

Abstract
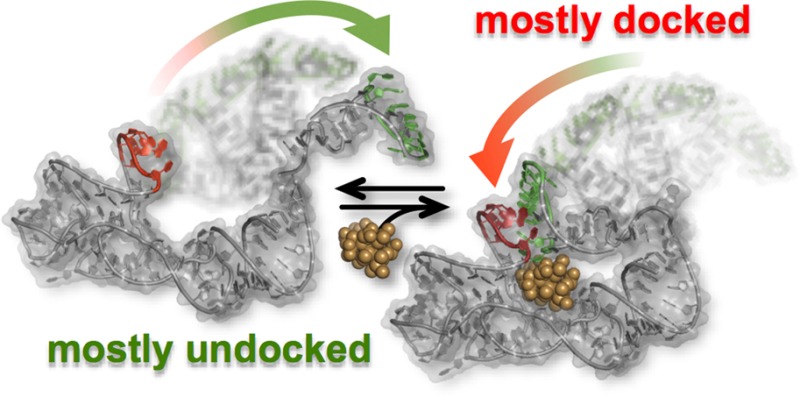
Riboswitches represent a family of highly structured regulatory elements found primarily in the leader sequences of bacterial mRNAs. They function as molecular switches capable of altering gene expression; commonly, this occurs via a conformational change in a regulatory element of a riboswitch that results from ligand binding in the aptamer domain. Numerous studies have investigated the ligand binding process, but little is known about the structural changes in the regulatory element. A mechanistic description of both processes is essential for deeply understanding how riboswitches modulate gene expression. This task is greatly facilitated by studying all aspects of riboswitch structure/dynamics/function in the same model system. To this end, single-molecule fluorescence resonance energy transfer (smFRET) techniques have been used to directly observe the conformational dynamics of a hydroxocobalamin (HyCbl) binding riboswitch (env8HyCbl) with a known crystallographic structure.1 The single-molecule RNA construct studied in this work is unique in that it contains all of the structural elements both necessary and sufficient for regulation of gene expression in a biological context. The results of this investigation reveal that the undocking rate constant associated with the disruption of a long-range kissing-loop (KL) interaction is substantially decreased when the ligand is bound to the RNA, resulting in a preferential stabilization of the docked conformation. Notably, the formation of this tertiary KL interaction directly sequesters the Shine-Dalgarno sequence (i.e., the ribosome binding site) via base-pairing, thus preventing translation initiation. These results reveal that the conformational dynamics of this regulatory switch are quantitatively described by a four-state kinetic model, whereby ligand binding promotes formation of the KL interaction. The results of complementary cell-based gene expression experiments conducted in Escherichia coli are highly correlated with the smFRET results, suggesting that KL formation is directly responsible for regulating gene expression.
1. Introduction
Riboswitches are noncoding structural elements in the leader sequence of select bacterial and eukaryal mRNAs that regulate downstream gene expression in a ligand-dependent fashion.2 The first experimentally verified riboswitch was found in mRNAs that code for the expression of a cobalamin (vitamin B12) transport protein (i.e., btuB in Escherichia coli).3,4 These structured nucleic acids function to maintain intracellular concentrations of vitamin B12, which is a critical cofactor for methyltransferase and isomerase enzymes associated with the production of S-adenosylmethionine and succinyl-CoA, respectively.5 The cobalamin riboswitch clan (Rfam: CL00101) is one of the most abundant and ubiquitous cis-acting mRNA regulatory elements throughout the bacterial domain of life.6 The importance of these RNAs and their associated effector molecule is further highlighted by the fact that only certain species of archaea and bacteria can biosynthesize cobalamin,7,8 and yet it is essential to many organisms, most notably humans.9
Generally, most riboswitches consist of two interacting structural elements that, together, result in biological functionality. The (i) aptamer domain of a riboswitch binds specific target molecules, which commonly promote formation of (or stabilizes) an alternative conformation in the (ii) regulatory element, or “expression platform”. These conformational transitions are associated with modulating production of the downstream gene.10,11 Single-molecule fluorescence resonance energy transfer12(smFRET) is, in principle, ideally suited to observe these ligand-induced conformational transitions.13−19 However, only a few investigations have successfully monitored conformational transitions associated with the regulatory element of a riboswitch,13,16,18 and, until now, none have been able to unambiguously link (i) ligand binding to (ii) conformational transitions within a biologically functional riboswitch. As one major result of the present work, these two processes have been observed both independently and simultaneously using smFRET techniques.
The novel X-ray crystal structure of a small, entirely biologically functional, cobalamin riboswitch, containing both the aptamer domain and expression platform, revealed that these two structural elements communicate via a tertiary interaction in order to confer biological functionality;1 such behavior has also been observed in other more extended cobalamin riboswitches.1,20 Complementary cell-based experiments conducted using this novel RNA construct demonstrated that a tertiary interaction modulates gene expression in a vitamin B12-dependent fashion.1 Specifically, it was shown that (i) this riboswitch selectively binds vitamin B12 cofactors with small β-axial ligands, like hydroxocobalamin (HyCbl), as opposed to the bulkier β-axial ligand of adenosylcobalamin,1 and (ii) binding of HyCbl, the photolysis product of adenosylcobalamin,21 facilitates docking between two loops of the RNA. It is this tertiary docking interaction that is responsible for regulating gene expression via formation of a kissing loop (KL) complex that directly sequesters the Shine-Dalgarno sequence, which is the ribosome binding site (RBS) of many mRNAs. Importantly, similar regulatory mechanisms have also been identified in other riboswitches like the SAM-III riboswitch.22
Although these recent structural and cell-based ensemble experiments, described above, have unveiled information about the mechanism of action associated with this riboswitch, many detailed questions regarding both ligand binding kinetics and conformational dynamics remain unanswered. To this end, a combination of fluorescence techniques, smFRET and cell-based fluorescent-reporter assays, has been used to offer the first, to our knowledge, detailed mechanistic account of a biologically functional riboswitch based on a collection of both cellular and smFRET data. The primary sequence (GenBank accession: AACY021350931.1/557–456) of the env8 hydroxocobalamin riboswitch (env8HyCbl, Figure 1a) used throughout these experiments comes from a comparative genomics analysis of structured noncoding RNAs.23 This RNA is a member of the AdoCbl-variant family (Rfam: RF01689), which is one of three families within the larger cobalamin riboswitch clan (Rfam: CL00101).24 Notably, this RNA contains all of the core characteristics associated with the entire cobalamin riboswitch clan, namely, a 4-way junction implicated in ligand recognition and a regulatory KL, making it a highly tractable model system that contains all of the structural elements necessary and sufficient for biological activity in a cellular context.
Figure 1.
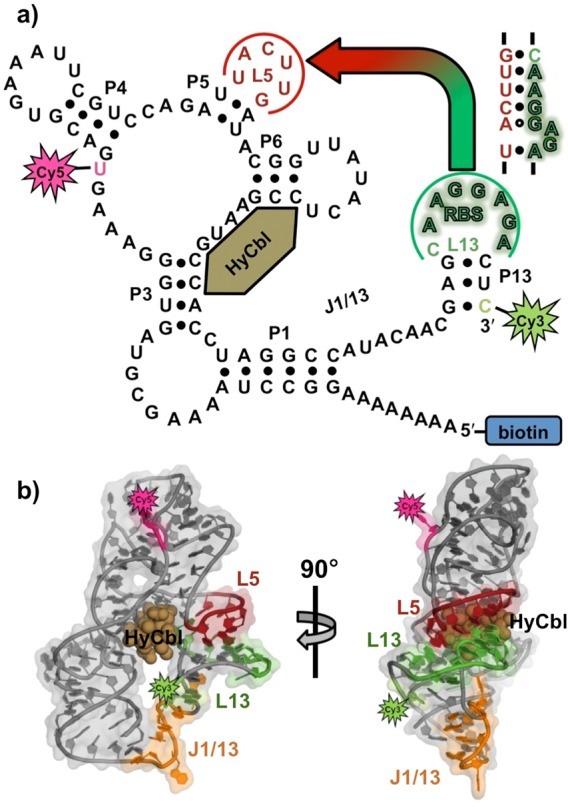
Schematic representation of the (a) secondary and (b) tertiary structure of the env8HyCbl riboswitch, complete with the Cy3 (green star), Cy5 (red star), and biotin (blue rectangle) synthetic moieties. The loops, L5 (red) and L13 (green), form a regulatory RNA kissing-loop (KL, see inset for sequence), which contains the purine-rich ribosome binding site (shadowed green nucleotides). Binding of hydroxocobalamin (bronze spheres, HyCbl) facilitates formation of the KL, which decreases the distance between the Cy3 (donor) and Cy5 (acceptor) fluorophores, and thereby results in increased energy transfer efficiency (EFRET).
Synthetic modifications to this RNA (Figure 1a) allow smFRET experiments to monitor, in real-time and under equilibrium conditions, the docking/undocking of the L5–L13 regulatory KL in the presence or absence of ligand (Figure 1b). Simultaneously, HyCbl-dependent quenching25 of Cy3 is used as an orthogonal experimental observable to monitor the kinetics of ligand binding independently of conformation. The results of these smFRET experiments provide substantial support for a four-state kinetic model that highlights several key biophysical features associated with this regulatory switch (Figure 2): (i) KL docking/undocking occurs in the absence of HyCbl and is dependent on Mg2+, (ii) HyCbl binding predominately occurs when the KL is undocked, (iii) KL docking/undocking also occurs while HyCbl is bound and is still dependent on Mg2+. Additionally, the single-molecule results reveal that, in the presence of Mg2+, HyCbl binding preferentially stabilizes the docked conformation. Finally, results from complementary cell-based reporter assays are used to relate these biophysical findings to the cellular function of this RNA. These comparisons reveal that formation of the regulatory switch (i.e., L5–L13 KL) in the single-molecule experiments is strongly correlated with repression of gene expression in the cell-based assay, regardless of whether the ligand is bound to the RNA. As a whole, these findings expose valuable new mechanistic and kinetic information about the dynamical relationship between structure and function in cobalamin riboswitches, which can also be applied to other functional RNAs.
Figure 2.
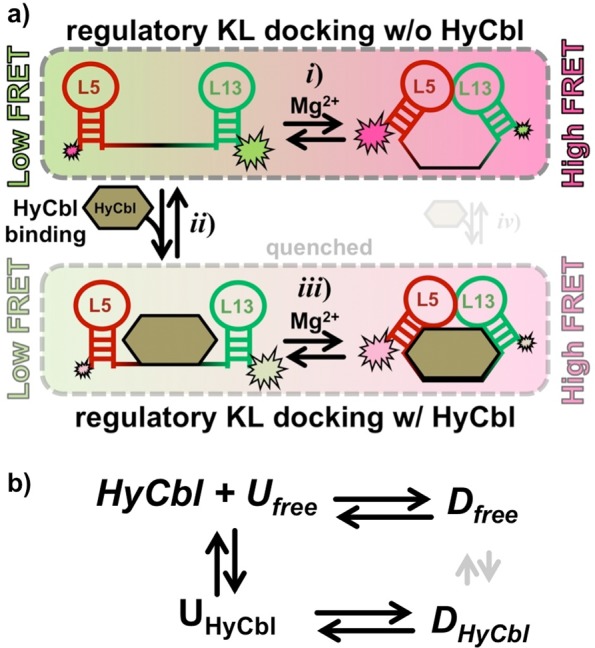
Four-state kinetic model for the env8HyCbl riboswitch represented (a) graphically and (b) symbolically. The four macroscopic conformations are linked by three coupled equilibria: (i) KL docking in the absence of ligand, (ii) ligand binding in the undocked conformation, and (iii) KL docking in the presence of ligand. Formation of the KL decreases the interdye distance, resulting in more efficient fluorescence resonance energy transfer (EFRET). Ligand binding quenches Cy3, which decreases the total fluorescence of Cy3 and Cy5 and can therefore be monitored independently of KL docking/undocking events. Although it is possible that a fourth (iv) equilibrium exists, we observe no experimental evidence for such a process (Section 3.3), indicating that this process is prevented by prohibitively large free energy barriers (as indicated by the small transparent arrows). Accordingly, this process is not considered for simplicity and ease of discussion; its inclusion would not alter the interpretations and conclusions derived from this work.
2. Results
Both freely diffusing and surface-immobilized experiments (see Materials and Methods) have been used to characterize the conformational transitions associated with the regulatory KL of the env8HyCbl riboswitch. Briefly, freely diffusing smFRET experiments26 make use of the subfemtoliter volumes associated with the diffraction-limited focus of a high numerical aperture objective. At ≈ 100 pM of Cy3–Cy5 labeled RNA, this volume is absent of fluorophores more than 99% of the time. Occasionally, an individual molecule will stochastically diffuse through this volume, resulting in a ≈ 1 ms burst of fluorescence limited in duration by RNA diffusion. The efficiency of fluorescence resonance energy transfer (EFRET) can be calculated for thousands of fluorescent bursts and compiled into a histogram to describe the probability of observing any specific EFRET value, which therefore reports on the conformational occupancy of the RNA. Alternatively, individual Cy3–Cy5 labeled RNAs can be immobilized to a microscope coverslip using biotin–streptavidin chemistry,27 which prevents the molecule from freely diffusing in solution. The diffraction-limited focus of the confocal fluorescence microscope can be used to continuously excite the fluorophores, allowing for collection of emitted photons. The collected signal is used to determine the EFRET for a single RNA as a function of time, whereby changes in EFRET result from discrete conformational transitions.28 The two single-molecule fluorescence experiments described above are used to rigorously characterize the in vitro behavior of the env8HyCbl riboswitch. Complementary cell-based fluorescence reporter assays are used to compare and contrast the in vitro and cellular behavior of this RNA. Specifically, these reporter assays are designed to monitor the regulated expression of a green fluorescent protein variant (GFPuv) provided by the env8HyCbl riboswitch.
2.1. KL Docking Equilibrium in the Absence of Ligand
As a first step, freely diffusing smFRET experiments have been used to explore the equilibrium behavior of the L5–L13 kissing loop (KL) interaction from the env8HyCbl riboswitch in solutions free of HyCbl. Under standard buffer conditions (see Materials and Methods), the distribution of EFRET values associated with thousands of individual fluorescently labeled RNA constructs reveals (Figure 3a) that the riboswitch resides entirely in a single low EFRET = 0.15(3) population. However, addition of MgCl2 is, by itself, sufficient to promote KL docking (Figure 3a), as indicated by the Mg2+-dependent increase in the relative probability of observing molecules in a second, higher EFRET = 0.58(3) population. This high EFRET value corresponds to an inter-dye distance of = 50(3) Å and, given the flexibility of the RNA–fluorophore linker, is entirely consistent with the ≈ 47 Å prediction based on the recent crystal structure.1 Accordingly, we attribute this high EFRET value to molecules residing in the docked KL conformation (Figure 2b, Dfree), with the low EFRET value corresponding to the undocked species (Figure 2b, Ufree), where the subscript “free” denotes that the ligand is not bound to the riboswitch. The necessity of Mg2+ for the formation of this interaction is supported by the X-ray crystal structure (PDB: 4FRN), which depicts divalent cations near the L5 and L13 loops.1 This is a common feature among other KL X-ray crystal structures29,30 and likely reflects the importance of Mg2+ in the formation of these types of tertiary interactions.
Figure 3.
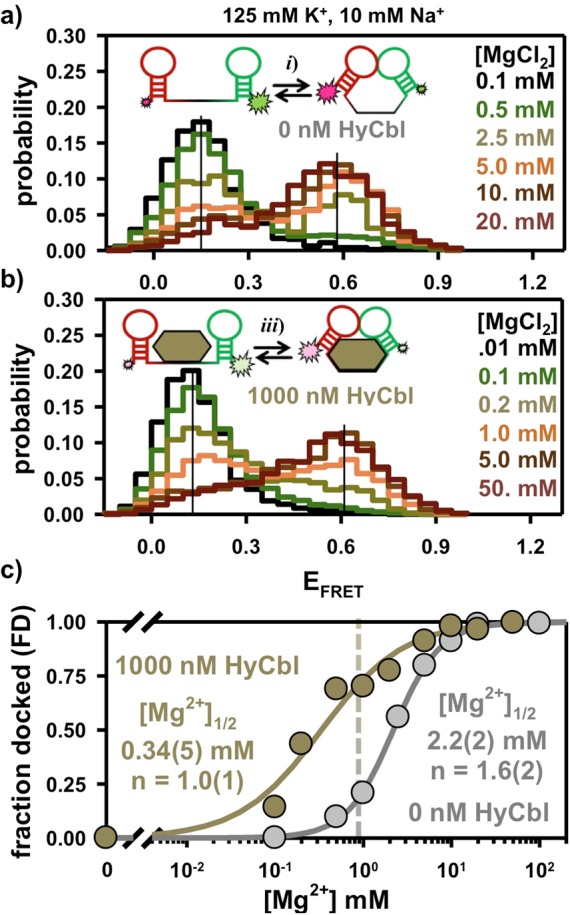
Freely diffusing smFRET experiments reveal the Mg2+-dependent formation of the docked conformation in the (a) absence and (b) presence of 1000 nM HyCbl. Note the color scheme in a and b is only intended to represent the increasing [HyCbl]; colors do not necessarily correlate one-to-one with [HyCbl]. (c) The presence of saturating HyCbl (1000 nM, bronze circles) decreases the concentration of Mg2+ required to achieve half-maximal occupancy of the docked conformation (i.e., [Mg2+]1/2) by ≈ 10-fold. The vertical dashed bar represents near-physiological salt conditions (see text for details). The error bars are smaller than the symbols used for each data point (Materials and Methods).
The residual probability associated with observing the low EFRET population at high [Mg2+] results from molecules that are unable to form the L5–L13 KL interaction (≈ 15%), as quantitatively determined in surface immobilized experiments described later (Section 2.5). The abundance of this population is dependent on the conditions associated with synthesis of the construct, perhaps, indicating the existence of misfolded species (see Materials and Methods). The normalized probability of observing the two EFRET distributions, when corrected for a ≈ 15% nonresponsive population, can be used to characterize the equilibrium behavior of the riboswitch. Specifically, the fractional occupancy of molecules in the docked or undocked conformation can be calculated using eqs 1a and 1b, respectively.
| 1a |
| 1b |
The parameters P(Dfree) and P(Ufree) describe the probability of observing the docked and undocked conformations, respectively, where the subscript “free” again denotes that the riboswitches are not bound to ligands. The [Mg2+] required to achieve half-maximal occupancy of the docked conformation (i.e., [Mg2+]1/2) is obtained by fitting the data to a Hill-type binding model. For data collected in the absence of HyCbl, such an analysis predicts [Mg2+]1/2 = 2.2(2) mM (Figure 3c), which is nearly twice normal physiological concentrations of free Mg2+.31 Although the regulatory KL can form in the absence of HyCbl, the FDfree is only ≈ 0.22 in solutions that appropriately mimic the ionic environment associated with the cellular milieu (e.g., 1 mM Mg2+, 125 mM K+, 10 mM Na+), suggesting that the undocked conformation is preferred under these conditions.
To confirm that the high EFRET population results from formation of the L5–L13 regulatory switch, a second, doubly labeled RNA construct has also been studied. This construct, referred to as the L5-mutant env8HyCbl riboswitch (Supporting Information Figure S1), contains a dinucleotide substitution at positions 48 and 49 in L5 (underlined nucleotides, 5′-UACUUG-′3 → 5′-UACAAG-′3). The UU to AA mutation prevents formation of two Watson–Crick base pairs that are otherwise present in the wild-type construct and preferentially destabilizes the docked conformation of the KL. As expected, this smFRET construct remains in the undocked (low EFRET) conformation even in solutions containing up to 30 mM MgCl2 (Supporting Information Figure S1), thus confirming that the donor and acceptor fluorophores accurately report on the formation of this regulatory KL interaction.
2.2. KL Docking Equilibrium in the Presence of Ligand
Analogous freely diffusing smFRET experiments have also been conducted in the presence of ligand in order to interrogate the equilibrium behavior of this interaction in solutions where the HyCbl concentration is more than 100 times larger than the previously reported1 binding affinity of 8(4) nM. Without any Mg2+ in solution, fluorescent molecules reside entirely in a single low EFRET = 0.12(3) population (Figure 3b) that is experimentally indistinguishable from the low EFRET = 0.15(3) population observed in the absence of ligand (Figure 3a). Notably, such behavior was also observed in recent smFRET investigations of a c-di-GMP riboswitch; however, complete folding required the presence of ligand, which is unlike the env8HyCbl riboswitch in this study. As was the case in the absence of ligand, Mg2+ is still required for the formation of the KL interaction, even at saturating [HyCbl]. Specifically, addition of MgCl2 into solutions containing 1000 nM HyCbl increases the probability of observing a second, higher EFRET = 0.61(4) population (Figure 3b) that is also experimentally indistinguishable from the high EFRET = 0.58(3) population noted previously in the absence of ligand (Figure 3a).
Under saturating HyCbl conditions, presumably every functional riboswitch is bound to a ligand. Therefore, the above observations support the existence of the L5–L13 KL equilibrium associated with the docked (Figure 2b, DHyCbl) and undocked (Figure 2b, UHyCbl) conformations of the RNA, where the “HyCbl” subscript now denotes the presence of a bound ligand. Notably, bound HyCbl reduces [Mg2+]1/2 by nearly an order of magnitude (Figure 3c), which indicates that under physiological free salt conditions (i.e., 1 mM Mg2+, 125 mM K+, 10 mM Na+) these ligand bound riboswitches reside predominantly in the docked conformation (FDHyCbl ≈ 0.75). In conjunction with the results of experiments performed in the absence of ligand, it is apparent that HyCbl binding increases the stability of the docked conformation in solutions containing Mg2+(e.g., at 1 mM Mg2+ FDfree ≈ 0.22 < FDHyCbl ≈ 0.75). Moreover, the increased occupancy of the docked conformation, where the L5–L13 KL is formed, demonstrates that ligand binding directly modulates the availability of the ribosome binding site within L13 of this regulatory switch.
2.3. Ligand Binding Properties
To directly explore the ligand-binding process, we exploit a unique photophysical property of hydroxocobalamin (HyCbl). Specifically, the absorbance spectrum of HyCbl overlaps with the fluorescence spectrum of Cy3, which allows this cofactor to effectively compete with Cy5 for acceptance of energy transfer (Supporting Information Figure S2). Because HyCbl is a nonfluorescent acceptor, energy transfer to it results in strongly quenched fluorescence.25 We can utilize this quenching phenomenon to monitor the ligand binding kinetics of the env8HyCbl riboswitch via time-dependent ensemble fluorometry. When HyCbl is dissociated from the RNA, the average intermolecular distance between it (acceptor) and Cy3 (donor) is large enough to neglect the effects of this quenching energy transfer process (Figure 4a). However, upon ligand binding, the intermolecular distance between the two is reduced to ≈ 23 Å,1 allowing for substantial energy transfer from the excited donor molecule (Cy3) to the nonfluorescent acceptor (HyCbl). Thus, energy transfer to HyCbl results in an additional nonradiative (knrad) component of the Cy3 fluorescence lifetime (τfluor = 1/(krad + knrad)), which leads to a decreased quantum yield (Q.Y. = krad/(krad + knrad)). As an attempt to quantify this ligand-dependent quenching, we have measured the fluorescence decay profiles32 of individual Cy3-only env8HyCbl RNAs in the presence and absence of 1000 nM HyCbl (Supporting Information Figure S2a) using the time-correlated single-photon counting capabilities associated with the experimental apparatus (see Materials and Methods). These lifetime measurements (i.e., Cy3-τfluor(free) = 1.40(2) ns and Cy3-τfluor(HyCbl) = 0.51(6) ns) effectively demonstrate the nonradiative quenching phenomenon associated with energy transfer from Cy3 to HyCbl.
Figure 4.
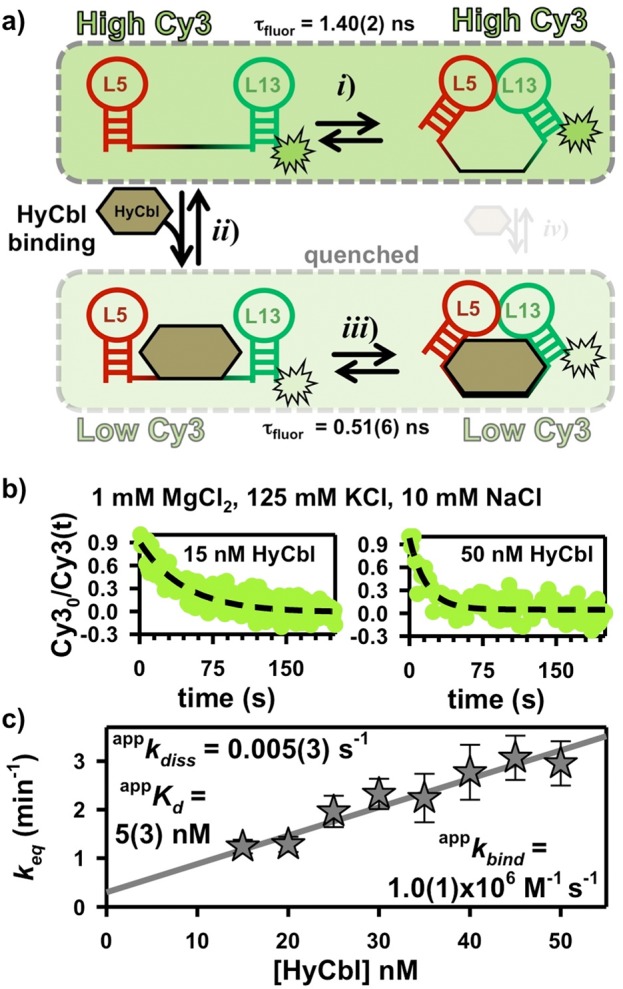
Time-dependent ensemble fluorometry. (a) HyCbl binding quenches Cy3 (see also Supporting Information Figure 2). (b) The single-exponential decay of the Cy3 fluorescence reports on the sum of the apparent binding (appkbind) and dissociation (appkdiss) rate constants associated with the interaction between HyCbl and the env8HyCbl riboswitch. Elevated concentrations of ligand increase the rate constant for establishing equilibrium. (c) A linear-fit of the equilibration rate constant as a function of [HyCbl] is used to measure the apparent ligand binding (appkbind) and dissociation (appkdiss) rate constants.
In the presence of the fluorescent acceptor, Cy5, it is important to stress that the energy transfer process to HyCbl competes equally with both the radiative decay process of Cy3 and the energy transfer process to Cy5 (Supporting Information Figure S2), resulting in uniform attenuation of both signals. Provided that the quenched fluorescence signals remain well above background, the ratiometric EFRET values associated with the docked (i.e., Dfree vs DHyCbl) and undocked (i.e., Ufree vs UHyCbl) populations remain unaltered after ligand binding (Supporting Information Figure S2b), as demonstrated experimentally (see Section 2.2).
This photophysical phenomenon is exploited in kinetic ensemble fluorescence experiments to monitor the HyCbl-induced reduction in fluorescence intensity of RNA constructs labeled with Cy3 upon addition of excess ligand ([HyCbl]/[RNA] > 10). The temporal decay of fluorescence intensity (Figure 4b) maps out the rate constant responsible for establishing a binding equilibrium (keq). For a single bimolecular process, this rate constant (keq) can simply be described as a function of ligand concentration using the ligand-binding (kbind) and dissociation (kdiss) rate constants (eq 2).
| 2 |
However, the ligand binding kinetics in the present system are further complicated by the two remaining KL equilibria (Figure 2), which limit the fraction of undocked molecules that can either bind to (FUfree) or dissociate from HyCbl (FUHyCbl). Thus, the observed rate constants represent apparent values. The [HyCbl]-dependence of appkeq is evident in Figure 4c and is well described by a straight line. Linear regression yields a slope of appkbind = 1.0(1) × 106 M–1 s–1 and an intercept of appkdiss = 0.005(3) s–1, where the ratio of these two values characterizes the apparent HyCbl dissociation equilibrium constant (appKd = 5(3) nM).
Using principles related to Michaelis–Menten enzyme kinetics,33 we can decouple these apparent kinetic parameters from the two KL docking equilibria by making the following assumption, which is experimentally validated later in surface-immobilized kinetic studies (Sections 2.5 and 2.6). Provided the docking/undocking kinetics associated with the KL equilibria are much faster than the ligand binding or dissociation processes, the apparent rate constants can be readily shown34 to equal true rate constants (i.e., kbind and kdiss), scaled by the fractional populations of the binding competent (FUfree) and dissociation competent (FUHyCbl) species, respectively (see eqs 3a and 3b).
| 3a |
| 3b |
The values of FUfree ≈ 0.78 and FUHyCbl ≈ 0.25 are obtained from the freely diffusing data depicted in Figure 3c at 1 mM MgCl2. These values are used to calculate the true rate and equilibrium constants, denoted by a lack of the superscript “app”, that characterize the ligand binding kinetics of the env8HyCbl riboswitch at 1 mM MgCl2 (kbind = 1.3(2) × 106 M–1 s–1, kdiss = 0.02(1) s–1, and Kd, = 15(8) nM).
2.4. HyCbl-Dependence of the KL Docking Equilibrium
The data sets described in the three previous sections (2.1, 2.2, and 2.3) quantitatively characterize all equilibrium constants required to predict the steady-state fractional occupancy of the docked conformation as a function of [HyCbl] for a given [MgCl2]. To experimentally test these predictions, the fractional occupancy of the docked conformation (FD) is determined using freely diffusing smFRET experiments wherein HyCbl is titrated into solutions at five different fixed MgCl2 concentrations (0.0, 0.5, 1.0, 2.5, 20 mM). In solutions lacking Mg2+, the addition of ligand is unable to promote formation of the docked conformation (Figure 5a), as Mg2+ is required for the formation of the regulatory KL interaction between L5 and L13. At intermediate concentrations of MgCl2 (e.g., 0.5, 1.0, and 2.5 mM), titration of HyCbl into solution facilitates formation of the docked conformation (Figure 5b). This observation is certainly consistent with the four-state model (Figure 2), wherein bound HyCbl, increases the stability of the regulatory KL interaction (i.e., FDfree < FDHyCbl). Titration of HyCbl into solutions with saturating concentrations of Mg2+ (e.g., 20 mM ≫ [Mg2+]1/2) reveals that ligand is unable to significantly increase the FD (Figure 5c), which nicely demonstrates that Mg2+ alone is sufficient to promote complete formation of the regulatory interaction responsible for sequestering the ribosome binding site.
Figure 5.
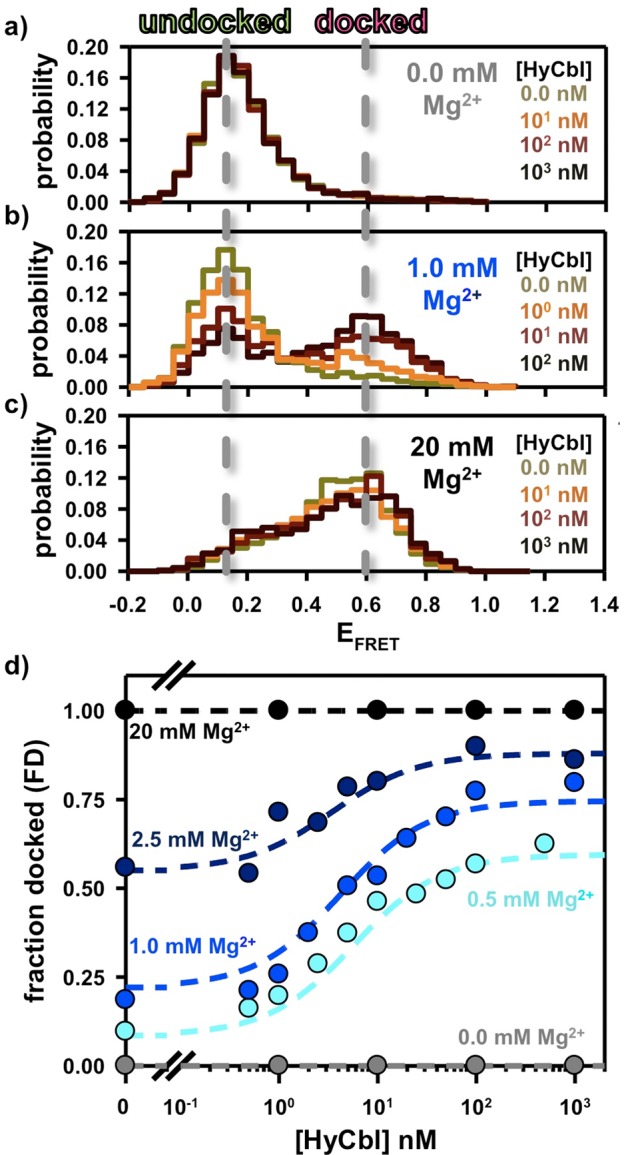
Freely diffusing single-molecule burst titrations of hydroxocobalamin (HyCbl) at various concentrations of MgCl2. (a) At 0 mM Mg2+, addition of ligand does not significantly influence the distribution of EFRET associated with the L5–L13 regulatory switch. (b) At intermediate concentrations of Mg2+(e.g., 2.5 mM), addition of HyCbl does significantly alter the distribution of EFRET. (c) Mg2+ (20 mM) is already sufficient to completely form the L5–L13 KL interaction, irrespective of [HyCbl]. Together, these results support the notion that docking of the env8HyCbl riboswitch is more favorable when the ligand is bound to the RNA under near-physiological salt conditions, but that HyCbl alone (i.e., 0 mM Mg2+) is insufficient to promote formation of the KL interaction. Note the color scheme in a–c is only intended to represent the increasing [HyCbl]; colors do not necessarily correlate one-to-one with [HyCbl]. (d) Experimental validation of the four-state kinetic model for the env8HyCbl riboswitch. The experimentally determined fractional occupancy of the docked conformation (FD, colored circles) is well described by the steady-state solution (dotted line) to the four-state model (Figure 2) over a wide range of Mg2+ and HyCbl concentrations.
Figure 5d compares the results of the single-molecule experiments with the model predictions (colored circles and dashed lines, respectively). This comparison demonstrates the remarkable quantitative accuracy achieved by the four-state kinetic model (Figure 2) used to describe conformational transitions associated with this biologically functional riboswitch. One noteworthy feature of this model is that the ligand-binding process is mostly insensitive to Mg2+, as indicated by transition midpoints (i.e., appKd) near 5 nM for [MgCl2] between 0.5 and 2.5 mM. Nucleic acid–ligand interactions have previously been shown to be strongly influenced by metal-ion-facilitated conformational rearrangements in many riboswitches, including the lysine,18 THF,35 c-di-GMP,19 SAH,36 and adenine37 sensing riboswitches. However, in the case of this HyCbl-sensing riboswitch, the two coupled KL docking equilibria result in a ligand binding process that is only subtly influenced by Mg2+. Specifically, addition of Mg2+ promotes docking, which, for ligand-free RNAs, decreases the fractional population of the binding competent state (i.e., FUfree). However, this is approximately offset by the corresponding decrease in the fractional population of HyCbl-bound species that can readily undergo dissociation (i.e., FUHyCbl).
2.5. KL Docking Kinetics in the Absence of Ligand
Fluorescence trajectories of individual surface-immobilized molecules have been recorded to observe the KL docking dynamics (Figure 6a–c) with 30 ms resolution. In the absence of HyCbl (Figure 6a), single RNAs display strongly anticorrelated fluctuations in donor and acceptor fluorescence, resulting from rapid transitions between the two well-separated FRET states. These FRET values are in excellent agreement with those measured in the freely diffusing experiments, which supports the conclusion that these time trajectories depict single-molecules transitioning between the docked (Dfree) and undocked (Ufree) conformations associated with the L5–L13 KL interaction. Kinetic analysis of the surface-immobilized data yields hundreds of individual dwell times that are compiled together and fit to single-exponential decays in order to yield the rate constants associated with docking and undocking. These rate constants reveal that, in ligand free solutions at 1 mM Mg2+, kdock,free = 1.2(3) s–1 and is slower than kundock,free = 6(2) s–1. Together, these rate constants result in FDfree = 0.17(7), which agrees well with results from the freely diffusing experiments (FDfree ≈ 0.22). A titration with respect to Mg2+, analogous to the ones from the freely diffusing smFRET experiments, reveals that increasing divalent cation concentration accelerates the docking and decelerates the undocking rate constants, respectively, both of which contribute nearly equally to shifts in the Mg2+-dependent FDfree (Figure 6c). As alluded to above, the fact that individual surface-immobilized molecules dock completely (FDfree ≈ 1.0) at high [MgCl2] confirms that the residual probability (≈ 15%) for observing the low EFRET population results from inactive molecules that cannot form the L5–L13 interaction. It is important to emphasize that the results for both freely diffusing and surface immobilized studies are indistinguishable, as indicated by the circles and stars in Figure 6d, respectively. Such a plot demonstrates convincingly that surface immobilization does not influence the biophysical behavior of this RNA.
Figure 6.

Kissing-loop docking kinetics in the absence of bound ligand. Representative (a) fluorescence and (b) FRET time-traces from a surface immobilized env8HyCbl riboswitch. Orange lines represent the results from a maximum-likelihood 2-state Bayesian model that bests describes the data. (c) The docking and undocking rate constants, as a function of [Mg2+], are used to calculate the fractional occupancy of the docked conformation (d) in the absence of HyCbl (FDfree), which reveals that the surface immobilized results (dark gray stars) are in quantitative agreement with the freely diffusing experiments (light gray circles). The error bars are often smaller than the symbols used for each data point (Materials and Methods).
2.6. KL Docking Kinetics in the Presence of Ligand
To complete the single-molecule kinetic characterization of this riboswitch, fluorescence time-trajectories of individual surface-immobilized molecules are recorded in the solutions containing 2.5 nM HyCbl and 1 mM Mg2+ (Figure 7). These results further support the proposed kinetic model (Figure 2) for ligand-facilitated formation of the regulatory switch. Specifically, the time-trajectories in the presence of HyCbl display distinct regions with vastly different fluorescence intensities. Some regions exhibit normal fluorescence signals for both the donor and the acceptor fluorophores, which are well resolved and anticorrelated (Figure 7b). In the other, intervening regions, the total fluorescence signal is significantly quenched (Figure 7c). The rate constants associated with the anticorrelated fluorescence fluctuations in the normal regions mimic those from molecules studied in the absence of HyCbl (Figure 7b vs 6b), as would be expected when the ligand is not bound to the RNA. Conversely, the quenched regions result from ligand binding events that localize HyCbl near Cy3, which introduces additional nonradiative relaxation pathways. Indeed, despite the substantially reduced signal in these regions, subtle fluctuations in acceptor intensity in excess of background noise are clearly observable and can be attributed to docking and undocking of the KL interaction during the long ligand-bound episodes.
Figure 7.

Surface immobilized KL docking kinetics in the presence of hydroxocobalamin (HyCbl). (a) Representative fluorescence trajectory from a surface immobilized molecule in the presence of 2.5 nM HyCbl. For a construct in the undocked conformation, ligand binding (arrow) significantly diminishes the fluorescence intensity, which returns back to normal after ligand release. (b) When the ligand is not bound, the docking/undocking kinetics of the regulatory KL are experimentally indistinguishable from those in the absence of HyCbl (see also Figure 6b). (c) When the ligand is bound, a maximum-likelihood 2-state model (black line) is used to quantify the dwell times associated with fluctuations in the acceptor fluorescence resulting from KL docking and undocking. A comparison between the two KL docking equilibria (b vs c) reveals that the effect of HyCbl binding significantly decreases the undocking rate constant (kundock).
To quantitatively measure the docked and undocked dwell times in the ligand-bound state, a Bayesian maximum-likelihood model38 is used to analyze the two-state fluctuations evident in the quenched regions of the fluorescence trajectories (Figure 7c). When the ligand is bound to the env8HyCbl riboswitch in solution containing 1 mM MgCl2, such an analysis reveals that the docking rate decreases only by 2-fold (kdock,HyCbl = 0.64(9) vs kdock,free = 1.2(3) s–1), whereas the undocking rate exhibits a much more significant HyCbl-sensitivity and decreases by ≈ 23-fold (kundock,HyCbl = 0.26(8) s–1 vs kundock,free = 6(2) s–1). The HyCbl-induced change to the KL undocking kinetics gives rise to substantially more favorable formation of the L5–L13 regulatory switch (FDfree = 0.17(7) vs FDHyCbl = 0.7(1)), as also observed in the corresponding freely diffusing experiments (FDfree ≈ 0.22 vs FDHyCbl ≈ 0.75). Further inspection of the fluorescence trajectories reveals that (i) HyCbl can only bind and quench fluorescence when the RNA is in the undocked (low EFRET) conformation (Supporting Information Figure S3) and (ii) ligand-bound dwell times can be longer than 20 s (Figure 7a). These bound-state lifetimes are consistent with those predicted from the ensemble time-dependent fluorescence experiments (Figure 4c), which yield appτHyCbl = (1/appkoff) = 200(120) s. Lastly, the time-traces of surface immobilized molecules clearly verify the previous assumption that the KL kinetics, both in the presence and absence of ligand, are much faster than those associated with establishing a ligand binding equilibrium. In total, the collection of results from the above biophysical fluorescence studies provide strong support for the proposed four-state kinetic model (Figure 2) associated with the regulatory conformational transitions of the env8HyCbl riboswitch.
2.7. Relevance of Kissing Loop Formation with Respect to Regulation of Gene Expression
To correlate the biophysical behavior of this riboswitch with its biological function, cell-based gene expression experiments (Figure 8) have been performed in an E. coli strain lacking cobalamin adenosyl transferase (ΔbtuR), thereby preventing the subsequent conversion of HyCbl to adenosylcobalamin. In these experiments, cells are transformed with vectors containing either the wild type or mutant riboswitches located upstream of a GFPuv reporter gene, termed WT env8HyCbl+GFPuv or L5 mutant env8HyCbl+GFPuv, respectively, and cultured in liquid media with or without 5000 nM HyCbl. The corresponding L5-mutant env8HyCbl+GFPuv vector contains a riboswitch with the exact same dinucleotide substitution used in the smFRET experiments (Supporting Information Figure S1). The results of our biophysical experiments suggest that this L5-mutant riboswitch should be incapable of regulating gene expression because it is unable to from the regulatory KL interaction that sequesters the ribosome-binding site. Provided that such a conservative, two nucleotide substitution mutation does not significantly alter the mRNA abundance associated with the L5-mutant env8HyCbl+GFPuv vector, the cells transformed with this vector can be used to provide a reasonable assessment of the maximum amount of GFPuv expression resulting from completely undocked riboswitches.
Figure 8.
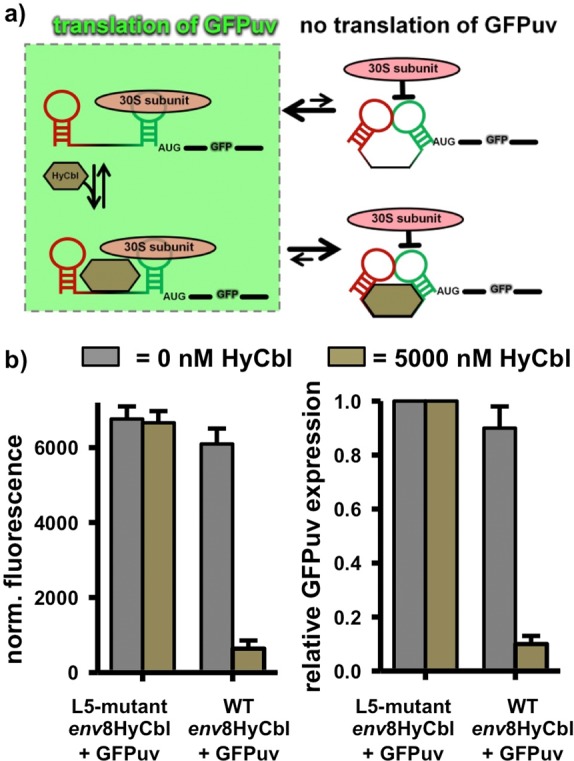
Cell-based regulation of gene expression via the env8HyCbl riboswitch. (a) Schematic diagram relating the four-state kinetic model to the regulation of gene expression via inhibition of translation initiation. (b) Histograms comparing the normalized fluorescence and relative GFPuv expression of cell cultures grown in the presence (bronze bars) or absence (gray bars) of 5000 nM HyCbl.
As expected, in growth media lacking HyCbl, cells transformed with WT env8HyCbl+GFPuv have normalized fluorescence values (Materials and Methods) lower than cells transformed with the L5-mutant env8HyCbl+GFPuv plasmid (gray bars, Figure 8b). This compares nicely with the single-molecule observation that the WT env8HyCbl FRET construct is mostly undocked (FUfree ≈ 0.78) at 1 mM Mg2+, whereas the L5-mutant is completely undocked (FUfree ≈ 1.0). Additionally, when cultured in media containing 5000 nM HyCbl, only cells harboring WT env8HyCbl+GFPuv show a substantial ligand-dependent reduction in GFPuv expression (bronze bars, Figure 8b), indicating that HyCbl decreases the expression of GFPuv, presumably via sequestration of the ribosome binding site within L13 of the env8HyCbl riboswitch. The pronounced similarities between the results of the in vitro and cell-based experiments suggests that (i) the fractional occupancy of the undocked conformation correlates nicely with the level of gene expression, regardless of whether or not ligand is bound and (ii) formation of the docked conformation, resulting from a long-range KL interaction between L5 and L13, sequesters the ribosome binding site within L13 and prevents initiation of translation.
3. Discussion
Ligand-induced conformational transitions are central to the function of many riboswitches. A significant body of structural work has revealed the numerous ways in which riboswitches can specifically interact with their cognate ligands. However, much less is known about how molecular recognition in the riboswitch aptamer domain translates into alternative conformations in the expression platform.10,11 The results of these single-molecule experiments mechanistically link: (i) ligand binding to (ii) conformational transitions in the expression platform of a fully functional riboswitch. Specifically, the fluorescently labeled RNA constructs examined in this work provide information about the equilibrium and kinetic behavior of a regulatory kissing loop (KL) interaction. Furthermore, the novel use of hydroxocobalamin-induced fluorescence quenching as an orthogonal experimental observable offers additional insights into the ligand binding process. Together, these results highlight several key findings about the functional mechanism of gene regulation for the env8HyCbl riboswitch, which can serve as a model system for the entire cobalamin riboswitch clan.
3.1. Kissing Loop Docking Dynamics
Both the freely diffusing and surface immobilized experiments clearly identify the L5–L13 kissing loop (KL) interaction as the structural motif that directly regulates biological function (i.e., gene expression). Additionally, this motif is also partly responsible for organizing the global conformation of the RNA. Many other functional nucleic acids make use of similar types of tertiary interactions to accomplish these same tasks. For example, KL interactions are important for organizing the three-dimensional structure of self-cleaving ribozymes39,40 and regulate a variety of biological processes,41 such as dimerization of retroviral genomes29 and plasmid replication.42,43 One characteristic of both intra- and intermolecular KL interactions is the formation of canonical Watson–Crick base pairs between the two loops that dock together. In addition to five complementary base pairs, the L5–L13 interaction contains a G–A mismatch adjacent to a dinucleotide (AG) bulge. Presumably, these structural defects serve to destabilize this interaction.44 In fact, recent kinetic studies of an intermolecular 6 bp RNA KL interaction associated with HIV-1 genome dimerization19 have reported rate constants for KL undocking (kundock ≈ 1.0–0.1 min–1) that are significantly slower than what is observed in the present study. The comparatively fast kundock for env8HyCbl in the absence of ligand likely results from the structural defects associated with the L5–L13 KL interaction. Notably, the presence of HyCbl partially compensates for these defects by slowing down the kundock, which results in ligand-dependent sequestration of the ribosome binding site. This nicely demonstrates how nucleotide sequence can be used to tune the stability of a RNA KL interaction. Other examples exist where localization of Mg2+ modulates the stability of these types of interactions, as is the case with intermolecular HIV (human immunodeficiency virus) genome dimerization.29 In the env8HyCbl riboswitch, divalent cations are essential for KL formation and likely play a similar role. Indeed, the X-ray crystal structure of this RNA depicts substantial electron density associated with Ba2+ (the more strongly X-ray scattering mimetic of Mg2+) near the interacting loops.1 It is important to mention that, for the env8HyCbl riboswitch, the presence of a bound ligand not only lowers the [Mg2+] required for half-maximal docking ([Mg2+]1/2), but that it also decreases the number of ions that condense onto the RNA during the conformational transition (Figure 3c). Together, these two observations indicate that HyCbl binding complements cation uptake, thus attenuating the Mg2+ requirements associated with this regulatory KL interaction.
3.2. KL Docking in the Presence of a Macromolecular Stabilizing Agent
While divalent cations and nucleotide sequence represent two effective means to adjust the stability of RNA kissing loops (KLs), there is also another pathway to accomplish such a task. In addition to distinctly identifying the existence of a KL docking equilibrium in the absence of ligand, the above experimental results reveal that KL docking and undocking also occurs when HyCbl is bound. Specifically, the presence of a bound ligand alters the energetics of this process to substantially favor the docked conformation. This is yet another example of how KL stability can be tuned to achieve a particular biological function. Careful inspection of the HyCbl-bound crystal structure reveals that most of the intermolecular contacts between the RNA and the ligand are van der Waals interactions governed by shape complementarity.1 These interactions significantly stabilize the docked conformation, as evidenced by the ≈ 23-fold slower kundock. This represents one example of how a specific macromolecular cosolute can stabilize the formation of a KL interaction directly responsible for a specific biological function.
Interestingly, a similar mechanism is employed in the Rop-RNA I-RNA II plasmid replication system.43 Specifically, the Rop protein acts as a molecular scaffold that stabilizes the intermolecular KL interaction between the loops of RNA I and RNA II45 and regulates formation of RNA primers required for DNA plasmid replication. In this system, the presence of Rop substantially reduces the rate constant describing RNA I dissociation from the ternary complex,46 similar to how the presence of HyCbl reduces the rate constant for KL undocking in the env8HyCbl riboswitch. Interestingly, the many ethanamide and propanamide functional groups in the corrin ring of HyCbl chemically recapitulate the amino acid side chains of asparagine and glutamine, respectively. This might suggest that the protein-like chemical functionality of HyCbl allows such a cofactor to imitate the stabilizing properties of Rop. Moreover, structural models of Rop revealed that these amino acids are localized to the proposed interface between the protein and the two kissing loops of RNA I and II,47 while binding studies have demonstrated that these amino acids are essential for recognizing the RNA KL.48 The functional similarities between the Rop-RNA I-RNA II system and the env8HyCbl riboswitch demonstrate how KL formation can be facilitated by a macromolecular binding partner (e.g., protein or ligand) in order to regulate a biologically relevant process (e.g., plasmid replication or gene expression).
3.3. Induced-Fit Ligand Recognition
A long-standing question in nucleic acid recognition of small molecule ligands is whether: (i) an induced-fit or (ii) a conformational-capture model most appropriately describes these aptamer–substrate interactions. The induced-fit model demands that ligand binding occurs in open (e.g., undocked) conformations of the aptamer, which then induces a structural transition that encapsulates the bound molecule.11,49,50 This is in contrast to the conformational-capture model,17,22 whereby ligand recognition occurs in transient, highly organized folded structures, similarly resulting in stabilization of the bound (e.g., docked) conformation; such a mechanism was recently identified via smFRET investigations of a c-di-GMP riboswitch.19 For the env8HyCbl riboswitch, the experimental validation of the four-state model (Figure 5d) and the surface-immobilized time-traces in the presence of HyCbl (Figure 7a, Supporting Information Figure S3) strongly suggest that ligand binding (i.e., fluorescence quenching) primarily occurs in the undocked conformation, thus favoring the induced-fit mechanism. This notion is further supported by existing structural data,1 which reveal that the env8HyCbl riboswitch conceals a majority (≈ 60%) of the solvent accessible surface area of the ligand when the KL interaction is formed. These steric constraints would make it difficult for the approximately spherical HyCbl to enter, or exit, the binding pocket when the L5–L13 interaction is formed, resulting in negligibly slow ligand association/dissociation from the docked conformation (Figure 2). As additional support for predominant ligand recognition in the undocked state, previous isothermal titration calorimetry experiments demonstrate that riboswitches lacking L13 can still effectively bind HyCbl, implying that ligand binding does not require the RNA to be in the docked conformation.1 Together, these observations indicate that ligand-recognition follows the induced-fit model, which reinforces the notion that this behavior is commonly associated with many small-molecule binding RNAs.51−53
The detailed kinetic characterization of this riboswitch enables comparisons with other biologically related processes. To do so, we draw on the following principles of chemical kinetics:(i) the rate constant describing the approach to equilibrium (keq) is given by the sum of the forward/reverse rate constants and (ii) the reciprocal of a first-order rate-constant represents the characteristic 1/e time-scale for that particular process. The ligand binding kinetics experiments reveal that the bimolecular appkbind is fast (≥106 M–1 s–1), relative to many other RNA–ligand interactions.54 However, the relatively slow (≤0.01 s–1) appkdiss mandates that, at HyCbl concentrations near appKd, the time-scale associated with ligand binding (i.e., [appkon × appKd]−1 ≈ 200 s) is much longer than that required to establish the two KL docking equilibria (i.e., [kdock,free + kundock,free]−1 ≈ 0.14 s and [kdock,HyCbl + kundock,HyCbl]−1 ≈ 1.1 s). Therefore, at [HyCbl] < 1000 nM (i.e., concentrations where ligand binding and KL docking time scales are comparable), ligand binding represents the rate limiting process in the four-state kinetic model (Figure 2).
The above conclusion raises a significant question with regard to genetic regulation: does the riboswitch-containing mRNA have enough time, after being transcribed, to bind the ligand before being degraded in the cell? Specifically, at [HyCbl] near appKd, the time-scale associated with ligand binding (i.e., ≈ 200 s) is indeed comparable to that of mRNA decay (≈ 300 s).55 Such a comparison suggests that ligand binding may be temporally limited, and thus that regulation of gene expression provided by the env8HyCbl riboswitch may not be completely thermodynamically controlled;54 further quantitative investigations focusing on both mRNA abundance and decay will be required to resolve such issues definitively.
3.4. Mechanism of Regulation for the env8 Hydroxocobalamin Riboswitch
Lastly, we make use of qualitative comparisons between the in vitro and cell-based results in order to obtain information about the relationship between the physical mechanism of the env8HyCbl riboswitch and the biologically relevant ligand-dependent modulation of gene expression. Provided that cells transformed with the L5-mutant env8HyCbl+GFPuv vectors do not suffer from any unforeseen changes in mRNA abundance, they can be used to define the maximum amount of gene expression (Figure 8b) due to their complete inability to form the regulatory KL. The normalized fluorescence (Materials and Methods) associated with maximal gene expression is used to approximate the relative gene expression associated with the WT env8HyCbl riboswitch in the presence or absence of 5000 nM ligand. The approximate quantitation of the cell-based experiments reveals that, in the absence of HyCbl, the env8HyCbl riboswitch encoded upstream of a GFPuv fluorescence reporter results in a relative gene expression value of 0.90(8). This cell-based value is quite similar to the in vitro values of FUfree = 0.83(7) and FUfree ≈ 0.78 (at 1 mM Mg2+) for surface-immobilized and freely diffusing smFRET experiments, respectively. The similarity of these values seems to suggest that gene expression is well correlated with FUfree. Furthermore, the relative gene expression for the WT env8HyCbl in the presence of 5000 nM HyCbl drops to a value of 0.10(3). Again, the HyCbl-dependent decrease in relative gene expression (0.90 → 0.10) observed in the cell-based experiments is comparable to the decrease in the fractional occupancy of the undocked conformation (FU) as determined via smFRET experiments (0.83 → 0.25). Together, the in vitro and cell-based experiments demonstrate that (i) the fractional occupancy of the undocked conformation at 1 mM Mg2+ is strongly correlated with the HyCbl-dependent expression of GFPuv and (ii) the L5-L13 KL interactions is certainly the physical switch associated with regulation of gene expression.
4. Conclusions
This work represents the first quantitative single-molecule investigations of a fully functional riboswitch, complete with complementary cell-based assays. The results of these experiments clearly identify the conformational mechanism responsible for regulation of gene expression by the env8HyCbl riboswitch, which serves as a model system for the entire clan of cobalamin riboswitches. Specifically, ligand binding primarily occurs when the L5–L13 kissing loop is absent and alters the energetics associated with this regulatory kissing-loop (KL), resulting in preferential stabilization of the docked state. In this conformation, the ribosome binding site within L13 makes canonical Watson–Crick base pairing interactions with L5. These base pairs conceal this region of the mRNA from the cellular machinery responsible for translation initiation, thus repressing expression of the downstream gene. Time-resolved ligand-binding experiments indicate that although this process is fast relative to other riboswitches, it is still much slower than the equilibration time-scale for the KL interaction. The high degree of correlation between cell-based GFPuv reporter assays and biophysical fluorescence experiments further supports the notion that formation of the KL interaction is directly responsible for repression of gene expression and identifies this region of the RNA as the physical regulatory switch. This KL interaction shares a number of biochemical features with other functional kissing loop systems, for example, the HIV genome dimerization system and the Rop-RNA I-RNA II plasmid replication system. Together, these similarities highlight the importance of (i) nucleotide sequence, (ii) divalent cations, and (iii) macromolecular binding partners in tuning the stability of this structural motif. One interesting direction for further study would be to more rigorously explore the importance of structural defects within the KL and their ability to confer ligand-facilitated docking. Most importantly, this work presents a detailed kinetic characterization of a ligand-facilitated conformational transition that serves as a point-of-comparison for other such studies of similar RNAs.
5. Materials and Methods
5.1. Fluorescent Construct Design
The env8 riboswitch constructs used in this work are based on a sequence originally identified from comparative metagenomic analyses of functional noncoding RNAs.23 The various constructs designed for single-molecule fluorescence resonance energy transfer (smFRET) experiments are prepared via nonsplinted ligation56 of two custom synthetically modified RNA oligonucleotides (Integrated DNA Technologies). The wild-type env8 HyCbl smFRET construct is assembled from the following two RNAs: WT strand 1 (5′-biotin-AAA AAA AAG GCC UAA AAG CGU AGU GGG AAA G[dT*]G ACG UGA AAU UCG UCC AGA UUA C-3′) and WT strand 2 (5′-phosphate-UUG AUA CGG UUA UAC UCC GAA UGC CAC CUA GGC CAU ACA ACG AGC AAG GAG ACU C-Cy3–3′). According to the X-ray crystal structure of env8HyCbl, nucleotide U24 is completely solvent exposed and lacks intramolecular contacts with other functional groups of the RNA.1 By way of additional confirmation, SHAPE chemical probing experiments reveal this nucleotide to be hyper-reactive, as is often the case with highly solvent exposed nucleotides. In conjunction with the X-ray crystal structure, this suggests that U24 is well suited for synthetic modification. Accordingly, this nucleotide is replaced by an amino-modified deoxynucleotide (dT*), which is subsequently chemically labeled with an NHS-functionalized Cy5 fluorophore (GE healthcare) following the manufacturer’s suggested protocol. Microcentrifuge size-exclusion columns (ThermoScientific) are used to remove the excess unreacted fluorophores from the labeling reaction. The WT strands 1 and 2 are annealed together by slowly cooling from 85 °C to room temperature in annealing buffer (50 mM HEPES, 800 μM hydroxocobalamin (Sigma-Aldrich), 100 mM KCl, pH 7.5) prior to enzymatic ligation via T4 RNA ligase I (New England Biolabs) Note, the slow cooling and lack of Mg2+ was chosen to maximize the abundance of RNA constructs that were in a ligand-responsive conformation (≈ 85%). Isolation of full-length doubly labeled RNA construct is achieved through the use of a reverse phase high-performance liquid chromatography (HPLC) column (Agilent).
Preparation of the L5-mutant construct is carried out using the same procedures, except that L5-mutant strand 2 (5′-phosphate-AAG AUA CGG UUA UAC UCC GAA UGC CAC CUA GGC CAU ACA ACG AGC AAG GAG ACU C-Cy3-3′) is used instead of WT strand 2, with the underlined nucleotides corresponding to the location of the desired mutations. Synthesis of the Cy3-only env8HyCbl construct used in the ensemble fluorometry experiments is performed using in vitro transcription by T7 RNA polymerase, as described previously.1 The resulting transcripts are exposed to NaIO4 and NaBH3CN for selective oxidation of the 3′-ribose57 and reacted with hydrazide-functionalized Cy3 to covalently attach the fluorophore to this position. Removal of excess dye and HPLC purification is performed, as described above, in order to isolate full-length fluorescently labeled RNA constructs.
5.2. Freely-Diffusing smFRET
Freely diffusing smFRET experiments are performed on a home-build inverted confocal fluorescence microscope, described previously,18 with a 1.2 N.A. water objective (Olympus Corporation) in epifluorescence configuration. When the concentration of fluorescently labeled RNA is sufficiently low (≈ 125 pM), individual molecules will transiently diffuse into the overlapping foci of two (532 nm,Time-Bandwidth products and 635 nm, PicoQuant GmbH) alternating laser excitation58 (ALEX) sources with ≈100 μW average power. This yields a short (<1 ms) burst of fluorescence photons dictated by the time the RNA spends within the overlapping excitation and detection volumes. The FRET efficiency (EFRET) resulting from 532 nm excitation is calculated for each fluorescent burst, where the use of ALEX techniques ensures that only dually labeled, nonphotophysically damaged constructs are considered for analysis. All experiments are performed at ambient temperatures (20–22 °C) in standard buffer (50 mM HEPES, 25 mM KOH, 10 mM NaOH, 100 mM KCl, 2 mM TROLOX, 100 nM PCD and 5 mM PCA, pH 7.7) consisting of the well-characterized enzymatic oxygen scavenging system, PCA/PCD,59 and various concentrations of MgCl2 and hydroxocobalamin (HyCbl). Error bars for freely diffusing experiments are often smaller than the associated data points and represent the propagated uncertainty associated with the fractional occupancy of the high FRET state as determined via a nonlinear least-squares (NLLS) fit to a sum of two-Gaussian distributions.
5.3. Ensemble Fluorometry
HyCbl-dependent Cy3 quenching ensemble experiments are performed on a fluorimeter (Jobin Yvon) with ≈500 pM fluorescent RNA in standard buffer containing 1 mM MgCl2 and 10–50 nM hydroxocobalamin (Figure 4). The excitation and emission filters are set to 532 ± 1 and 570 ± 7 nm, respectively, with each data point representing one second of integrated fluorescence. The narrow, off-peak excitation filter ensures that photobleaching of Cy3 represents a negligible contribution to the time-dependent reduction in the fluorescence intensity. Error bars for the ensemble fluorometry experiments represent the standard deviation associated with triplicate measurements of each data point.
5.4. Surface-Immobilized smFRET
Surface-immobilzed smFRET experiments are performed on the same inverted confocal fluorescence microscope used for the freely diffusing experiments. Immobilization is accomplished using biotin–streptavidin chemistry, resulting in a surface coverage of <1 molecule per μm2. Average laser powers are ≈2 μW in order to reduce the effects of photobleaching and prolong the observation of surface immobilized molecules. Fluorescence trajectories are constructed from 30 ms time bins of detected photons. Docking and undocking rate constants are determined using a previously described thresholding routine28 and a Bayesian maximum-likelihood model.38 By way of validating each of these approaches, the two analyses result in rate constants that are experimentally indistinguishable when applied to the same data set. Error bars for surface immobilized experiments are often smaller than the associated data points. They represent the standard deviation derived from NLLS fits to decaying exponentials for three unique cumulative dwell time distributions,60 each containing approximately the same number of dwell times (≈100–500 each).
5.5. Cell-Based GFP Reporter Assays
Riboswitch constructs are amplified via recombinant overlapping PCR and cloned into a pBR327 derivative between NsiI and HindIII restriction sites. For the cell-based assays, plasmids are transformed into E. coli Keio strain Δ△btuR (Keio collection(61) JW1262). The cells are plated onto LB agar plates supplemented with 100 μg/mL ampicillin, and incubated at 37 °C for 14–16 h to facilitate colony formation. Three colonies are picked for each construct and grown to saturation in a rich, chemically defined medium (CSB media: NaH2PO4 (4.6 mg/mL), K2HPO4 (11.7 mg/mL), and (NH4)2SO4 (2.0 mg/mL), d-glucose (0.6%), sodium citrate (5 mM), and MgSO4 (492 μM), FeCl3·6H2O (109 μM), ZnCl2·4H2O (28.3 μM), CoCl2·6H2O (24.8 μM), Na2MoO4 (13.8 μM), CaCl2·2H2O (20.1 μM), CuCl2 (21.9 μM), MnCl2 (23.4 μM), and H3BO3 (23.9 μM)) supplemented with 100 μg/mL ampicillin to maintain plasmid selection. To measure cellular fluorescence resulting from GFPuv expression, 5 μL of saturated overnight culture has been added to 5 mL of CSB medium supplemented with 100 μg/mL ampicillin and 5 μM hydoxocobalamin, and grown to mid-log phase via incubation at 37 °C in a rotating drum. For fluorescence measurements, 300 μL of cells from each biological replicate is added to the wells of a Greiner 96 well, half-area microplate. GFPuv expression is monitored at an excitation wavelength of 395 nm and a 510 nm emission wavelength using a Tecan Infinite M200 PRO plate-reader. The average and standard deviation for the fluorescence of each individual well is determined using three biological replicates measured in triplicate and normalized to cell density as determined by the OD600. Normalized fluorescence values for each construct are background corrected via subtraction of cell-density normalized fluorescence from a pBR327 empty vector control. Error bars represent the propagated uncertainty in the relative gene expression as determined by the standard deviations of the normalized fluorescence values.
Acknowledgments
Support for this work has been provided by the National Science Foundation (CHE 1266416, PHY 1125844), the National Institute of Standards and Technology, and the National Institutes of Health (GM-073850) and their Molecular Biophysics Training Program (T32 GM-065103).
Supporting Information Available
Figures related to the L-5 mutant env8HyCbl riboswitch, the HyCbl-induced fluorescence quenching, and the preference for ligand binding in the undocked conformation. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Johnson J. E. Jr.; Reyes F. E.; Polaski J. T.; Batey R. T. Nature 2012, 492, 133–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker B. J.; Breaker R. R. Curr. Opin. Struct. Biol. 2005, 15, 342–348. [DOI] [PubMed] [Google Scholar]
- Nahvi A.; Sudarsan N.; Ebert M. S.; Zou X.; Brown K. L.; Breaker R. R. Chem. Biol. 2002, 9, 1043. [DOI] [PubMed] [Google Scholar]
- Nou X.; Kadner R. J. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 7190–7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R.; Ragsdale S. W. Annu. Rev. Biochem. 2003, 72, 209–247. [DOI] [PubMed] [Google Scholar]
- Nahvi A.; Barrick J. E.; Breaker R. R. Nucleic Acids Res. 2004, 32, 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov D. A.; Vitreschak A. G.; Mironov A. A.; Gelfand M. S. J. Biol. Chem. 2003, 278, 41148–41159. [DOI] [PubMed] [Google Scholar]
- Martens J. H.; Barg H.; Warren M. J.; Jahn D. Appl. Microbiol. Biotechnol. 2002, 58, 275–285. [DOI] [PubMed] [Google Scholar]
- Roth J. R.; Lawrence J. G.; Bobik T. A. Annu. Rev. Microbiol. 1996, 50, 137–181. [DOI] [PubMed] [Google Scholar]
- Breaker R. R. Cold Spring Harbor Perspect. Biol. 2012, 4, a003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garst A. D.; Edwards A. L.; Batey R. T. Cold Spring Harbor Perspect. Biol. 2011, 3, a003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R.; Hohng S.; Ha T. Nat. Methods 2008, 5, 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemay J. F.; Penedo J. C.; Tremblay R.; Lilley D. M.; Lafontaine D. A. Chem. Biol. 2006, 13, 857–868. [DOI] [PubMed] [Google Scholar]
- Suddala K. C.; Rinaldi A. J.; Feng J.; Mustoe A. M.; Eichhorn C. D.; Liberman J. A.; Wedekind J. E.; Al-Hashimi H. M.; Brooks C. L. 3rd; Walter N. G. Nucleic Acids Res. 2013, 41, 10462–10475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souliere M. F.; Altman R. B.; Schwarz V.; Haller A.; Blanchard S. C.; Micura R. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, E3256–3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller A.; Altman R. B.; Souliere M. F.; Blanchard S. C.; Micura R. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 4188–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller A.; Rieder U.; Aigner M.; Blanchard S. C.; Micura R. Nat. Chem. Biol. 2011, 7, 393–400. [DOI] [PubMed] [Google Scholar]
- Fiegland L. R.; Garst A. D.; Batey R. T.; Nesbitt D. J. Biochemistry 2012, 51, 9223–9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood S.; Ferre-D’Amare A. R.; Rueda D. ACS Chem. Biol. 2012, 7, 920–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peselis A.; Serganov A. Nat. Struct. Mol. Biol. 2012, 19, 1182–1184. [DOI] [PubMed] [Google Scholar]
- Taylor R. T.; Smucker L.; Hanna M. L.; Gill J. Arch. Biochem. Biophys. 1973, 156, 521–533. [DOI] [PubMed] [Google Scholar]
- Wilson R. C.; Smith A. M.; Fuchs R. T.; Kleckner I. R.; Henkin T. M.; Foster M. P. J. Mol. Biol. 2011, 405, 926–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg Z.; Wang J. X.; Bogue J.; Yang J.; Corbino K.; Moy R. H.; Breaker R. R. Genome Biol. 2010, 11, R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S.; Bateman A.; Marshall M.; Khanna A.; Eddy S. R. Nucleic Acids Res. 2003, 31, 439–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.; Li Y.; Liu C.; Wu Q.; Zhao Y.; Lu L.; Tang H. Talanta 2008, 77, 176–181. [DOI] [PubMed] [Google Scholar]
- Deniz A. A.; Dahan M.; Grunwell J. R.; Ha T.; Faulhaber A. E.; Chemla D. S.; Weiss S.; Schultz P. G. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 3670–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamichhane R.; Solem A.; Black W.; Rueda D. Methods 2010, 52, 192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodak J. H.; Downey C. D.; Fiore J. L.; Pardi A.; Nesbitt D. J. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 10505–10510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennifar E.; Walter P.; Ehresmann B.; Ehresmann C.; Dumas P. Nat. Struct. Biol. 2001, 8, 1064–1068. [DOI] [PubMed] [Google Scholar]
- Horiya S.; Li X.; Kawai G.; Saito R.; Katoh A.; Kobayashi K.; Harada K. Chem. Biol. 2003, 10, 645–654. [DOI] [PubMed] [Google Scholar]
- Tyrrell J.; McGinnis J. L.; Weeks K. M.; Pielak G. J. Biochemistry 2013, 52, 8777–8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz J. R.Principles of Fluorescence Spectroscopy; Springer: New York, 2006. [Google Scholar]
- Johnson K. A.; Goody R. S. Biochemistry 2011, 50, 8264–8269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbitt D. J.; Leone S. R. J. Chem. Phys. 1981, 75, 4949–4959. [Google Scholar]
- Trausch J. J.; Ceres P.; Reyes F. E.; Batey R. T. Structure 2011, 19, 1413–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards A. L.; Reyes F. E.; Heroux A.; Batey R. T. RNA 2010, 16, 2144–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reining A.; Nozinovic S.; Schlepckow K.; Buhr F.; Furtig B.; Schwalbe H. Nature 2013, 499, 355–359. [DOI] [PubMed] [Google Scholar]
- Bronson J. E.; Fei J.; Hofman J. M.; Gonzalez R. L. Jr.; Wiggins C. H. Biophys. J. 2009, 97, 3196–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen A. A.; Collins R. A. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 7730–7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toor N.; Keating K. S.; Fedorova O.; Rajashankar K.; Wang J.; Pyle A. M. RNA 2010, 16, 57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunel C.; Marquet R.; Romby P.; Ehresmann C. Biochimie 2002, 84, 925–944. [DOI] [PubMed] [Google Scholar]
- Wagner E. G.; Brantl S. Trends Biochem. Sci. 1998, 23, 451–454. [DOI] [PubMed] [Google Scholar]
- Eguchi Y.; Tomizawa J. Cell 1990, 60, 199–209. [DOI] [PubMed] [Google Scholar]
- Salim N.; Lamichhane R.; Zhao R.; Banerjee T.; Philip J.; Rueda D.; Feig A. L. Biophys. J. 2012, 102, 1097–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmer-Citterich M.; Anceschi M. M.; Banner D. W.; Cesareni G. EMBO J. 1988, 7, 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Primo C. J. Mol. Recognit. 2008, 21, 37–45. [DOI] [PubMed] [Google Scholar]
- Banner D. W.; Kokkinidis M.; Tsernoglou D. J. Mol. Biol. 1987, 196, 657–675. [DOI] [PubMed] [Google Scholar]
- Predki P. F.; Nayak L. M.; Gottlieb M. B.; Regan L. Cell 1995, 80, 41–50. [DOI] [PubMed] [Google Scholar]
- Ottink O. M.; Rampersad S. M.; Tessari M.; Zaman G. J.; Heus H. A.; Wijmenga S. S. RNA 2007, 13, 2202–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang K.; Rieder R.; Micura R. Nucleic Acids Res. 2007, 35, 5370–5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson J. R. Nat. Struct. Biol. 2000, 7, 834–837. [DOI] [PubMed] [Google Scholar]
- Noeske J.; Buck J.; Furtig B.; Nasiri H. R.; Schwalbe H.; Wohnert J. Nucleic Acids Res. 2007, 35, 572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batey R. T.; Gilbert S. D.; Montange R. K. Nature 2004, 432, 411–415. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Lau M. W.; Ferre-D’Amare A. R. Biochemistry 2010, 49, 9123–9131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein J. A.; Khodursky A. B.; Lin P. H.; Lin-Chao S.; Cohen S. N. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 9697–9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M. J., Query C. C.. Uses of Site-Specifically Modified RNAs Constructed by RNA Ligation. In RNA:Protein Interactions: A Practical Approach; Smith C. W. J., Ed.; Oxford University Press, Inc.: New York, 1998; pp 75–140. [Google Scholar]
- Proudnikov D.; Mirzabekov A. Nucleic Acids Res. 1996, 24, 4535–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapanidis A. N.; Laurence T. A.; Lee N. K.; Margeat E.; Kong X.; Weiss S. Acc. Chem. Res. 2005, 38, 523–533. [DOI] [PubMed] [Google Scholar]
- Aitken C. E.; Marshall R. A.; Puglisi J. D. Biophys. J. 2008, 94, 1826–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco M.; Walter N. G. Methods Enzymol. 2010, 472, 153–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T.; Ara T.; Hasegawa M.; Takai Y.; Okumura Y.; Baba M.; Datsenko K. A.; Tomita M.; Wanner B. L.; Mori H. Mol. Syst. Biol. 2006, 220060008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.