications. Specifically, 100 ng of each plasmid was combined into the appropriate combinations in a total volume of 7 μL or less, and 60 μL Dulbecco’s Modified Eagle Medium (DMEM) was added. To this mixture, 1.5 μL Attractene was added, and each sample was mixed by vortexing. The samples were incubated at room temperature for 10 min and then added to ∼8 × 104 cells in 0.5 mL DMEM that had been supplemented with penicillin, streptomycin, and amino acids (referred to as media complete) in a 24-well culture plate (Corning, product number 3473). For cells transfected with plasmids containing the pTRE-tight promoter, doxycycline was supplemented at the time of transfection. For cells containing plasmids harboring the DAPG-inducible system, DAPG was added 6 h post-transfection. Transfections were supplemented with 0.5 mL media complete 24 h post-transfection and doxycycline where appropriate. Cells were trypsinized 48 h post-transfection and subjected to flow cytometry (see below). Specifically, cells were trypsinized by aspirating the growth medium and applying 0.5 mL 0.25% trypsin-EDTA (Corning, product number 25-053) to adherent cells. Once cells were liberated from the plate, 2 mL media complete was added to each sample to halt trypsinization. Trypsinized cells were then spun down at 950 rpm for 10 min at 25 °C, the supernatant removed, and resuspended in 300 μL 1× phosphate buffered saline (PBS). From here, the trypsinized, PBS suspended cells were subjected to flow cytometry.
Flow Cytometry
Cells were analyzed by flow cytometry using a BD Biosciences LSRII flow cytometer. eBFP2 was measured using a 405 nm laser and a 450/50 filter, and eYFP with a 488 nm laser and a 530/30 filter. Cells were analyzed using FlowJo (TreeStar Inc., Ashland, OR), and populations were selected by gating out the background BFP signal of untransfected cells. Specifically, a gate was applied to encompass those cells that did not correspond to background (or where no signal was present on a BFP histogram of untransfected cells). The resulting gate was applied to all samples, to ensure that only cells expressing the eBFP2 transfection control were included in the analysis. Gated populations of >25 000 cells were used to calculate the geometric mean of the FITC-A fluorescence. When used to assay a promoter, this is referred to as the “Promoter Activity.”
Fold Change and Circuit Copy Number Calculations
All circuit plasmids were cotransfected with the transfection control marker, P-constitutive-eBFP. BFP-positive cells were separated into 360 logarithmically spaced bins based on raw fluorescence, referred to as the “Transfection Marker”. Fold-activation was calculated by dividing FITC-A fluorescence values from fully induced cells (20 μM DOX and 30 μM DAPG) by uninduced cells within each bin.
Hill Equation Curve Fitting
Response curves parameters for all activators and repressors were calculated by fitting to their respective Hill equations (equations 1 and 2). For each input, average fluorescence values from biological triplicates (collected on different days) were fit to the appropriate form of the Hill equation. Nonlinear least-squares regression was used to determine values for the Hill coefficient (n) and dissociation constant (K), and to minimize the error between the fitted and actual values.
Calculation of Fold-Change
The fold-change was determined by dividing the background subtracted YFP fluorescence values for cells containing the reporter plasmid alone (P-pTFx-reporter) by that of cells containing both the reporter and the transcription factor (either P-constitutive TFx, P-TRE-tight/TFx, or P-PhlFR/TFx) encoding plasmids, in the case of the repressors (where both transfections contained plasmids P-constitutive-Gal4-VP16 and P-constitutive-eBFP). For the activators, fold-change was calculated by taking the inverse of the equation used to calculate the fold-change for the repressors (where both transfections contained the P-constitutive-eBFP plasmid).
Microscope Imaging
Images were taken using an EVOS Digital inverted microscope (containing a 3MP color digital camera and LCD display). The excitation and emission wavelengths to obtain fluorescent images were as follows: 357 nm excitation 447 nm emission for eBFP, and 500 nm excitation 542 nm emission for eYFP. Images were taken at a 10× objective.
Acknowledgments
This work was provided by DARPA-BAA-11-23 and National Institutes of Health (NIH) P50 (P50GM098792) to R.W. and C.A.V., and a Life Technologies research contract (A114510), an ONR MURI (N00014-13-1-0074) and a NIH GMS “Light Gradients” (R01 GM095765) award to C.A.V.
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
B.C.S., A.G., and C.A.V. designed experiments, performed experiments, analyzed data, and wrote the manuscript. V.S. and L.W. designed and performed experiments. K.C., A.T., and J.C. designed experiments. R.W. designed experiments, analyzed data, and edited the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Fischbach M. A.; Bluestone J. A.; Lim W. A. (2013) Cell-based therapeutics: The next pillar of medicine. Sci. Transl. Med. 5, 179ps177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacchus W.; Aubel D.; Fussenegger M. (2013) Biomedically relevant circuit-design strategies in mammalian synthetic biology. Mol. Syst. Biol. 9, 691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. Y.; Smolke C. D. (2011) From DNA to targeted therapeutics: Bringing synthetic biology to the clinic. Sci. Transl. Med. 3, 106ps142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slusarczyk A. L.; Lin A.; Weiss R. (2012) Foundations for the design and implementation of synthetic genetic circuits. Nat. Rev. Genet. 13, 406–420. [DOI] [PubMed] [Google Scholar]
- Fussenegger M.; Schlatter S.; Datwyler D.; Mazur X.; Bailey J. E. (1998) Controlled proliferation by multigene metabolic engineering enhances the productivity of Chinese hamster ovary cells. Nat. Biotechnol. 16, 468–472. [DOI] [PubMed] [Google Scholar]
- Wurm F. M. (2004) Production of recombinant protein therapeutics in cultivated mammalian cells. Nat. Biotechnol. 22, 1393–1398. [DOI] [PubMed] [Google Scholar]
- Weber W.; Fussenegger M. (2007) Inducible product gene expression technology tailored to bioprocess engineering. Curr. Opin. Biotechnol. 18, 399–410. [DOI] [PubMed] [Google Scholar]
- Wieland M.; Fussenegger M. (2012) Reprogrammed cell delivery for personalized medicine. Adv. Drug Delivery Rev. 64, 1477–1487. [DOI] [PubMed] [Google Scholar]
- Weber W.; Fussenegger M. (2009) The impact of synthetic biology on drug discovery. Drug Delivery Rev. 14, 956–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen A. A.; Segall-Shapiro T. H.; Voigt C. A. (2013) Advances in genetic circuit design: Novel biochemistries, deep part mining, and precision gene expression. Curr. Opin. Chem. Biol. 17, 878–892. [DOI] [PubMed] [Google Scholar]
- Ruder W. C.; Lu T.; Collins J. J. (2011) Synthetic biology moving into the clinic. Science 333, 1248–1252. [DOI] [PubMed] [Google Scholar]
- Ramos J. L.; Martinez-Bueno M.; Molina-Henares A. J.; Teran W.; Watanabe K.; Zhang X.; Gallegos M. T.; Brennan R.; Tobes R. (2005) The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev.: MMBR 69, 326–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbertson L.; Nodwell J. R. (2013) The TetR family of regulators. Microbiol. Mol. Biol. Rev.: MMBR 77, 440–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton B. C.; Nielsen A. A.; Tamsir A.; Clancy K.; Peterson T.; Voigt C. A. (2014) Genomic mining of prokaryotic repressors for orthogonal logic gates. Nat. Chem. Biol. 10, 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen M.; Bujard H. (1992) Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. U.S.A. 89, 5547–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen M.; Freundlieb S.; Bender G.; Muller G.; Hillen W.; Bujard H. (1995) Transcriptional activation by tetracyclines in mammalian cells. Science 268, 1766–1769. [DOI] [PubMed] [Google Scholar]
- Agha-Mohammadi S.; Lotze M. T. (2000) Regulatable systems: Applications in gene therapy and replicating viruses. J. Clin. Invest. 105, 1177–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urlinger S.; Baron U.; Thellmann M.; Hasan M. T.; Bujard H.; Hillen W. (2000) Exploring the sequence space for tetracycline-dependent transcriptional activators: Novel mutations yield expanded range and sensitivity. Proc. Natl. Acad. Sci. U.S.A. 97, 7963–7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieyra D. S.; Goodell M. A. (2007) Pluripotentiality and conditional transgene regulation in human embryonic stem cells expressing insulated tetracycline-ON transactivator. Stem Cells 25, 2559–2566. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Chen J.; Ren J.; Bao L.; Liao J.; Cui C.; Rao L.; Li H.; Gu Y.; Dai H.; Zhu H.; Teng X.; Cheng L.; Xiao L. (2009) Generation of pig induced pluripotent stem cells with a drug-inducible system. J. Mol. Cell Biol. 1, 46–54. [DOI] [PubMed] [Google Scholar]
- Zhao X.; Yu Y.; Zhao Z.; Guo J.; Fu L.; Yu T.; Hou L.; Yi S.; Chen W. (2012) Establishment of tetracycline-inducible, survivin-expressing CHO cell lines by an optimized screening method. Biosci., Biotechnol., Biochem. 76, 1909–1912. [DOI] [PubMed] [Google Scholar]
- Jones J.; Nivitchanyong T.; Giblin C.; Ciccarone V.; Judd D.; Gorfien S.; Krag S. S.; Betenbaugh M. J. (2005) Optimization of tetracycline-responsive recombinant protein production and effect on cell growth and ER stress in mammalian cells. Biotechnol. Bioeng. 91, 722–732. [DOI] [PubMed] [Google Scholar]
- Forster K.; Helbl V.; Lederer T.; Urlinger S.; Wittenburg N.; Hillen W. (1999) Tetracycline-inducible expression systems with reduced basal activity in mammalian cells. Nucleic Acids Res. 27, 708–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Wang C.; Ke N.; Bliesath J.; Chionis J.; He Q. S.; Li Q. X.; Chatterton J. E.; Wong-Staal F.; Zhou D. (2007) A more efficient RNAi inducible system for tight regulation of gene expression in mammalian cells and xenograft animals. RNA 13, 1375–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez E.; No D.; West A.; Evans R. M. (1997) Inducible gene expression in mammalian cells and transgenic mice. Curr. Opin. Biotechnol. 8, 608–616. [DOI] [PubMed] [Google Scholar]
- Auslander S.; Fussenegger M. (2013) From gene switches to mammalian designer cells: Present and future prospects. Trends Biotechnol. 31, 155–168. [DOI] [PubMed] [Google Scholar]
- Weber W.; Fux C.; Daoud-el Baba M.; Keller B.; Weber C. C.; Kramer B. P.; Heinzen C.; Aubel D.; Bailey J. E.; Fussenegger M. (2002) Macrolide-based transgene control in mammalian cells and mice. Nat. Biotechnol. 20, 901–907. [DOI] [PubMed] [Google Scholar]
- Fussenegger M.; Morris R. P.; Fux C.; Rimann M.; von Stockar B.; Thompson C. J.; Bailey J. E. (2000) Streptogramin-based gene regulation systems for mammalian cells. Nat. Biotechnol. 18, 1203–1208. [DOI] [PubMed] [Google Scholar]
- Mullick A.; Xu Y.; Warren R.; Koutroumanis M.; Guilbault C.; Broussau S.; Malenfant F.; Bourget L.; Lamoureux L.; Lo R.; Caron A. W.; Pilotte A.; Massie B. (2006) The cumate gene-switch: A system for regulated expression in mammalian cells. BMC Biotechnol. 6, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber W.; Rimann M.; Spielmann M.; Keller B.; Daoud-El Baba M.; Aubel D.; Weber C. C.; Fussenegger M. (2004) Gas-inducible transgene expression in mammalian cells and mice. Nat. Biotechnol. 22, 1440–1444. [DOI] [PubMed] [Google Scholar]
- Malphettes L.; Weber C. C.; El-Baba M. D.; Schoenmakers R. G.; Aubel D.; Weber W.; Fussenegger M. (2005) A novel mammalian expression system derived from components coordinating nicotine degradation in arthrobacter nicotinovorans pAO1. Nucleic Acids Res. 33, e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber W.; Schoenmakers R.; Spielmann M.; El-Baba M. D.; Folcher M.; Keller B.; Weber C. C.; Link N.; van de Wetering P.; Heinzen C.; Jolivet B.; Sequin U.; Aubel D.; Thompson C. J.; Fussenegger M. (2003) Streptomyces-derived quorum-sensing systems engineered for adjustable transgene expression in mammalian cells and mice. Nucleic Acids Res. 31, e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neddermann P.; Gargioli C.; Muraglia E.; Sambucini S.; Bonelli F.; De Francesco R.; Cortese R. (2003) A novel, inducible, eukaryotic gene expression system based on the quorum-sensing transcription factor TraR. EMBO Rep. 4, 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M.; Hafner M.; Sontag E.; Davidsohn N.; Subramanian S.; Purnick P. E.; Lauffenburger D.; Weiss R. (2012) Modular design of artificial tissue homeostasis: Robust control through synthetic cellular heterogeneity. PLoS Comput. Biol. 8, e1002579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartenbach S.; Daoud-El Baba M.; Weber W.; Fussenegger M. (2007) An engineered l-arginine sensor of Chlamydia pneumoniae enables arginine-adjustable transcription control in mammalian cells and mice. Nucleic Acids Res. 35, e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacchus W.; Weber W.; Fussenegger M. (2013) Increasing the dynamic control space of mammalian transcription devices by combinatorial assembly of homologous regulatory elements from different bacterial species. Metab. Eng. 15, 144–150. [DOI] [PubMed] [Google Scholar]
- Gitzinger M.; Kemmer C.; Fluri D. A.; El-Baba M. D.; Weber W.; Fussenegger M. (2012) The food additive vanillic acid controls transgene expression in mammalian cells and mice. Nucleic Acids Res. 40, e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitzinger M.; Kemmer C.; El-Baba M. D.; Weber W.; Fussenegger M. (2009) Controlling transgene expression in subcutaneous implants using a skin lotion containing the apple metabolite phloretin. Proc. Natl. Acad. Sci. U.S.A. 106, 10638–10643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber W.; Lienhart C.; Baba M. D.; Fussenegger M. (2009) A biotin-triggered genetic switch in mammalian cells and mice. Metab. Eng. 11, 117–124. [DOI] [PubMed] [Google Scholar]
- Weber W.; Stelling J.; Rimann M.; Keller B.; Daoud-El Baba M.; Weber C. C.; Aubel D.; Fussenegger M. (2007) A synthetic time-delay circuit in mammalian cells and mice. Proc. Natl. Acad. Sci. U.S.A. 104, 2643–2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemmer C.; Gitzinger M.; Daoud-El Baba M.; Djonov V.; Stelling J.; Fussenegger M. (2010) Self-sufficient control of urate homeostasis in mice by a synthetic circuit. Nat. Biotechnol. 28, 355–360. [DOI] [PubMed] [Google Scholar]
- Weber W.; Schoenmakers R.; Keller B.; Gitzinger M.; Grau T.; Daoud-El Baba M.; Sander P.; Fussenegger M. (2008) A synthetic mammalian gene circuit reveals antituberculosis compounds. Proc. Natl. Acad. Sci. U.S.A. 105, 9994–9998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland M.; Fussenegger M. (2012) Engineering molecular circuits using synthetic biology in mammalian cells. Annu. Rev. Chem. Biomol. Eng. 3, 209–234. [DOI] [PubMed] [Google Scholar]
- Deans T. L.; Cantor C. R.; Collins J. J. (2007) A tunable genetic switch based on RNAi and repressor proteins for regulating gene expression in mammalian cells. Cell 130, 363–372. [DOI] [PubMed] [Google Scholar]
- Kramer B. P.; Fussenegger M. (2005) Hysteresis in a synthetic mammalian gene network. Proc. Natl. Acad. Sci. U.S.A. 102, 9517–9522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer B. P.; Viretta A. U.; Daoud-El-Baba M.; Aubel D.; Weber W.; Fussenegger M. (2004) An engineered epigenetic transgene switch in mammalian cells. Nat. Biotechnol. 22, 867–870. [DOI] [PubMed] [Google Scholar]
- Kramer B. P.; Weber W.; Fussenegger M. (2003) Artificial regulatory networks and cascades for discrete multilevel transgene control in mammalian cells. Biotechnol. Bioeng. 83, 810–820. [DOI] [PubMed] [Google Scholar]
- Zhao W.; Bonem M.; McWhite C.; Silberg J. J.; Segatori L. (2014) Sensitive detection of proteasomal activation using the Deg-On mammalian synthetic gene circuit. Nat. Commun. 5, 3612. [DOI] [PubMed] [Google Scholar]
- Auslander S.; Auslander D.; Muller M.; Wieland M.; Fussenegger M. (2012) Programmable single-cell mammalian biocomputers. Nature 487, 123–127. [DOI] [PubMed] [Google Scholar]
- Leisner M.; Bleris L.; Lohmueller J.; Xie Z.; Benenson Y. (2010) Rationally designed logic integration of regulatory signals in mammalian cells. Nat. Nanotechnol. 5, 666–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z.; Wroblewska L.; Prochazka L.; Weiss R.; Benenson Y. (2011) Multi-input RNAi-based logic circuit for identification of specific cancer cells. Science 333, 1307–1311. [DOI] [PubMed] [Google Scholar]
- Tigges M.; Marquez-Lago T. T.; Stelling J.; Fussenegger M. (2009) A tunable synthetic mammalian oscillator. Nature 457, 309–312. [DOI] [PubMed] [Google Scholar]
- Tigges M.; Denervaud N.; Greber D.; Stelling J.; Fussenegger M. (2010) A synthetic low-frequency mammalian oscillator. Nucleic Acids Res. 38, 2702–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrill D. R.; Inniss M. C.; Boyle P. M.; Silver P. A. (2012) Synthetic memory circuits for tracking human cell fate. Genes Dev. 26, 1486–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacchus W.; Lang M.; El-Baba M. D.; Weber W.; Stelling J.; Fussenegger M. (2012) Synthetic two-way communication between mammalian cells. Nat. Biotechnol. 30, 991–996. [DOI] [PubMed] [Google Scholar]
- Weber W.; Schuetz M.; Denervaud N.; Fussenegger M. (2009) A synthetic metabolite-based mammalian inter-cell signaling system. Mol. BioSyst. 5, 757–763. [DOI] [PubMed] [Google Scholar]
- Weber W.; Kramer B. P.; Fussenegger M. (2007) A genetic time-delay circuitry in mammalian cells. Biotechnol. Bioeng. 98, 894–902. [DOI] [PubMed] [Google Scholar]
- Kramer B. P.; Fischer C.; Fussenegger M. (2004) BioLogic gates enable logical transcription control in mammalian cells. Biotechnol. Bioeng. 87, 478–484. [DOI] [PubMed] [Google Scholar]
- Folcher M.; Xie M.; Spinnler A.; Fussenegger M. (2013) Synthetic mammalian trigger-controlled bipartite transcription factors. Nucleic Acids Res. 41, e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurt J. A.; Thibodeau S. A.; Hirsh A. S.; Pabo C. O.; Joung J. K. (2003) Highly specific zinc finger proteins obtained by directed domain shuffling and cell-based selection. Proc. Natl. Acad. Sci. U.S.A. 100, 12271–12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boch J.; Scholze H.; Schornack S.; Landgraf A.; Hahn S.; Kay S.; Lahaye T.; Nickstadt A.; Bonas U. (2009) Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326, 1509–1512. [DOI] [PubMed] [Google Scholar]
- Qi L. S.; Larson M. H.; Gilbert L. A.; Doudna J. A.; Weissman J. S.; Arkin A. P.; Lim W. A. (2013) Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert L. A.; Larson M. H.; Morsut L.; Liu Z.; Brar G. A.; Torres S. E.; Stern-Ginossar N.; Brandman O.; Whitehead E. H.; Doudna J. A.; Lim W. A.; Weissman J. S.; Qi L. S. (2013) CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil A. S.; Lu T. K.; Bashor C. J.; Ramirez C. L.; Pyenson N. C.; Joung J. K.; Collins J. J. (2012) A synthetic biology framework for programming eukaryotic transcription functions. Cell 150, 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Cong L.; Lodato S.; Kosuri S.; Church G. M.; Arlotta P. (2011) Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat. Biotechnol. 29, 149–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A.; Lohmueller J. J.; Silver P. A.; Armel T. Z. (2012) Engineering synthetic TAL effectors with orthogonal target sites. Nucleic Acids Res. 40, 7584–7595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiani S.; Beal J.; Ebrahimkhani M. R.; Huh J.; Hall R. N.; Xie Z.; Li Y.; Weiss R. (2014) CRISPR transcriptional repression devices and layered circuits in mammalian cells. Nat. Methods 11, 723–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaber R.; Lebar T.; Majerle A.; Ster B.; Dobnikar A.; Bencina M.; Jerala R. (2014) Designable DNA-binding domains enable construction of logic circuits in mammalian cells. Nat. Chem. Biol. 10, 203–208. [DOI] [PubMed] [Google Scholar]
- Maeder M. L.; Linder S. J.; Cascio V. M.; Fu Y.; Ho Q. H.; Joung J. K. (2013) CRISPR RNA-guided activation of endogenous human genes. Nat. Methods 10, 977–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P.; Aach J.; Stranges P. B.; Esvelt K. M.; Moosburner M.; Kosuri S.; Yang L.; Church G. M. (2013) CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 31, 833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pinera P.; Kocak D. D.; Vockley C. M.; Adler A. F.; Kabadi A. M.; Polstein L. R.; Thakore P. I.; Glass K. A.; Ousterout D. G.; Leong K. W.; Guilak F.; Crawford G. E.; Reddy T. E.; Gersbach C. A. (2013) RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 10, 973–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzadfard F.; Perli S. D.; Lu T. K. (2013) Tunable and multifunctional eukaryotic transcription factors based on CRISPR/Cas. ACS Synth. Biol. 2, 604–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera V. M.; Clackson T.; Natesan S.; Pollock R.; Amara J. F.; Keenan T.; Magari S. R.; Phillips T.; Courage N. L.; Cerasoli F. Jr.; Holt D. A.; Gilman M. (1996) A humanized system for pharmacologic control of gene expression. Nat. Med. 2, 1028–1032. [DOI] [PubMed] [Google Scholar]
- Beerli R. R.; Schopfer U.; Dreier B.; Barbas C. F. 3rd. (2000) Chemically regulated zinc finger transcription factors. J. Biol. Chem. 275, 32617–32627. [DOI] [PubMed] [Google Scholar]
- Li Y.; Moore R.; Guinn M.; Bleris L. (2012) Transcription activator-like effector hybrids for conditional control and rewiring of chromosomal transgene expression. Sci. Rep. 2, 897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S.; Brigham M. D.; Trevino A. E.; Hsu P. D.; Heidenreich M.; Cong L.; Platt R. J.; Scott D. A.; Church G. M.; Zhang F. (2013) Optical control of mammalian endogenous transcription and epigenetic states. Nature 500, 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polstein L. R.; Gersbach C. A. (2012) Light-inducible spatiotemporal control of gene activation by customizable zinc finger transcription factors. J. Am. Chem. Soc. 134, 16480–16483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; J Y.; Chen H.; Liao W.; Weiss R.; Xie Z.. Modular construction of synthetic circuits using TALE transcriptional repressors in mammalian cells. Nat. Chem. Biol. 2014, under review [DOI] [PMC free article] [PubMed]
- Banaszynski L. A.; Chen L. C.; Maynard-Smith L. A.; Ooi A. G.; Wandless T. J. (2006) A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 126, 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski I.; Ma J.; Triezenberg S.; Ptashne M. (1988) GAL4-VP16 is an unusually potent transcriptional activator. Nature 335, 563–564. [DOI] [PubMed] [Google Scholar]
- Kim D. W.; Uetsuki T.; Kaziro Y.; Yamaguchi N.; Sugano S. (1990) Use of the human elongation factor 1 α promoter as a versatile and efficient expression system. Gene 91, 217–223. [DOI] [PubMed] [Google Scholar]
- Guye P.; Li Y.; Wroblewska L.; Duportet X.; Weiss R. (2013) Rapid, modular, and reliable construction of complex mammalian gene circuits. Nucleic Acids Res. 41, e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loser P.; Jennings G. S.; Strauss M.; Sandig V. (1998) Reactivation of the previously silenced cytomegalovirus major immediate-early promoter in the mouse liver: Involvement of NFkappaB. J. Virol. 72, 180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livet J.; Weissman T. A.; Kang H.; Draft R. W.; Lu J.; Bennis R. A.; Sanes J. R.; Lichtman J. W. (2007) Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 450, 56–62. [DOI] [PubMed] [Google Scholar]
- Schnider-Keel U.; Seematter A.; Maurhofer M.; Blumer C.; Duffy B.; Gigot-Bonnefoy C.; Reimmann C.; Notz R.; Defago G.; Haas D.; Keel C. (2000) Autoinduction of 2,4-diacetylphloroglucinol biosynthesis in the biocontrol agent Pseudomonas fluorescens CHA0 and repression by the bacterial metabolites salicylate and pyoluteorin. J. Bacteriol. 182, 1215–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangera M. G.; Thomashow L. S. (1999) Identification and characterization of a gene cluster for synthesis of the polyketide antibiotic 2,4-diacetylphloroglucinol from Pseudomonas fluorescens Q2–87. J. Bacteriol. 181, 3155–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree B. (1976) Theoretical considerations of the sensitivity conferred by substrate cycles in vivo. Biochem. Soc. Trans. 4, 999–1002. [DOI] [PubMed] [Google Scholar]
- Weiss R. (2001) Cellular Computation and Communications Using Engineered Genetic Regulatory Networks, p 1, Massachusetts Institute of Technology, Cambridge, MA. [Google Scholar]
- Yokobayashi Y.; Weiss R.; Arnold F. H. (2002) Directed evolution of a genetic circuit. Proc. Natl. Acad. Sci. U.S.A. 99, 16587–16591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamsir A.; Tabor J. J.; Voigt C. A. (2011) Robust multicellular computing using genetically encoded NOR gates and chemical ‘wires’. Nature 469, 212–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly J. R.; Rubin A. J.; Davis J. H.; Ajo-Franklin C. M.; Cumbers J.; Czar M. J.; de Mora K.; Glieberman A. L.; Monie D. D.; Endy D. (2009) Measuring the activity of BioBrick promoters using an in vivo reference standard. J. Biol. Eng. 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon T. S.; Lou C.; Tamsir A.; Stanton B. C.; Voigt C. A. (2012) Genetic programs constructed from layered logic gates in single cells. Nature 491, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou C.; Stanton B.; Chen Y. J.; Munsky B.; Voigt C. A. (2012) Ribozyme-based insulator parts buffer synthetic circuits from genetic context. Nat. Biotechnol. 30, 1137–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidsohn N.; Beal J.; Kiani S.; Adler A.; Yaman F.; Li Y.; Xie Z.; Weiss R. (2014) Accurate predictions of genetic circuit behavior from part characterization and modular composition. ACS Synth. Biol. 10.1021/sb500263b. [DOI] [PubMed] [Google Scholar]
- Anderson J. C.; Voigt C. A.; Arkin A. P. (2007) Environmental signal integration by a modular AND gate. Mol. Syst. Biol. 3, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin A. P. (2001) Synthetic cell biology. Curr. Opin. Biotechnol. 12, 638–644. [DOI] [PubMed] [Google Scholar]
- Purcell O.; Lu T. K. (2014) Synthetic analog and digital circuits for cellular computation and memory. Curr. Opin. Biotechnol. 29C, 146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z. L.; Shao S. L.; Lv J. W.; Wang C. H.; Yuan C. Z.; Zhang W. W.; Xu X. J. (2011) Co-transfection and tandem transfection of HEK293A cells for overexpression and RNAi experiments. Cell Biol. Int. 35, 187–192. [DOI] [PubMed] [Google Scholar]
- Atchison M. L. (1988) Enhancers: Mechanisms of action and cell specificity. Annu. Rev. Cell Biol. 4, 127–153. [DOI] [PubMed] [Google Scholar]
- Garnett A. T.; Square T. A.; Medeiros D. M. (2012) BMP, Wnt, and FGF signals are integrated through evolutionarily conserved enhancers to achieve robust expression of Pax3 and Zic genes at the zebrafish neural plate border. Development 139, 4220–4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erceg J.; Saunders T. E.; Girardot C.; Devos D. P.; Hufnagel L.; Furlong E. E. (2014) Subtle changes in motif positioning cause tissue-specific effects on robustness of an enhancer’s activity. PLoS Genet. 10, e1004060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossett L. A.; Kelvin D. J.; Sternberg E. A.; Olson E. N. (1989) A new myocyte-specific enhancer-binding factor that recognizes a conserved element associated with multiple muscle-specific genes. Mol. Cell. Biol. 9, 5022–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtha M.; Tokcaer-Keskin Z.; Tang Z.; Strino F.; Chen X.; Wang Y.; Xi X.; Basilico C.; Brown S.; Bonneau R.; Kluger Y.; Dailey L. (2014) FIREWACh: High-throughput functional detection of transcriptional regulatory modules in mammalian cells. Nat. Methods 11, 559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickel D. E.; Zhu Y.; Nord A. S.; Wylie J. N.; Akiyama J. A.; Afzal V.; Plajzer-Frick I.; Kirkpatrick A.; Gottgens B.; Bruneau B. G.; Visel A.; Pennacchio L. A. (2014) Function-based identification of mammalian enhancers using site-specific integration. Nat. Methods 11, 566–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuan D. Y.; Solomon W. B.; London I. M.; Lee D. P. (1989) An erythroid-specific, developmental-stage-independent enhancer far upstream of the human “beta-like globin” genes. Proc. Natl. Acad. Sci. U.S.A. 86, 2554–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser P.; Hurst J.; Collis P.; Grosveld F. (1990) DNaseI hypersensitive sites 1, 2, and 3 of the human beta-globin dominant control region direct position-independent expression. Nucleic Acids Res. 18, 3503–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug B. A.; Moon A. M.; Ley T. J. (1992) Structure and function of the murine beta-globin locus control region 5′ HS-3. Nucleic Acids Res. 20, 5771–5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way J. C.; Collins J. J.; Keasling J. D.; Silver P. A. (2014) Integrating biological redesign: Where synthetic biology came from and where it needs to go. Cell 157, 151–161. [DOI] [PubMed] [Google Scholar]
- Fath S.; Bauer A. P.; Liss M.; Spriestersbach A.; Maertens B.; Hahn P.; Ludwig C.; Schafer F.; Graf M.; Wagner R. (2011) Multiparameter RNA and codon optimization: A standardized tool to assess and enhance autologous mammalian gene expression. PloS One 6, e17596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley J. L.; Temple G. F.; Brasch M. A. (2000) DNA cloning using in vitro site-specific recombination. Genome Res. 10, 1788–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A. T.; Zhou X.; Vink M.; Klaver B.; Verhoef K.; Marzio G.; Berkhout B. (2004) Viral evolution as a tool to improve the tetracycline-regulated gene expression system. J. Biol. Chem. 279, 18776–18782. [DOI] [PubMed] [Google Scholar]
- Nolden L.; Ngouoto-Nkili C. E.; Bendt A. K.; Kramer R.; Burkovski A. (2001) Sensing nitrogen limitation in Corynebacterium glutamicum: The role of glnK and glnD. Molecular Microbiol. 42, 1281–1295. [DOI] [PubMed] [Google Scholar]
- Shaw G. C.; Fulco A. J. (1993) Inhibition by barbiturates of the binding of Bm3R1 repressor to its operator site on the barbiturate-inducible cytochrome P450BM-3 gene of Bacillus megaterium. J. Biol. Chem. 268, 2997–3004. [PubMed] [Google Scholar]
- Jeng W. Y.; Ko T. P.; Liu C. I.; Guo R. T.; Liu C. L.; Shr H. L.; Wang A. H. (2008) Crystal structure of IcaR, a repressor of the TetR family implicated in biofilm formation in Staphylococcus epidermidis. Nucleic Acids Res. 36, 1567–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata M.; Ohno S.; Kumano M.; Yamane K.; Ohki R. (2003) Multidrug resistant phenotype of Bacillus subtilis spontaneous mutants isolated in the presence of puromycin and lincomycin. Can. J. Microbiol. 49, 71–77. [DOI] [PubMed] [Google Scholar]
- Rey D. A.; Puhler A.; Kalinowski J. (2003) The putative transcriptional repressor McbR, member of the TetR-family, is involved in the regulation of the metabolic network directing the synthesis of sulfur containing amino acids in Corynebacterium glutamicum. J. Biotechnol. 103, 51–65. [DOI] [PubMed] [Google Scholar]
- Grkovic S.; Hardie K. M.; Brown M. H.; Skurray R. A. (2003) Interactions of the QacR multidrug-binding protein with structurally diverse ligands: Implications for the evolution of the binding pocket. Biochemistry 42, 15226–15236. [DOI] [PubMed] [Google Scholar]
- Bryan L. (1984) Antimicrobial Drug Resistance, Elsevier Science, Amsterdam. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.