

Abstract
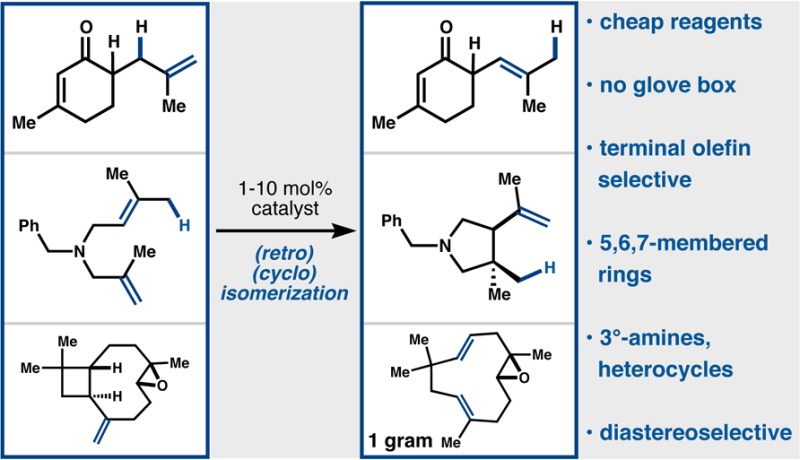
Catalytic amounts of Co(SaltBu,tBu)Cl and organosilane irreversibly isomerize terminal alkenes by one position. The same catalysts effect cycloisomerization of dienes and retrocycloisomerization of strained rings. Strong Lewis bases like amines and imidazoles, and labile functionalities like epoxides, are tolerated.
Metal-catalyzed alkene isomerization1−3 is a powerful chemical transformation that achieves what classical techniques cannot—a [1,3]-hydrogen shift—but can be limited by the ability of carbophilic Lewis acids to coordinate weakly Lewis basic and weakly back-bonding alkenes.4 Similarly, metal-catalyzed alkene cycloisomerization5 can achieve the equivalent of a pericyclic ene reaction under generally mild conditions but usually requires one or more alkyne, allene, or butadiene functions due to the facile coordination of these species with transition metals, especially Pd.6 Cycloisomerizations7,8 involving only nonconjugated alkenes are thus inhibited by strong Lewis bases like nitrogenous heterocycles or aliphatic amines.9,10 Here we report alkene isomerizations, polyene cycloisomerizations, and alkene retrocycloisomerizations that tolerate Lewis basic functionality, including amines, imidazoles, and epoxides, all triggered by a Co(SaltBu,tBu)Cl/silane catalyst system.11 Experimental observations combined with literature precedent suggest a radical isomerization12,13 via reversible hydrogen atom transfer (HAT).14,15 Notably, the efficiency of the reaction appears to depend on the persistence of an intermediate carbon-centered radical, which is tied to the stability of a metal-ligand “counter-radical”. These observations are consistent with the effect of cobalt-ligand electronics on polydispersity and chain length in reversibly terminated radical polymerization.15,16
We recently reported Mn- and Co-catalyzed alkene hydrogenations that deliver thermodynamically preferred stereoisomers via a radical mechanism.17 We proposed that the first step of the reaction involves HAT from a metal hydride,18,19 which may be operative in analogous Mukaiyama-type20 radical hydrofunctionalization reactions.21−26 Since mechanistic studies of known HAT-mediated reactions implicate reversible carbon radical formation,14a,14b it seemed reasonable that an appropriate ligand sphere on Mn or Co might promote an alkene isomerization along a radical reaction path and prevent reduction (Figure 1).27
Figure 1.
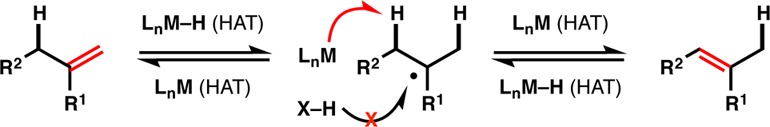
Can isomerization predominate over hydrogenation in a HAT equilibrium?
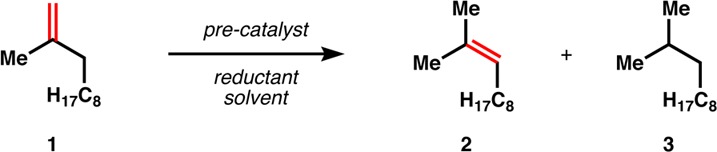
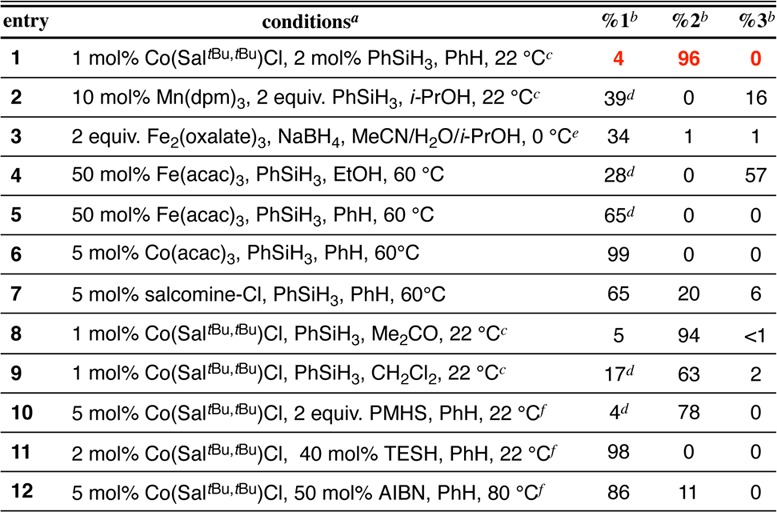
A screen of metal catalysts, reductants, and solvents that are known to effect Mukaiyama-type radical hydrofunctionalization was conducted in search of isomerization reactivity (1→2, Table 1). We found that Co-salen complexes28,29 in benzene with phenylsilane as a H source under anaerobic conditions afforded a high yield of isomer and little to no hydrogenation (entries 1,8–10), whereas most other metal complexes known to participate in radical hydrofunctionalization gave either reduction or poor conversion (entries 2–6). Phenylsilane proved to be the best reductant. Polymethylhydrosiloxane (PMHS) could also be used, but Et3SiH (TESH) failed (entries 10,11).
Table 1. Products from Common Mukaiyama Conditions.
Under Ar, 1 h unless noted.
According to GC-FID.
After 3 h.
Multiple unidentified products were observed.
Under air.
After 24 h.
Since the metal complex abstracts a H-atom from the reaction intermediate, silane can be used catalytically (2–10 mol%). In a similar way, AIBN functioned as a viable source of H-atom at elevated temperature (entry 12), in support of a radical mechanism for reversible HAT and analogous to the mechanism associated with Co-porphyrin/AIBN living radical polymerization.30,31 Exclusion of either AIBN or the Co catalyst resulted in no isomerized product (see Supporting Information).
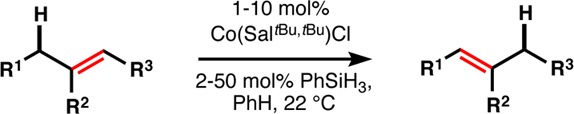
Due to ease of handling, Co(SaltBu,tBu)Cl—a crystalline, air- and moisture-stable complex—formed the basis of an exploration of substrate scope (Table 2).32 The choice of axial ligand (Cl, F, OAc) had only a minor influence on yield in a few cases. We quickly found that 1,1-disubstituted alkenes exhibited rapid reaction rates at room temperature (entry 1), whereas monosubstituted alkenes required elevated temperatures for high conversion (entry 2). The basis for the difference in temperature is discussed below. Other substitution patterns were less reactive, which allowed isomerizations to occur over a single position and then stop (six other minor decene isomers33 totaled 14%, av 2% each). Thus, the isomerization appears not to be an equilibrium to the most stable alkene, but rather an irreversible reaction that discriminates against steric bulk, so that isomerization to a skipped diene (entry 3) proceeds without further isomerization to the conjugated diene, and isomerization to a homostyrene is possible (entry 4). Nevertheless, isomerization of unsymmetrical alkenes (entry 5) selectively provides the tetrasubstituted alkene instead of the trisubstituted isomer, probably reflective of a lower activation energy for methine C–H abstraction compared to the methylene C–H.34 Electron-deficient alkenes are cleanly isomerized to greater substitution (entry 6), which may be useful given the high prevalence but promiscuity of methylene lactone and ester motifs in complex, bioactive molecules. Homoallylic ethers may be isomerized to allyl ethers without further isomerization (entry 7), but given the proper substitution patterns, isomerization into conjugation with oxygen is possible (entry 8). Consequently, isomerization of a bis-allylic silyl ether can give rise to an aldol enolsilane (entry 9), and the mildness of the conditions prevents hydrolysis or elimination. Similarly, allyl ketones can be isomerized into vinyl ketones with no conjugation (entry 10).
Table 2. Survey of Terminal Alkene Isomerization.
2 mol% [Co], 2 mol% [Si].
10 mol% [Co], 50 mol% [Si] at 60 °C.
86:14 2-decene:decenes.
5 mol% [Co], 10 mol% [Si].
3 mol% [Co], 6 mol% [Si].
1 mol% [Co], 2 mol% [Si].
Given the established radical nature of intermediates in Mukaiyama hydrofunctionalization reactions and prior work in radical clock reactions,18b,21f,24,35 we wondered if cyclization could outcompete HAT to the metal, thus providing a mechanistically unique cycloisomerization of a polyene and the equivalent of a pericyclic ene reaction. Indeed, these reactions work well to provide cycloisomerized products in good yield and diastereoselectivity. For instance, allyl prenyl and methallyl prenyl malonates cyclize cleanly to the disubstituted cyclopentanes 4 and 5. Six-membered rings like 6 are also kinetically viable, although an inseparable linear isomer was produced as a minor product (this was optimized, see Figure 3, below). Polyenes (7) and rings (8, 9) are competent acceptors, as are polarized alkenes like enolsilanes (10), enabling a mild Conia-ene-like reaction that is difficult to achieve with Brønsted acid catalysis. Similarly, cycloisomerization is effective on aromatic rings (11, 12), unlike most metal-catalyzed processes, and under exceptionally mild conditions compared to Friedel-Crafts reactions. Diesters are not crucial, as 13 can be produced, albeit as a mixture of diastereomers. Thorpe-Ingold acceleration is also not necessary to achieive competitive cyclization vs isomerization, as tetrahydrofurans 14–17 are produced cleanly. Remarkably, tertiary amines (18) and imidazoles (19) show no inhibition of catalysis. Also, reflecting the radical nature of the reaction, 4.1.0-bicycle 20 undergoes isomerization to 21 via cleavage of the cyclopropane36 and addition to the pendant aromatic ring.
Figure 3.
Effects of electronics and temperature.
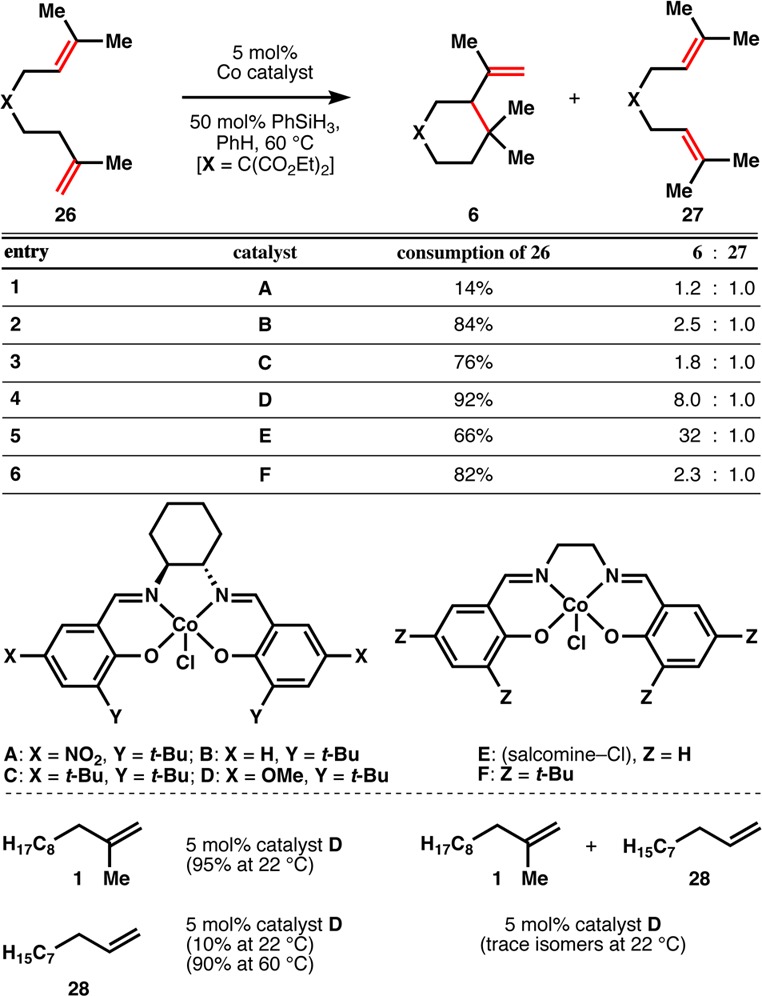
Analogous to this last example is the retrocycloisomerization of (−)-caryophyllene oxide (22, Figure 2), which is carried out under our standard conditions and provides a high yield of (−)-humulene oxide II (23). This showcase reaction enables the production of an expensive fragrance and flavor (23, $600/5 mg, Cheminstock) from a cheap commercial source (22, $0.28/g, SigmaAldrich) and offers this HAT isomerization as a useful general method for modification of feedstock chemicals to high-value materials. The strained terpene β-funebrene (24) can also be cleanly isomerized to α-funebrene (25) with no decomposition, in contrast to acidic conditions.37
Figure 2.
Modification of complex molecules.
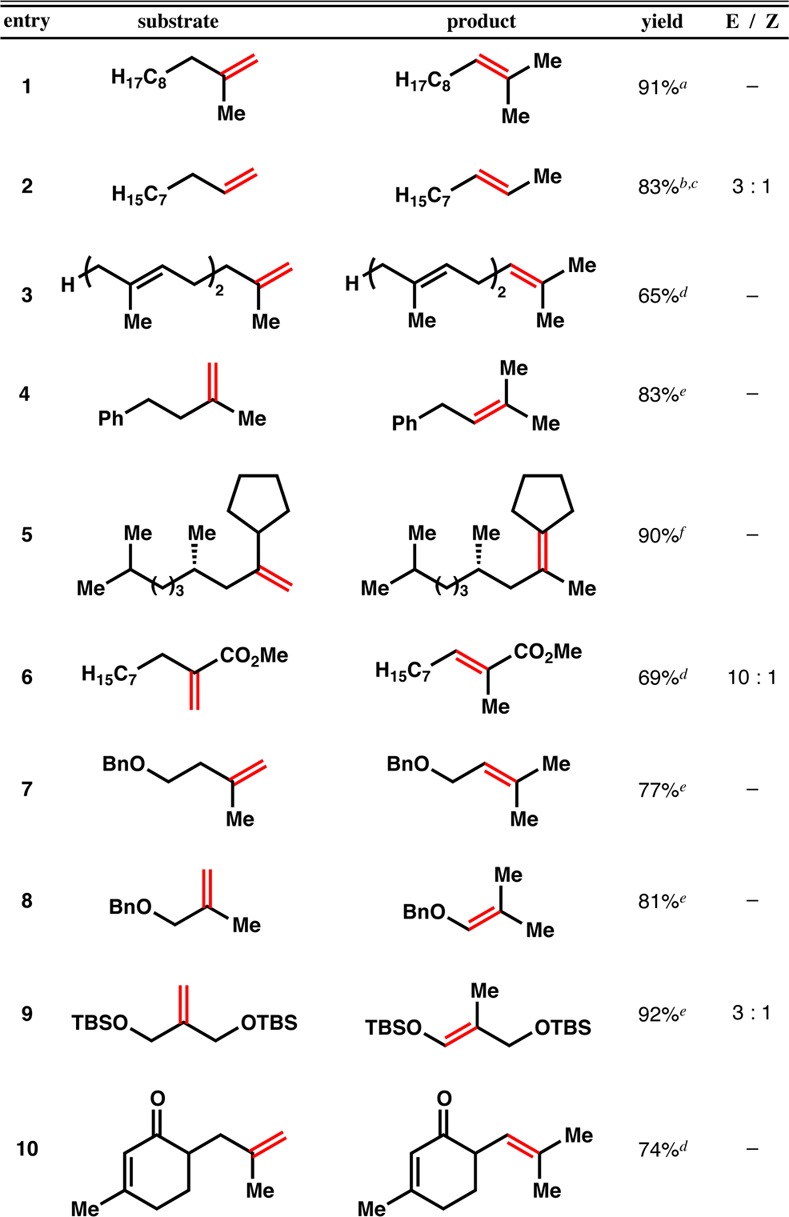
The efficiency of cycloisomerization under these conditions is related to the persistence of the intermediate C-centered radical, as measured by unimolecular cyclization, i.e., a radical clock competition against HAT to the metal-ligand complex. Since the cycloisomerization of 6 under standard conditions provided ∼21% of the uncyclized isomer (Table 3 and Figure 3), we used this substrate to probe the effects of ligand electronics on radical persistence. An electron-deficient ligand (complex A) shows poor conversion, but more importantly it effects a low ratio of cycloisomerization to linear isomerization. Electron-neutral ligands (B,C) show moderate selectivity for cyclization, and an electron-rich ligand (D) amplifies cyclization. These effects of ligand electronics are consistent with equivalent changes in reversible HAT and metal-carbon radical collapse in polymerizations mediated by Co-salen complexes.15c That is, electron-deficient complexes terminate the carbon radical, whereas electron-rich complexes allow its persistence. Salcomine-Cl (E) is highly selective for cyclization, although we found it to be a poor catalyst for linear isomerization (Table 1, entry 7), consistent with radical capture (see Figure 4). The t-Bu groups of catalyst F increase conversion but decrease cyclization.
Table 3. Survey of Diene Cycloisomerization.
At 60 °C.
5 mol% [Co], 10 mol% [Si].
3 mol% [Co], 6 mol% [Si].
at 100 °C.
6 mol% [Co], 12 mol% [Si].
Stereoisomer tentatively assigned by NOE.
Figure 4.
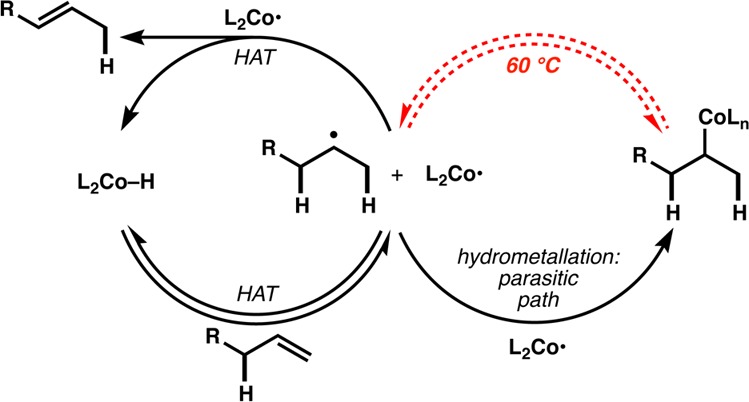
Hypothetical catalytic cycle.
In essence, our work is a unimolecular variant of catalytic chain transfer and living radical polymerization reactions,16 as well as a link between Mukaiyama hydrofunctionalizations17−26 and HAT studies.14 Polymerizations using Co-salen catalysts implicate the intermediacy of a reactive, transient, and unobservable metal hydride as a product of reversible termination,15,38 and Nojima observed metal hydrides in Mukaiyama-type reactions.35 It remains unclear in individual reports whether hydrofunctionalization proceeds via initial hydrometalation or HAT,39 but the low turnover number associated with the isomerization of 28 with D at ambient temperature, and its inhibition of the isomerization of 1 (Figure 3), are consistent with carbon radical/metal collapse as an off-cycle, parasitic pathway; i.e., the hydrometalation product does not lie within the catalytic cycle (see Figure 4). Elevated temperature provides the activation energy to promote C–Co bond homolysis15c,38 and re-engage the catalytic cycle. It is unknown whether collapse occurs in the solvent cage.40 HAT is further supported by the absence of a proximal, empty valence for hydrometalation or cyclometalation on the putative Co-salen hydride complex.31 The isomerization appears to be under kinetic, not thermodynamic control, since isopentenyl prenyl malonate 26 cycloisomerizes, whereas diprenyl malonate 27 does not, even though both species would give rise to the same radical intermediate subject to the same unimolecular cyclization rate. A tentative catalytic cycle based on these observations is shown in Figure 4.
In summary, we have shown that catalytic amounts of Co(salent-Bu,t-Bu)Cl and organosilanes effect the isomerization of terminal alkenes, in the form of cycloisomerization if a pendant alkene is available and retrocycloisomerization if the alkene is adjacent to a strained ring. These reactions likely proceed by reversible hydrogen atom transfer to generate a transient carbon-centered radical. Given the tolerance of these reactions to functional groups, and the number of permutations available to salen ligand substitution, application and expansion of these tools are likely.
Acknowledgments
Dedicated to Carlos F. Barbas, III, in memoriam. Financial support for this work was provided by the NIH (GM104180) and FRQNT (fellowship to F.B.). We thank Carla Obradors for early contributions, Professors Jin-Quan Yu and Phil Baran for helpful conversations, and Laura Pasternack for help with NMR. We are grateful to Eli Lilly, Boehringer Ingelheim, Amgen, the Baxter Foundation, and the Sloan Foundation for additional financial support.
Supporting Information Available
Experimental procedures and spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
‡ S.W.M.C. and F.B. contributed equally.
The authors declare the following competing financial interest(s): A provisional patent (U.S. no. 62/078,140) has been filed.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Yus M.; Foubelo F.. Science of Synthesis, Vol. 47b; Georg Thieme Verlag KG: Stuttgart, 2010; p 1067. [Google Scholar]
- Leading references on isomerization of electron-neutral alkenes:; a Larionov E.; Li H.; Mazet C. Chem. Commun. 2014, 50, 9816. [DOI] [PubMed] [Google Scholar]; b Larsen C. R.; Grotjahn D. B. J. Am. Chem. Soc. 2012, 134, 10357. [DOI] [PubMed] [Google Scholar]; c Chen C.; Dugan T. R.; Brennessel W. W.; Weix D. J.; Holland P. L. J. Am. Chem. Soc. 2014, 136, 945. [DOI] [PubMed] [Google Scholar]; d Gauthier D.; Lindhardt A. T.; Olsen E. P. K.; Overgaard J.; Skrydstrup T. J. Am. Chem. Soc. 2010, 132, 7998. [DOI] [PubMed] [Google Scholar]
- For isomerization of alkenes allylic to heteroatoms, see the following reviews. Allylic ethers and alcohols:; a Uma R.; Crévisy C.; Grée R. Chem. Rev. 2003, 103, 27. [DOI] [PubMed] [Google Scholar]; Allylic amines:; b Krompiec S.; Krompiec M.; Penczek R.; Ignasiak H. Coord. Chem. Rev. 2008, 252, 1819. [Google Scholar]
- Collman J. P.; Hegedus L. S.; Norton J. R.; Finke R. G.. Principles and Applications of Organotransition Metal Chemistry; University Science Books: Mill Valley, CA, 1987; p 527. [Google Scholar]
- a Trost B. M. Angew. Chem., Int. Ed. Engl. 1995, 34, 259. [Google Scholar]; b Trost B. M. Acc. Chem. Res. 2002, 35, 695. [DOI] [PubMed] [Google Scholar]
- Michelet V.; Toullec P. Y.; Genêt J.-P. Angew. Chem., Int. Ed. 2008, 47, 4268. [DOI] [PubMed] [Google Scholar]
- Comprehensive review of diene cycloisomerizations:Yamamoto Y. Chem. Rev. 2012, 112, 4736. [DOI] [PubMed] [Google Scholar]
- Widenhoefer R. A. Acc. Chem. Res. 2002, 35, 905. [DOI] [PubMed] [Google Scholar]
- Ojima I.; Donovan R. J.; Shay W. R. J. Am. Chem. Soc. 1992, 114, 6580. [Google Scholar]
- For example, see:; a Okamoto S.; Livinghouse T. J. Am. Chem. Soc. 2000, 122, 1223. [Google Scholar]; For exceptions, see:; b Takacs J. M.; Weidner J. J.; Newsome P. W.; Takacs B. E.; Chidambaram R.; Shoemaker R. J. Org. Chem. 1995, 60, 3473. [Google Scholar]; c Piers W. E.; Shapiro P. J.; Bunel E. E.; Bercaw J. E. Synlett 1990, 74. [Google Scholar]
- Olefin isomerization using HCo(CO)4:Heck R. F.; Breslow D. S. J. Am. Chem. Soc. 1961, 83, 4023. [Google Scholar]
- Halogen atom-transfer cycloisomerization:; a Clark A. J. Chem. Soc. Rev. 2002, 31, 1. [DOI] [PubMed] [Google Scholar]; b Muñoz-Molina J. M.; Belderrain T. R.; Peŕez P. J. Eur. J. Inorg. Chem. 2011, 3155. [Google Scholar]
- Alkene isomerization mediated by sulfinyl radical HAT (radical abstraction):; a Vogel P.; Turks M.; Bouchez L.; Marković D.; Varela-Álvarez A.; Sordo J. A. Acc. Chem. Res. 2007, 40, 931. [DOI] [PubMed] [Google Scholar]; Thiol-catalyzed radical isomerization:; b Fielding A. J.; Roberts B. P. Tetrahedron Lett. 2001, 42, 4061. [Google Scholar]
- Norton observed a reversible, initial HAT in hydrogenation:; a Tang L.; Papish E. T.; Abramo G. P.; Norton J. R.; Baik M.-H.; Friesner R. A.; Rappé A. J. Am. Chem. Soc. 2003, 125, 10093. [DOI] [PubMed] [Google Scholar]; b Choi J.; Tang L.; Norton J. R. J. Am. Chem. Soc. 2007, 129, 234. [DOI] [PubMed] [Google Scholar]; Isomerization and cycloisomerization were also observed in HAT hydrogenation and reductive cyclization studies:; c Li G.; Pulling M. E.; Estes D. P.; Norton J. R. J. Am. Chem. Soc. 2012, 134, 14662. [DOI] [PubMed] [Google Scholar]; d Hartung J.; Pulling M. E.; Smith D. M.; Yang D. X.; Norton J. R. Tetrahedron 2008, 64, 11822. [Google Scholar]; e Han A.; Spataru T.; Hartung J.; Li G.; Norton J. R. J. Org. Chem. 2014, 79, 1938. [DOI] [PubMed] [Google Scholar]
- a Nakano T.; Okamoto Y. ACS Symp. Ser. 1998, 685, 451. [Google Scholar]; b Liao C.-M.; Hsu C.-C.; Wang F.-S.; Wayland B. B.; Peng C.-H. Polym. Chem. 2013, 4, 3098. [Google Scholar]; c Chiang L.; Allan L. E. N.; Alcantara J.; Wang M. C. P.; Storr T.; Shaver M. P. Dalton Trans. 2014, 43, 4295. [DOI] [PubMed] [Google Scholar]
- Gridnev A. A.; Ittel S. D. Chem. Rev. 2001, 101, 3611. [DOI] [PubMed] [Google Scholar]
- Iwasaki K.; Wan K. K.; Oppedisano A.; Crossley S. W. M.; Shenvi R. A. J. Am. Chem. Soc. 2014, 136, 1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger also proposed hydrogen radical addition as the initiating step in Fe-mediated radical hydrofunctionalizations:; a Ishikawa H.; Colby D. A.; Seto S.; Va P.; Tam A.; Kakei H.; Rayl T. J.; Hwang I.; Boger D. L. J. Am. Chem. Soc. 2009, 131, 4904. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Leggans E. K.; Barker T. J.; Duncan K. K.; Boger D. L. Org. Lett. 2012, 14, 1428. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Barker T. J.; Boger D. L. J. Am. Chem. Soc. 2012, 134, 13588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzon insightfully invoked HAT in alkenyl halide reduction:King S. M.; Ma X.; Herzon S. B. J. Am. Chem. Soc. 2014, 136, 6884. [DOI] [PubMed] [Google Scholar]
- Mukaiyama T.; Yamada T. Bull. Chem. Soc. Jpn. 1995, 68, 17. [Google Scholar]
- a Gaspar B.; Waser J.; Carreira E. M. Org. Synth. 2010, 87, 88. [Google Scholar]; b Gaspar B.; Carreira E. M. J. Am. Chem. Soc. 2009, 131, 13214. [DOI] [PubMed] [Google Scholar]; c Gaspar B.; Carreira E. M. Angew. Chem., Int. Ed. 2008, 47, 5758. [DOI] [PubMed] [Google Scholar]; d Gaspar B.; Waser J.; Carreira E. M. Synthesis 2007, 3839. [Google Scholar]; e Gaspar B.; Carreira E. M. Angew.Chem., Int. Ed. 2007, 46, 4519. [DOI] [PubMed] [Google Scholar]; f Waser J.; Gaspar B.; Nambu H.; Carreira E. M. J. Am. Chem. Soc. 2006, 128, 11693. [DOI] [PubMed] [Google Scholar]; g Waser J.; Gonzaĺez-Goḿez J. C.; Nambu H.; Huber P.; Carreira E. M. Org. Lett. 2005, 7, 4249. [DOI] [PubMed] [Google Scholar]; h Waser J.; Nambu H.; Carreira E. M. J. Am. Chem. Soc. 2005, 127, 8294. [DOI] [PubMed] [Google Scholar]; i Waser J.; Carreira E. M. Angew. Chem., Int. Ed. 2004, 43, 4099. [DOI] [PubMed] [Google Scholar]; j Waser J.; Carreira E. M. J. Am. Chem. Soc. 2004, 126, 5676. [DOI] [PubMed] [Google Scholar]
- a Magnus P.; Payne A. H.; Waring M. J.; Scott D. A.; Lynch V. Tetrahedron Lett. 2000, 41, 9725. [Google Scholar]; b Magnus P.; Waring M. J.; Scott D. A. Tetrahedron Lett. 2000, 41, 9731. [Google Scholar]
- Wang L.-C.; Jang H.-Y.; Roh Y.; Lynch V.; Schultz A. J.; Wang X.; Krische M. J. J. Am. Chem. Soc. 2002, 124, 9448. [DOI] [PubMed] [Google Scholar]
- Shigehisa H.; Aoki T.; Yamaguchi S.; Shimizu N.; Hiroya K. J. Am. Chem. Soc. 2013, 135, 10306. [DOI] [PubMed] [Google Scholar]
- Lo J. C.; Yabe Y.; Baran P. S. J. Am. Chem. Soc. 2014, 136, 1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girijavallabhan V.; Alvarez C.; Njoroge F. G. J. Org. Chem. 2011, 76, 6442. [DOI] [PubMed] [Google Scholar]
- Formation of an alkene and cobalt hydride to terminate a radical Heck coupling:Weiss M. E.; Kreis L. M.; Lauber A.; Carreira E. M. Angew. Chem., Int. Ed. 2011, 50, 11125. [DOI] [PubMed] [Google Scholar]
- Tokunaga M.; Larrow J. F.; Kakiuichi F.; Jacobsen E. N. Science 1997, 277, 936. [DOI] [PubMed] [Google Scholar]
- See Supporting Information for more entries.
- Li S.; Peng C.-H.; Fryd M.; Wayland B. B.; de Bruin B. J. Am. Chem. Soc. 2008, 130, 13373. [DOI] [PubMed] [Google Scholar]
- De Bruin B.; Dzik W. I.; Li S.; Wayland B. B. Chem.—Eur. J. 2009, 15, 4312. [DOI] [PubMed] [Google Scholar]
- Ford D. D.; Nielsen L. P. C.; Zuend S. J.; Musgrave C. B.; Jacobsen E. N. J. Am. Chem. Soc. 2013, 135, 15595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The commercial sample of 1-decene also contained 4% isomers.
- Zavitsas A. A.; Melikian A. A. J. Am. Chem. Soc. 1975, 97, 2757. [Google Scholar]
- Tokuyasu T.; Kunikawa S.; Masuyama A.; Nojima M. Org. Lett. 2002, 4, 3595. [DOI] [PubMed] [Google Scholar]
- Bullock R. M.; Samsel E. G. J. Am. Chem. Soc. 1987, 109, 6542. [Google Scholar]
- Pronin S. V.; Shenvi R. A. Nat. Chem. 2012, 4, 915. [DOI] [PubMed] [Google Scholar]
- Morrison D. A.; Davis T. P.; Heuts J. P.; Messerle B.; Gridnev A. A. J. Polym. Sci. A: Polym. Chem. 2006, 6171. [Google Scholar]
- The mechanism may also depend on the electronics of the alkene.
- Garr C. D.; Finke R. G. Inorg. Chem. 1993, 32, 4414. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.