

Abstract

Lanthipeptides are a class of ribosomally synthesized and posttranslationally modified peptide natural products (RiPPs) that typically harbor multiple intramolecular thioether linkages. For class II lanthipeptides, these cross-links are installed in a multistep reaction pathway by a single enzyme (LanM). The multifunctional nature of LanMs and the manipulability of their genetically encoded peptide substrates (LanAs) make LanM/LanA systems promising targets for the engineering of new antibacterial compounds. Here, we report the development of a semiquantitative mass spectrometry-based assay for kinetic characterization of LanM-catalyzed reactions. The assay was used to conduct a comparative kinetic analysis of two LanM enzymes (HalM2 and ProcM) that exhibit drastically different substrate selectivity. Numerical simulation of the kinetic data was used to develop models for the multistep HalM2- and ProcM-catalyzed reactions. These models illustrate that HalM2 and ProcM have markedly different catalytic efficiencies for the various reactions they catalyze. HalM2, which is responsible for the biosynthesis of a single compound (the Halβ subunit of the lantibiotic haloduracin), catalyzes reactions with higher catalytic efficiency than ProcM, which modifies 29 different ProcA precursor peptides during prochlorosin biosynthesis. In particular, the rates of thioether ring formation are drastically reduced in ProcM, likely because this enzyme is charged with installing a variety of lanthipeptide ring architectures in its prochlorosin products. Thus, ProcM appears to pay a kinetic price for its relaxed substrate specificity. In addition, our kinetic models suggest that conformational sampling of the LanM/LanA Michaelis complex could play an important role in the kinetics of LanA maturation.
Introduction
Class II lanthipeptide synthetases (generically known as LanM enzymes) are multifunctional enzymes that catalyze the posttranslational modification of genetically encoded precursor peptides (termed LanA peptides) into products (lanthipeptides) containing lanthionine (Lan, 1) or methyllanthionine (MeLan, 2) moieties (or both) (Scheme 1). The LanA precursor peptides are composed of an N-terminal leader peptide and a C-terminal core peptide that harbors the posttranslational modifications. Lanthipeptides belong to a growing class of ribosomally synthesized, and posttranslationally modified peptide (RiPP) natural products, many of which possess antibacterial or other biological activities.1−3 From in vitro studies of the LanM enzymes characterized to date,4−6 the chemical mechanism leading to the biosynthesis of the (Me)Lan moieties (Scheme 1) is known to involve ATP-dependent phosphorylation of serine (Ser) or threonine (Thr) residues located in the C-terminal core peptide portion of the LanA precursor peptide, followed by elimination of the phosphates to generate dehydroalanine (Dha) or dehydrobutyrine (Dhb) residues, respectively. Intramolecular Michael-type addition of Cys thiols within the core peptide onto these Dha and Dhb residues completes the biosynthesis of the (methyl)lanthionine moiety. Remarkably, the multifunctional LanM enzymes catalyze all of these reactions, usually at multiple sites within the LanA core peptide, and typically with regio- and stereospecific control over the cyclization reactions.
Scheme 1. Chemical Mechanism of a Typical LanM-Catalyzed Reaction.
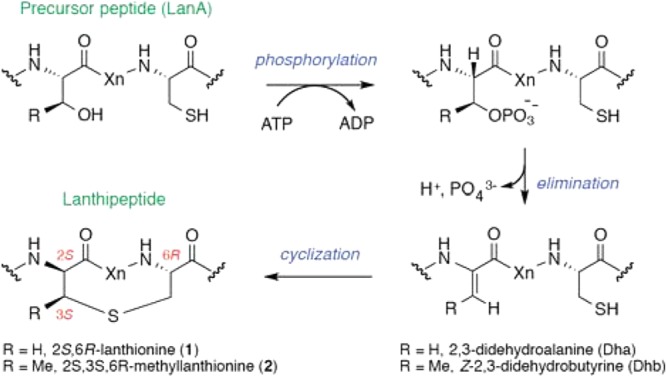
Despite our steadily increasing understanding of the chemical mechanisms of lanthipeptide biosynthesis and of the substrate specificity and evolution of LanM enzymes,7 the complexity in the maturation pathways of lanthipeptides has severely hindered development of quantitative methods to compare the kinetics of different lanthipeptide biosynthetic enzymes. As a result, very little is known regarding how LanM enzymes orchestrate their multiple activities in time and space to achieve biosynthesis of structures with defined (Me)Lan ring architectures, which can be critical for maintaining biological activity of the final compound.8−12 Furthermore, as more LanM/LanA systems are being investigated, it is becoming apparent that they often have subtle differences in their catalytic properties that make mechanistic generalizations between biosynthetic systems difficult. These differences include variable directionality of the LanM-installed modifications within the LanA substrate,6,13 different roles for the N-terminal leader peptide portion of the LanA substrate,14−16 and differences in the stereochemistry of the Lan and MeLan rings.17
Clearly, kinetic assays capable of the simultaneous quantitation of time-dependent changes in the concentrations of substrate, partially modified LanA reaction intermediates, and final product will be helpful to better identify, quantify, and define these interesting differences within LanM enzymes. In addition, a quantitative kinetic assay would enable molecular details of LanM catalyzed reactions to be elucidated and could perhaps illuminate specific features that contribute to leader peptide-dependent enzyme activation,16,18,19 substrate selectivity (or flexibility), and catalysis. A more thorough understanding of LanM function could also facilitate the engineering of novel, biologically active lanthipeptide derivatives,10,12,20−22 and a general kinetic method could also be applied to answer similar questions for other in vitro reconstituted class I–IV lanthipeptide synthetases.23−27
Because of its high sensitivity and potential ability to detect and distinguish most of the relevant peptide species in a typical LanM/LanA reaction, we reasoned that electrospray ionization mass spectrometry (ESI-MS) would be the analytical method of choice for the development of a general kinetic assay for LanM synthetases. Tandem ESI-MS has been previously employed to establish the order and directionality of the posttranslational modifications installed by several classes of lanthipeptide synthetases,6,13,24,28,29 but no attempt was made in these previous studies to quantify the relative concentrations of the various intermediates that were detected or how they interconverted in a time-dependent manner.
In the present work, we expand on these previous studies and describe a semiquantitative top-down liquid chromatography electrospray ionization mass spectrometry (LC/ESI-MS)-based assay that was used for the characterization of the kinetic properties of two LanM synthetases. The assay operates under the hypothesis that the LanM-installed post-translational modifications (phosphorylation, elimination, and cyclization) will not drastically alter the ionization properties of full-length LanA peptides during electrospray, such that a linear relationship between LC/ESI-MS signal and peptide concentration is maintained.30−34 To test this hypothesis, we designed several validation experiments that can be readily applied to develop semiquantitative mass spectrometry-based kinetic assays for other RiPP biosynthetic enzymes. The optimized assay, combined with numerical simulation of the kinetic data, was then used to compare the kinetic properties of the class II lanthipeptide synthetases, HalM2 and ProcM, in reactions with their substrates (His6P-HalA2 and ProcA2.8, respectively). Collectively, these studies mark the first measurement of kinetic parameters for any lanthipeptide synthetase and have revealed several stark differences in the kinetic properties of HalM2 and ProcM, which we interpret in terms of the putative evolutionary pressures placed on these enzymes. More detailed characterization of HalM2, ProcM, and other lanthipeptide biosynthetic enzymes is possible now that a reliable and semiquantitative kinetic assay is in hand.
Results and Discussion
Overview of a Mass Spectrometry-Based Kinetic Assay for LanM Synthetases
We will first describe several key aspects of the approach that allowed quantitative measurements of the myriad reactions catalyzed by LanM enzymes. Briefly, 100 μL aliquots were removed at desired time points from small-scale (1.5 mL) LanM/LanA reaction mixtures containing 1 μM LanM and 40 μM LanA and were quenched and diluted 10-fold into acidic media (1 mM EDTA, 10 mM TCEP, 100 mM citrate, pH 3.5) to inactivate the synthetase (Supporting Information (SI) Figure S1) and to maintain Cys residues in their reduced state.35 The conjugate addition reaction of a Cys to a dehydroamino acid does not result in a change of mass; therefore, cyclized and uncyclized peptides cannot be distinguished by MS. However, the differential reactivity of thiols and thioethers toward electrophiles can be used to differentiate uncyclized from cyclized peptides. Therefore, following a 10 min incubation period in the quench buffer, free Cys residues in the sample were alkylated with 10 mM N-ethylmaleimide (NEM). NEM was chosen as the alkylation agent primarily for its small size (125.13 Da), fast thiolate alkylation kinetics, and its selectivity for thiolates at pH values below 7.0.36 To facilitate discussion of the observed ions, all kinetic assays in this study included this alkylation step unless otherwise indicated. Each alkylated time point aliquot was then desalted by C4 solid phase extraction (C4-SPE) and analyzed by LC/ESI-MS. The relative concentrations of peptides present in the sample were quantified from the integrated peak areas of the extracted ion chromatograms (EICs) of the ions of interest. Successful implementation of this kinetic assay for LanM/LanA systems required an efficient and chemoselective method for alkylation of the Cys residues of LanA peptides, a linear relationship between the concentration and ESI-MS signals of the relevant peptide species, and similar ionization efficiencies of the LanA peptide species that were quantified (starting material, intermediates, and final product). Validation experiments that show that the assay fulfills these criteria are presented in the following section for both the HalM2 and ProcM-catalyzed reactions.
Validation of the LC/ESI-MS Kinetic Assay
The reactions catalyzed by HalM2 and ProcM that were investigated in this work are shown in Scheme 2. HalM2 catalyzes the post-translational maturation of the His-tagged HalA2 precursor peptide 3 into the fully modified HalA2 species 4. Removal of the leader peptide leader from species 4 yields the Halβ peptide of the two-component lantibiotic haloduracin. In total, the HalM2-catalyzed reaction involves seven phosphoryl transfers, seven phosphate eliminations, and the formation of four nonoverlapping thioether rings. The ProcM/ProcA2.8 reaction is less complex, involving only two dehydrations and the formation of two nonoverlapping Lan rings (5 → 6; Scheme 2).
Scheme 2. Posttranslational Maturation of His6P-HalA2 and ProcA2.8 Catalyzed by HalM2 and ProcM, Respectively.
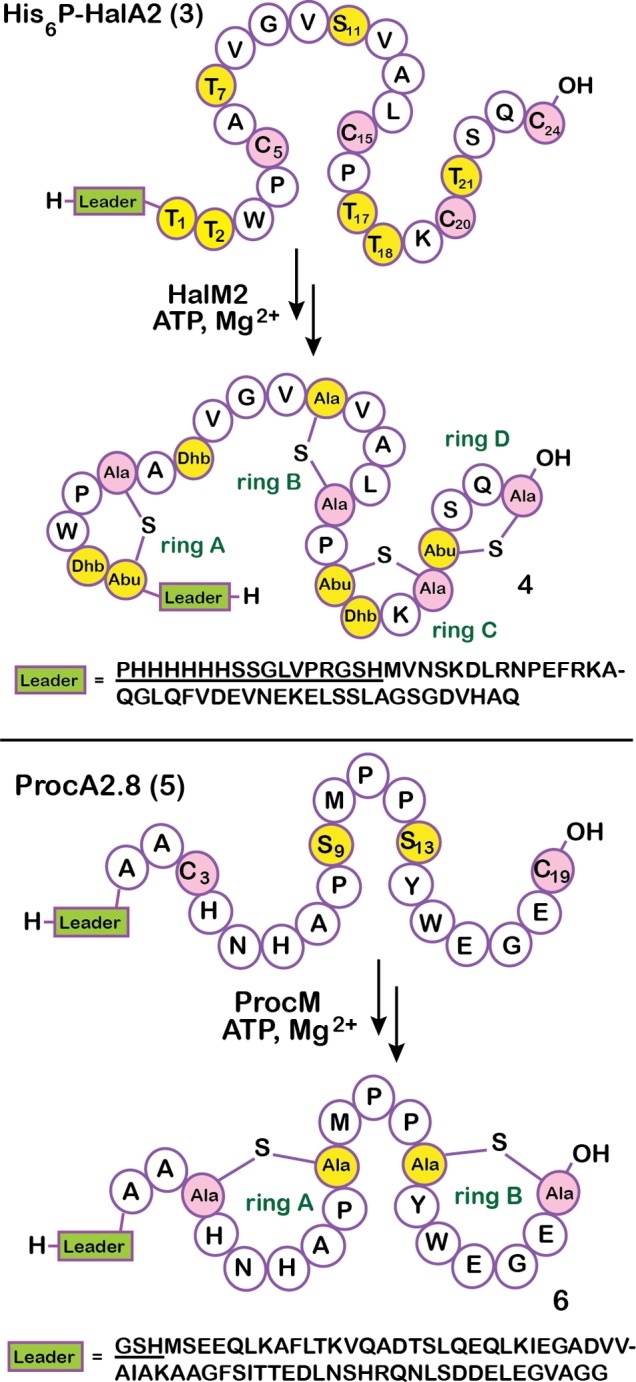
The Ser and Thr residues to be dehydrated are yellow, and the Cys residues involved in cyclization are pink. Amino acid residues are numbered here and throughout the text according to their position relative to the start of the core peptide. The underlined portion of the leader sequence for each peptide is derived from the vector used for expression of the peptide and is not part of the native amino acid sequence. Dhb, dehydrobutyrine; Abu, 1-α-aminobutric acid.
ProcA2.8 is the precursor peptide to one of 29 prochlorosins produced by the marine cyanobacterium, Prochlorococcus MIT9313.37 In an attempt to compare and contrast the catalytic properties of HalM2 and ProcM, we conducted in vitro kinetic assays of the reactions depicted in Scheme 2. The time points were analyzed by top-down LC/ESI-MS to detect and quantify full-length (nonproteolyzed) reaction intermediates. The His6P-HalA2- and ProcA2.8-derived peptides eluted from the LC column as single broad peaks (SI Figure S2). A comparison of the mass spectra for selected time points in the HalM2 reaction (Figure 1A,B) shows that the starting material (detected as its 4-fold alkylated derivative 7) is converted through a series of intermediates (8–24) to the final, fully modified His6P-HalA2 product (4). For ease of discussion, throughout the text, we will refer to peptide species by referencing their corresponding NEM-alkylated derivatives, which, as discussed in the previous section, reports on the cyclization state. Similarly, the ProcA2.8 starting material (detected as its 2-fold alkylated derivative 26) was converted by ProcM via intermediates (27–41) to the fully modified ProcA2.8 product (6, Figure 1C,D). Close inspection of the t = 0 time points for these reactions shows very efficient NEM-mediated alkylation of Cys residues in both the His6P-HalA2 and ProcA2.8 precursor peptides (Figure 1B,D, respectively); neither nonalkylated nor partially alkylated forms of the His6P-HalA2 and ProcA2.8 starting materials were observed. Both enzymatic reactions proceeded to near completion (Figure 1B,D, bottom panels), with the vast majority of the detectable His6P-HalA2- and ProcA2.8-derived peptides corresponding to the expected reaction products of the HalM2- and ProcM-catalyzed reactions: compounds 4 and 6, respectively (Scheme 2). Samples derived from these reactions were proteolytically digested and reanalyzed by LC/ESI-MS. The smaller size of the proteolytic fragments generated by these digestions enabled isotopic resolution and more confident identification of the major peptide ion signals present in these reactions (SI Figures S3 and S4, and Tables S1 and S2).
Figure 1.

Mass spectral time courses for the HalM2/His6P-HalA2 and ProcM/ProcA2.8 reactions. Both reactions were conducted with 1 μM LanM and 40 μM LanA. Time-dependent changes in the electrospray ionization mass spectra are shown for peptide samples recovered from the HalM2- and ProcM-catalyzed reactions (panels A–B and C–D, respectively) at the indicated time points. The peptide samples were alkylated with N-ethylmaleimide. All peptides from both reactions were detected in several different multiply charged ionic states (panels A and C). A closer view of the 8+ ion families is shown in panels B and D. The starting material, reaction intermediates, and final products for the two reactions are numbered. The assignment of these MS signals to specific peptide ions is detailed in Figures S3 and S4 and in Tables S1 and S2 of the Supporting Information. The t = 0 min spectra show that partially alkylated precursor peptide derivatives could not be detected following alkylation of the samples with NEM under our conditions. Instead, only the fully NEM-alkylated forms of His6P-HalA2 and ProcA2.8 (7 and 26, respectively) were observed.
One critical feature of the LC/ESI-MS data is depicted in Figure 1A,C. Several different multiply charged ionic states of each peptide species are visible in the time-of-flight mass spectrum. This permitted calculation of the relative abundance of each peptide species using every charge state that made a significant contribution to the mass spectrum and effectively provided multiple measurements of the fractional abundance of each peptide species in every sample that was analyzed. To mitigate the slight charge state-dependent differences in relative ion intensities that are often observed in mass spectra, the fractional abundances calculated at each charge state were weighted by the contribution of that charge state to the total spectrum. In doing this, a charge-state-weighted average for the fractional abundance of each peptide species was calculated. The procedure for determining the relative concentrations of LanM/LanA reaction intermediates is described further in the Methods section and is adapted from previous work on non-RiPP systems.30,31
Before detailed kinetic analysis of the data shown in Figure 1 could be performed, several important validation experiments were necessary. First, the range over which the LC/ESI-MS signals varied linearly as a function of peptide concentration had to be determined (Figure 2). Serial dilutions of the unmodified ProcA2.8 and His6P-HalA2 peptides ranging from 0.31 to 20 μM were analyzed in duplicate by LC/ESI-MS under conditions that were identical to those used for analysis of the kinetic assay samples. This experiment yielded excellent linearity between the peptide concentration and the total EIC signal for both peptides (R2 > 0.99). In a similar fashion, the linearity of the signals associated with reaction intermediates was assessed by analyzing serial dilutions of the 8 and 90 min time points from the HalM2 and ProcM reactions, respectively. These time points were chosen because most of the relevant peptide species were represented in the corresponding mass spectra (Figure 1).
Figure 2.
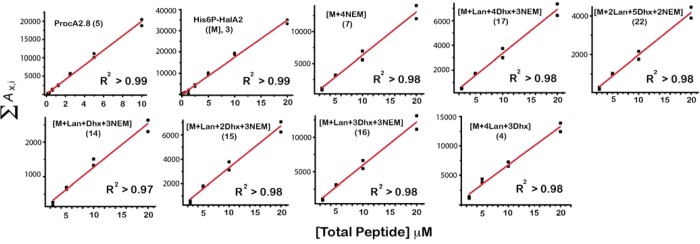
Linear relationship between LC/ESI-MS signals and peptide concentration for the ProcA2.8 and His6P-HalA2 precursor peptides and for a series of intermediates formed during catalysis by HalM2. Peptide samples were serially diluted in duplicate and analyzed by LC/ESI-MS, and the total extracted ion chromatogram peak area for each peptide (∑Ax,i, as defined in the Methods) was plotted vs concentration and fitted with a line. The charge states used for the calculation of ∑Ax,i for the His6P-HalA2- and ProcA2.8-derived peptides are shown in Figure 1A and C, respectively. The ProcA2.8 (5) and His6P-HalA2 (3) samples were pure preparations of the respective, unmodified precursor peptides. The linear ranges for His6P-HalA2-derived compounds 4, 7, 14, 15, 16, 17, and 22 were determined by serial dilution and LC/ESI-MS analysis of the 8 min time point from the HalM2/His6P-HalA2 reaction, as described in the SI, supporting methods. The linear range for all additional HalM2/His6P-HalA2 and ProcM/ProcA2.8 reaction intermediates included in the kinetic analysis are shown in SI Figures S5 and S6, respectively.
As is shown in Figure 2 for the HalM2/His6P-HalA2 reaction, a linear response between the LC/ESI-MS signal and peptide concentration was obtained for all of the major ion signals. Similarly, each of the major species in the ProcM-catalyzed reaction as well as other minor species in both reactions exhibited linear signal responses in this concentration range (SI Figures S5 and S6). To remain within the linear signal range of the assay, all kinetic samples were injected into the LC/ESI-MS instrument at total peptide concentrations of 10 and 20 μM for the ProcM- and HalM2-catalyzed reactions, respectively. These data suggest that suppression of peptide ionization during LC/ESI-MS is either minimal or is equally exerted on all species under our conditions, despite the coelution of the relevant peptide species from the LC column (SI Figure S2).
Finally, the LC/ESI-MS kinetic assay requires the ionization efficiencies of the LanA substrate, reaction intermediates, and product to be similar because the method is a relative quantitation technique in which the signal for each peptide in the reaction mixture is normalized by the sum of the signals for all of the peptides. To test this critical requirement, we devised a simple experiment that should be generally applicable to other lanthipeptide biosynthetic systems. We carried out HalM2 and ProcM reactions under the standard reaction conditions given in the Methods and quenched the samples after 8 or 90 min, respectively. Half of each quenched sample was alkylated with NEM under the standard conditions given in the Methods, and the other half was left untreated. As expected, the LC/ESI-MS signals are different in the alkylated and nonalkylated samples for the HalM2 and ProcM reactions (Figure 3), even though the MS signals reflect the same underlying distribution of reaction intermediates.
Figure 3.
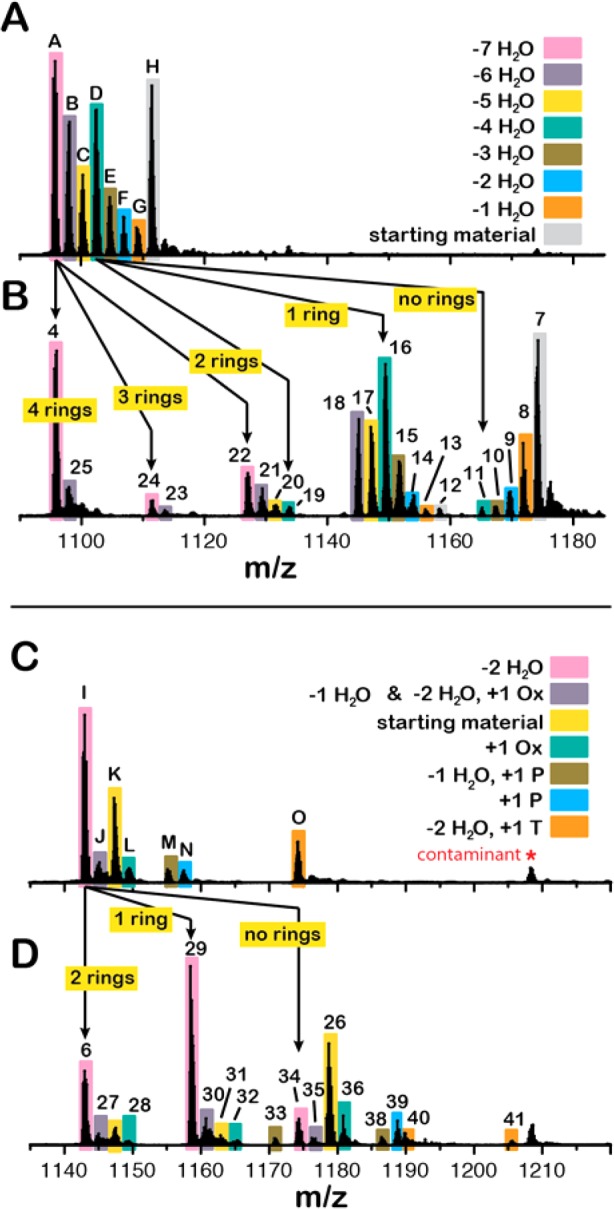
Effect of NEM alkylation on peptide ionization efficiency. A HalM2/His6P-HalA2 reaction was quenched after 8 min and split into two portions that were either untreated or alkylated with NEM. LC/ESI-MS analysis of these samples allowed for comparison of the relative abundances of the nonalkylated peptide signals (panel A) with their respective NEM-alkylated daughter signals (panel B) to determine the extent to which NEM alkylation alters peptide ionization efficiency (SI Table S3). The plots in panels A and B are shown on the same m/z scale to illustrate the shift in peaks observed upon NEM treatment. Each addition of NEM indicates that a free Cys was present and, hence, that a thioether ring was not formed. The resolution of peaks A (into 4, 24, and 22) and D (into 19, 16, 11) afforded by NEM treatment is illustrated. Similarly, the relative abundances of nonalkylated peptides derived from the 90 min time point of a ProcM/ProcA2.8 reaction (panel C) were compared with their respective daughter signals present in the NEM alkylated sample (panel D). In this reaction, phosphorylated peptides (P), oxidized species (Ox), and TCEP adducts (T) were observed in addition to dehydration, cyclization, and NEM-alkylation. The relative abundance values for these data were calculated as described in the text and are shown in SI Table S3. For an additional description of the grouping of the ProcA2.8 peptides given in panel C, see Table S3 in the Supporting Information.
Because the cyclization reaction does not involve a change in mass, the signals in the nonalkylated samples could theoretically consist of multiple, isobaric peptide species that differ only in the number of (methyl)lanthionine rings. In the alkylated samples, these otherwise isobaric signals are resolved into a set of MS signals that reflect the addition of NEM moieties onto Cys thiols and, hence, the extent of thioether ring formation. As long as NEM alkylation does not drastically alter the ionization efficiencies of the peptides, then the fractional abundance of a given species in the nonalkylated sample should be identical to the fractional abundance of the sum of its alkylated and nonalkylated species in the NEM-treated sample. Indeed, excellent agreement was found between the fractional abundances of parent and NEM-treated peptide signals for both the HalM2- and ProcM-catalyzed reactions (SI Table S3). These data strongly suggest that NEM alkylation has a minimal effect on the ionization efficiency of HalM2/His6P-HalA2 and ProcM/ProcA2.8 reaction intermediates.
To test whether phosphorylation of ProcA2.8 affects the LC/ESI-MS signal, we enzymatically synthesized a phosphorylated ProcA2.8 variant38 and compared its ionization efficiency with that of the nonphosphorylated peptide in an LC/ESI-MS coinjection experiment (SI Figure S7). This experiment suggested that the effects of phosphorylation on ProcA2.8 ionization under our experimental conditions are negligible. The relatively large sizes of the His6P-HalA2- and ProcA2.8-derived peptide intermediates (all >8500 Da) likely helps to mask potential differences in peptide ionization efficiency. These findings suggest that similar top-down mass spectrometry-based methods could likely be applied to study the kinetic properties of many other lanthipeptide biosynthetic systems as well as the maturation of other classes of RiPPs. Having validated several key features of the LC/ESI-MS assay, we turned our attention to using this analytical method to characterize the multistep reaction pathways mediated by HalM2 and ProcM.
Overview of the HalM2-Catalyzed Reaction
In an attempt to characterize the kinetics of the HalM2-catalyzed reactions (3 → 4, Scheme 2), time-dependent changes in the concentrations of the HalM2/His6P-HalA2 reaction intermediates were determined. Time courses for the 14 different species included in the kinetic analysis are shown in Figure 4 along with progress curves simulated using the mechanism shown in Scheme 3. The relevant His6P-HalA2-derived species for this analysis include the starting material (7), species with zero (8–10), one (14–18), or two (19–22) thioether rings that differ in the number of dehydrated Ser/Thr residues, the final product containing four thioether rings and three Dhb residues (4) and adducts thereof (SI Figure S3), whose fractional abundance was summed with compound 4 for analysis. Cumulatively, these species account for more than 95% of the total peptide signal at all time points in the reaction.
Figure 4.
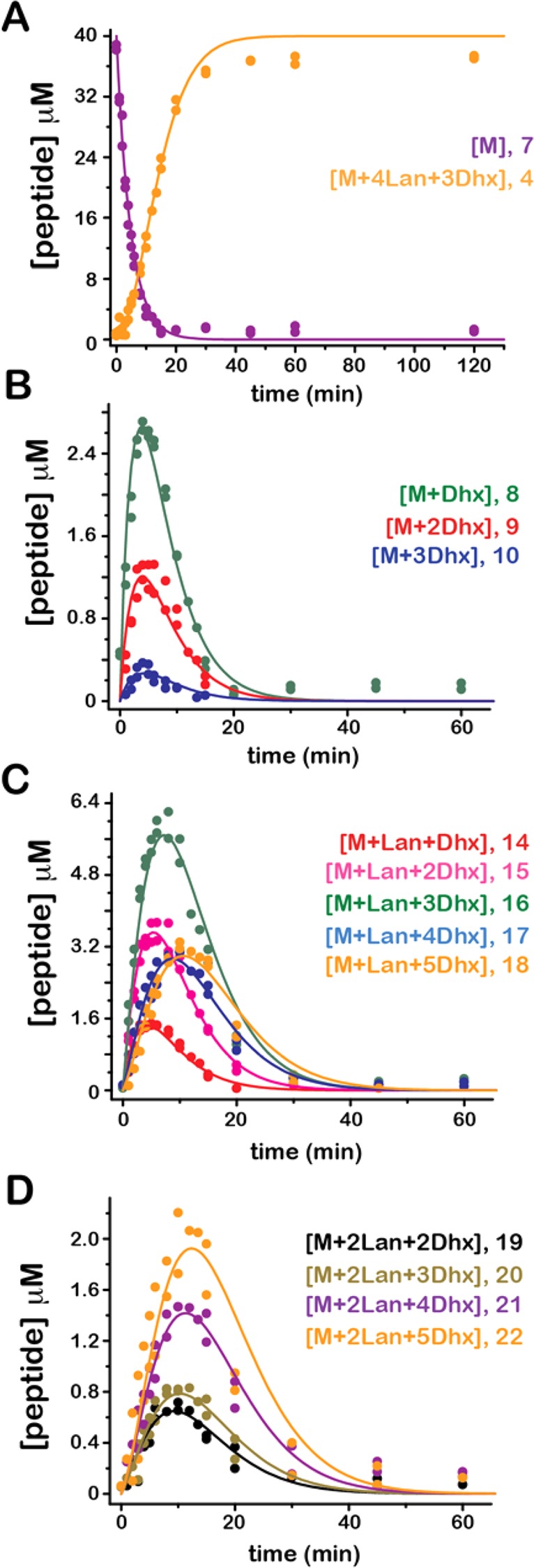
Time courses for peptide species involved in the HalM2/His6P-HalA2 reaction. The charge-state-weighted fractional abundances of peptide species were determined as described in the methods, converted to peptide concentration, and plotted vs reaction time. The major species included the starting material and final product (7 and 4, respectively, panel A), species with no thioether ring and 1–3 dehydrations (8–10, panel B), species with 1 thioether ring and 1–5 additional dehydrations (14–18, panel C), and species with two thioether rings and 2–5 additional dehydrations (19–22, panel D). The progress curves overlying the data were generated by numerical simulation with the kinetic model shown in Scheme 3 and the rate constants given in Table 1. The reaction does not go to completion, leading to slight discrepancies between the model and the data at longer time points. The FitSpace Explorer calculation for this global fit, showing the extent to which the variable parameters in the kinetic model are constrained by the data, is shown in Figure S13 of the Supporting Information. Two additional low-abundance species, each containing three thioether rings (compounds 23 and 24) were detected in the mass spectra for the reaction but were not included in this model. A preliminary kinetic model that accounts for these two species is presented in the Supporting Information.
Scheme 3. Kinetic Model for HalM2-Catalyzed Maturation of His6P-HalA2.

For clarity, peptide binding and dissociation steps from the enzyme are not shown. These steps were included in the model (Scheme 4) and were held constant for each species as described in the text. Rate constants are color-coded: black, allowed to vary freely during nonlinear regression; all other colors, held at fixed ratio relative to each other (e.g. k9→14/k10→15 = 1.0). The progress curves simulated for each species in this mechanism are shown along with the experimental data in Figure 4. Compound numbering corresponds to the NEM-alkylated forms of these peptides, which were observed in the mass spectra for the reaction (Figure 1B) and are assigned in the Supporting Information (Figure S3, Table S1). Species 24 was not included in the simulated kinetic model and is shown here only to illustrate that it is likely an intermediate during the conversion of 22 → 4 that is consumed at a net rate faster than it is formed.
A minimal kinetic model for the HalM2-catalyzed reaction is depicted in Scheme 3. Rate constants for this mechanism were determined by numerical simulation of the data shown in Figure 4 with KinTek Explorer39,40 using several simplifying assumptions. Because it is not possible to obtain the reaction intermediates in pure form, binding constants were not determined experimentally. Instead, the bimolecular HalM2 binding kinetics with each His6P-HalA2-derived species were assumed to be leader-peptide-dependent and identical, such that the peptide binding and dissociation rates (kon and koff, respectively) could be held fixed during nonlinear regression. This assumption is reasonable given the established role of LanA leader peptide sequences in directing LanM binding and activation14,15,18,41 and the fact that the leader peptide is not modified during HalA2 maturation. The value for Kd used in the simulations (1.8 μM) was derived from fluorescence polarization measurements of the HalM2/HalA2 binding interaction (SI Figure S8). The peptide dissociation rate used in the simulations (koff = 18 min–1) was derived from single molecule fluorescence binding measurements that are described in the Supporting Information (Figures S9–S11, Table S4).
The second-order binding rate constant used in the simulations (kon = 10 μM–1 min–1) is derived from the estimated values of Kd, koff, and the relation: Kd = koff/kon. For our second major assumption, the dehydration and cyclization reactions were assumed to be irreversible. The dehydration is coupled to the energetically favorable cleavage of the Pγ moiety of ATP, which should strongly favor the net dehydration of Ser/Thr residues.
Regarding the cyclization reaction, the opening of a LanM-installed thioether ring to regenerate the free Cys and the Dhx residue has never been reported,13 and LanM reactions are typically driven to near completion under in vitro conditions (e.g., Figure 1). This observation suggests that, even if reversible, the equilibrium likely strongly favors the cyclized thioether for most lanthipeptides. Phosphorylated His6P-HalA2 species were not observed in the reaction mass spectra, so phosphorylation rates were not explicitly included in the model. Finally, the 5 mM ATP concentration was assumed to be constant and at saturating levels for the duration of the reaction. In support of this assumption, a control reaction performed at lower ATP concentration (500 μM) resulted in similar kinetics for the formation of the fully modified product (SI Figure S12).
Simulated progress curves for each of the major species according to this mechanism are shown along with the data in Figure 4, revealing an overall satisfactory fit (χ2/degrees of freedom (DoF) = 1.187; see the Methods section for a description of the nonlinear fitting procedure). To achieve a fit of the data with this kinetic model, rate constants were first manually adjusted using the dynamic simulation capabilities of KinTek Explorer to give progress curves that approximated the experimental data as closely as possible. Preliminary analysis using the FitSpace Explorer suite of KinTek Explorer revealed that some of the rate constants in the HalM2 kinetic model were not well constrained by the data; however, these steps had to be included in the mechanism to account for the peptide species that were observed in the mass spectra for the reaction. To overcome this limitation of the model, the magnitudes of these poorly constrained rate constants were first estimated by dynamic simulation and were then held at fixed ratios relative to another rate constant during nonlinear least-squares regression. The rate constants that were linked at fixed ratios with other rate constants are indicated in both Scheme 3 and Table 1, and the rationale for choosing how to link these poorly constrained rate constants during fitting is discussed further below. After a reasonable nonlinear fit was obtained, FitSpace Explorer was used to verify that the magnitude of each variable rate constant in the kinetic model was well-constrained by the data and to calculate upper and lower boundaries for each parameter (SI Figure S13). The best-fit values and boundaries for each variable rate constant in the mechanism are summarized in Table 1. Several additional kinetic models for the HalM2/His6P-HalA2 reaction were also considered and are discussed in the Supporting Information (Scheme S1). We emphasize that these alternative models produced sets of rate constants that were very similar to those given by the model shown in Scheme 3 (SI Table S5).
Table 1. Summary of Simulated Rate Constants for the HalM2/His6P-HalA2 Reaction.
| kx | best fit ± SE (min–1) | FitSpace boundaries (min–1)b | knet (μM–1 min–1)c | max concn (μM)d | processivity factore |
|---|---|---|---|---|---|
| kon | 10a | held constant | |||
| koff | 18 | held constant | |||
| k7→8 | 17 ± 0.8 | 15.1–18.8 | 4.9 | 0.94 | |
| k8→9 | 61 ± 3 | 57.1–70.2 | 7.7 | 2.7 | 3.4 |
| k9→10 | 25 ± 3 | 15.7–38.4 | 5.8 | 1.3 | 1.4 |
| k9→14 | 104 ± 5 | 90.7–117 | 8.5 | 1.3 | 5.8 |
| k10→15 | 104 | fixed relative to k9→14 | 8.5 | 0.4 | 5.8 |
| k14→15 | 81 ± 4 | 70.3–89.1 | 8.2 | 1.5 | 4.5 |
| k15→16 | 36 ± 1.3 | 32.9–40.3 | 6.7 | 3.7 | 2.0 |
| k16→17 | 16 ± 0.8 | 13.9–17.6 | 4.7 | 6.2 | 0.89 |
| k17→18 | 23 ± 1.7 | 18.5–28.9 | 5.6 | 3.1 | 1.3 |
| k16→19 | 2.5 ± 0.26 | 1.88–3.18 | 1.2 | 6.2 | 0.14 |
| k17→20 | 5.0 | fixed relative to k16→19 | 2.2 | 3.1 | 0.28 |
| k18→21 | 20 ± 1.8 | 16.1–25.2 | 5.3 | 3.3 | 1.1 |
| k19→20 | 19 ± 2.7 | 12–29.3 | 5.1 | 0.8 | 1.1 |
| k20→21 | 32 ± 1.9 | 26.6–39.5 | 6.4 | 0.8 | 1.8 |
| k21→22 | 58 | fixed relative to k20→21 | 7.6 | 1.5 | 3.2 |
| k22→4 | 41 | fixed relative to k20→21 | 6.9 | 2.2 | 2.3 |
kon is in units of μM–1 min–1.
FitSpace boundaries are given as 1.12 times the χ2 minimum of the best fit. See Methods for additional details.
knet = (konkx)/(koff + kx). knet is a second-order rate constant that serves as an approximation of kcat/Km.
Refers to the maximum observed concentration of the substrate for the indicated reaction. The total peptide concentration was 40 μM.
Processivity factor is defined as kx/koff
A Description of the HalM2-Catalyzed Reaction
In the initial phase of the reaction, the starting material is converted to a twice-dehydrated species (7 → 8 → 9, Scheme 3) and then to a species with one thioether ring and three dehydrations (15). The observed intermediates show that the pathway leading from 9 to 15 can occur through one of two routes (9 → 14 → 15 or 9 → 10 → 15, Scheme 3), but the simulations suggest that the bulk of the reaction flux (81%) follows the path (9 → 14 → 15). The very small quantities of peptide containing more than three dehydrations and no thioether rings suggests that the first cyclization event typically occurs prior to the fourth dehydration. Tandem MS studies have localized the first three dehydrations to Thr1, Thr2, and Thr7 and the first cyclization to the attack of Cys5 on Dhb1 to form methyllanthionine ring A (Scheme 2).6 The steps that result in the net conversion of 9 → 15 include the three fastest reactions in the model (k9→14 = k10→15 = 104 min–1, and k14→15 = 81 min–1). The rates for the two possible cyclization steps leading to ring A (k9→14 and k10→15) were not individually well constrained by the data. Since the fitted values for these constants tended to be similar in magnitude and faster than the other rate constants in the model during our initial simulations, we held the magnitudes of these two rate constants equal to each other during fitting. As indicated by the estimated processivity factors for these steps (Table 1), much of the reaction flux from 9 → 15 may occur in a processive manner without dissociation of the intermediates from HalM2. The His6P-HalA2 residues involved in these fast phase reactions (Thr1, Thr2, Cys5, and Thr7) are all closely spaced on the N-terminus of the His6P-HalA2 core peptide (Scheme 2) and, as such, can perhaps be modified rapidly by the active site(s) of the synthetase without the need for extensive conformational sampling within the HalM2/His6P-HalA2 Michaelis complex or for enzyme–substrate dissociation.
After the installation of MeLan ring A, the majority of the remaining dehydrations appear to be carried out by HalM2 prior to formation of the second thioether ring. This order is evidenced by the relatively large quantities of species 15–18 that accumulate during the reaction and by the low-level accumulation of partially dehydrated intermediates containing more than one thioether ring (19, 20, 21, and 23, Figure 1, Table 1). Tandem MS studies indicated that the fourth dehydration occurs at Ser11, followed by the fifth and sixth dehydrations at Thr17 and Thr18, which are not strongly discriminated by HalM2.6 Among peptide species containing MeLan ring A (14–18), the kinetic model suggests there is a general decrease in the dehydration rate at sites more distant from the leader peptide. Interestingly, despite the accumulation of relatively large amounts of the 6-fold dehydrated species, [M + Lan + 5Dhx] (18), the abundance of a fully dehydrated peptide with one thioether ring ([M + Lan + 6Dhx]) was below the quantitation limit of our assay. This suggests that the seventh and final dehydration (of Thr21)6 catalyzed by HalM2 is kinetically significant only after the second thioether ring is installed, possibly because ring formation shortens the distance from the leader peptide to Thr21.
Tandem MS studies demonstrated that ring B is the second ring formed during HalA2 maturation (by attack of Cys15 on Dha11, Scheme 2).6 As with the conversion of species 9 → 15 during the fast phase of the reaction, several routes are available for the conversion of species 16 ([M + Lan + 3Dhx]) to the 6-fold dehydrated species with two thioether rings ([M + 2Lan + 4Dhx], species 21). The two slowest steps in the simulated model involve the formation of the second thioether ring from intermediates with either four or five total dehydrations (k16→19 = 2.5 min–1 and k17→20 = 5.0 min–1, respectively). The rates for these two minor routes for ring B cyclization were not individually well constrained and were held fixed relative to each other. In this model, ring B formation is fastest from species 18, [M + Lan + 5Dhx] (k18→21 = 20 min–1), which suggests that the majority (71%) of the flux from 16 → 21 occurs via the route 16 → 17 → 18 → 21 and that more extensive dehydration of the His6P-HalA2 core may facilitate the second cyclization step. Following the formation of ring B, the final dehydration occurs to give a fully dehydrated species with two thioether rings (k21→22 = 58 min–1). The rate constants for the dehydration of the 2-fold cyclized His6P-HalA2 species (k19→20, k20→21, and k21→22) were not all well constrained by the data, likely because reaction flux through species 19 and 20 represents a minor pathway. In our model, k19→20 was allowed to vary freely while the magnitudes for k20→21, and k21→22 were held fixed relative to the final step in the mechanism (k22→4), as discussed below.
The HalM2-catalyzed reaction is completed by the formation of the final two MeLan rings (22 → 24 → 4) at a net rate of k22→4 = 41 min–1. As noted above, the fully dehydrated peptides (22, 24, and 4) are the most abundant forms of the 2-, 3-, and 4-fold cyclized His6P-HalA2 species, respectively. Thus, by the time the third and fourth thioether rings are formed, His6P-HalA2 appears to be nearly completely dehydrated, suggesting that the bulk of the reaction flux to the final product moves from 21 → 22 → 24 → 4. Small amounts of peptide containing three thioether rings could be detected in the reaction spectra (23 and 24, Figure 1B); however, the time courses for these low-abundance species were difficult to simulate using the model in Scheme 3 (SI Figure S14), and FitSpace Explorer indicated that the kinetic data could be adequately described with a single net rate constant for the conversion of 22 → 4. For these reasons, species 23 and 24 were excluded from the kinetic model presented in Scheme 3. We were able to simulate the kinetics of these species using a more complex kinetic model and altered assumptions regarding the binding rates (SI Scheme S1, model 4) but additional experiments will be needed to validate this model. Regardless, it is clear from these data that the final cyclization step (24 → 4) is likely faster than the penultimate cyclization (22 → 24), such that the accumulation of the 3-fold cyclized intermediate 24 is limited.
General Summary of the HalM2-Catalyzed Reaction
A few key features of the HalM2-catalyzed maturation of His6P-HalA2 should be emphasized. First, as noted in previous work,6 the reaction is not strictly processive, meaning that reaction intermediates can be released into solution by HalM2 prior to further enzymatic processing. The evidence for this distributive mechanism is that most of the relevant peptide species accumulate to concentrations greater than the HalM2 concentration used in the assay (1 μM), which is possible only if the intermediates can be released into solution between successive modification events. Distributive mechanisms appear common for RiPP biosynthesis and have also been observed for class I and III lanthipeptide systems25,28,29 as well as for the enzymes involved in the biosynthesis of cyanobactins and linear azole-containing peptides.42−44 Because of this distributive behavior, we modeled the HalM2-catalyzed maturation of His6P-HalA2 according to Scheme 4, in which each intermediate was allowed to dissociate and rebind the enzyme during the course of the reaction. Although it is clear from these data that the reaction is not strictly processive, it is possible that certain segments of the His6P-HalA2 maturation process are more processive than others. Most notably, phosphorylated His6P-HalA2 species were not detectable in the mass spectra for the reaction, suggesting that these species are transient intermediates in the dehydration process (Scheme 1) and are not typically released by HalM2 during turnover. In addition, simulation of the HalM2/His6P-HalA2 kinetic data suggests that certain HalM2-catalyzed chemical transformations are, indeed, faster than the peptide dissociation rate (18 min–1) estimated by single molecule fluorescence binding assays, providing the necessary kinetic basis for processivity. Several notable examples of this apparent processivity include the conversion of [M + 2Dhx] to [M + Lan + 2Dhx] (9 → 14 → 15) during the early stages of the reaction, the three successive dehydrations of [M + 2Lan + 2Dhx] to give [M + 2Lan + 5Dhx] (19 → 20 → 21 → 22), and the rapid conversion of [M + 2Lan + 5Dhx] to the final product involving cyclization of rings C and D (22 → 4).
Scheme 4. Schematic Representation of the Models Used for Simulation of the HalM2 and ProcM Kinetic Data.
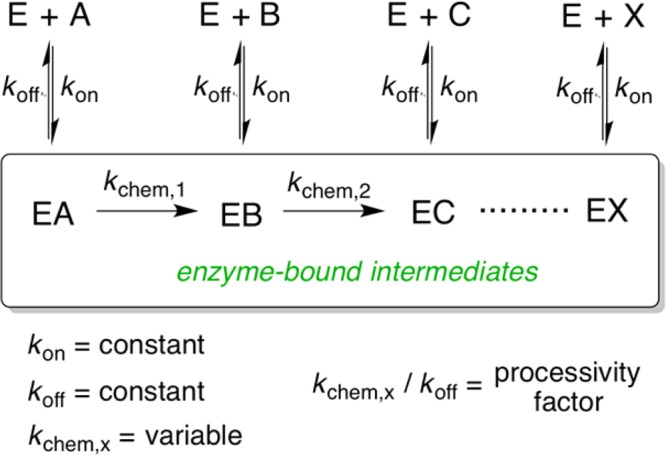
A second general feature of the overall process is that, with the exception of some of the rapid modifications at the N-terminus of the core peptide (8 → 9, 9 → 14, 10 → 15, and 14 → 15) and perhaps the final dehydration (k21→22 = 58 min–1) and cyclization (k24→4 > 41 min–1), most of the rate constants in the simulated model are similar in magnitude, such that there is <10-fold variation in the calculated values of the bimolecular rate constant for each reaction in the model (knet, Table 1). Consequently, there is no clear rate-limiting step in the maturation pathway under these conditions, and the two slowest steps in the model (16 → 19 and 17 → 20) can be circumvented by a more kinetically efficient route (16 → 17 → 18 → 21). When considering the different chemical modifications catalyzed by HalM2 (Scheme 1) as well as the different local amino acid contexts and dramatically changing structure of the His6P-HalA2 peptide that must be accommodated by the HalM2 active site(s), the similarity in knet values in our model suggests that something other than the intrinsic rate of the chemical transformations may be limiting the kinetics of the individual HalM2-catalyzed reactions. One possibility is that conformational changes in the HalM2/HalA2 complex (such as docking of the core peptide into the dehydratase/cyclase active sites) gate the chemical steps. The extent to which chemical steps limit turnover in LanM enzymes can be probed now that a reliable and semiquantitative kinetic assay is in hand.
Finally, it appears as if certain His6P-HalA2 modifications have significant effects on the kinetics of subsequent steps. For example, the installation of the B ring seems to be a critical event in His6P-HalA2 maturation, because many of the subsequent chemical conversions (20 → 21 → 22 → 24 → 4) are some of the most efficient in the pathway according to our model. Interestingly, it has been shown that disrupting the formation of ring B by mutagenesis of Cys11 alters the ability of HalM2 to install rings C and D,45 suggesting that ring B indeed forms a critical structural element for the efficient processing of the remaining thioether rings. A recent study on the class III lanthipeptide synthetase involved in curvopeptin biosynthesis also reported that some cyclization steps required prior installments of other modifications.24 A second example of how His6P-HalA2 intermediate structure may affect the kinetics of subsequent steps can be seen when comparing the rates of a given dehydration reaction as a function of the number of thioether rings in the peptide. In general, thioether rings appear to enhance the rate of a given dehydration reaction (e.g., compare k9→10 with k14→15 and k17→18 with k20→21, Scheme 3). In the extreme case, the final dehydration does not even seem to occur until the second thioether ring is installed. Perhaps the reduced flexibility of the cyclized species facilitates productive docking of the core peptide in the dehydratase active site. This apparent effect of substrate structure on reaction kinetics provides additional evidence for a kinetic model that is limited mainly by conformational changes of the HalM2/His6P-HalA2 complex, rather than by chemical transformations. This hypothesis is discussed further below.
Overview of the ProcM-Catalyzed Reaction
ProcM is a remarkable enzyme from the marine cyanobacterium Prochlorococcus strain MIT9313 that is responsible for the biosynthesis of the prochlorosins, a group of 29 different lanthipeptides encoded in four separate regions of the MIT9313 genome.37 Both in vitro activity assays and in vivo heterologous coexpression experiments have shown that ProcM (the only LanM encoded in the Prochlorococcus MIT9313 genome) is capable of modifying all of the ProcA precursor peptides that have been tested to date into products with defined thioether ring topologies.37,46,47 The exact functions of the resulting prochlorosins are not currently known, but the transcription levels of procM and several procA genes in cultured Prochlorococcus MIT9313 cells respond to environmental changes, and several mature prochlorosins were detected in the spent media used to culture MIT9313 cells.37 Collectively, these observations suggest that the prochlorosin biosynthetic machinery is expressed in the native producer and that the prochlorosins are functional molecules. Thus, ProcM appears to be a rare example of a biosynthetic enzyme with extremely relaxed substrate specificity.
Arguably the most interesting feature of prochlorosin biosynthesis is the extreme variability in the core peptide sequences of the ProcA substrate peptides and the diverse final thioether ring topologies in the mature prochlorosins,37 begging the question as to how (or whether) a single enzyme can orchestrate the biosynthesis of so many different final structures. As expected, recent mechanistic studies of ProcM using synthetically prepared ProcA peptides containing selectively protected Cys residues, demonstrated that ProcM indeed plays a direct role in ProcA cyclization.13 Although the mechanism(s) governing the regioselectivity of thioether ring formation by lanthipeptide synthetases is currently unknown, an emerging hypothesis is that the ring topology in some lanthipeptides can be determined in part by features of the primary sequence of the core peptide.7,17,19 For example, ProcM was recently shown to install the wild type lacticin 481 ring topology when presented with a chimeric substrate containing the ProcA3.2 leader peptide fused to the lacticin 481 core peptide.7 This result is difficult to explain unless the lacticin 481 core peptide sequence plays some role in dictating the preferred cyclization pattern. ProcM is also unusual among LanM enzymes in that the catalytic Zn2+ ion in the cyclase domain is coordinated by three Cys residues rather than by the typical Cys2His coordination geometry found in most LanMs.7 This unusual Zn coordination could influence the reactivity of ProcA-derived thiolate nucleophiles in a manner that accommodates the many different cyclization patterns installed by ProcM. Clearly, a better understanding of the ProcM-catalyzed reactions could help to elucidate the features that govern cyclization by this versatile catalyst and could help to better manipulate enzyme-mediated thioether ring biosynthesis for engineering purposes.
A Kinetic Model for the ProcM/ProcA2.8 Reaction
We conducted kinetic studies of the ProcM/ProcA2.8 reaction using the LC/ESI-MS assay and methods of analysis discussed above. ProcM catalyzes the dehydration of Ser9 and Ser13 in the ProcA2.8 core peptide en route to biosynthesis of two nonoverlapping lanthionine rings (formed by attack of Cys3 on Dha9 and by attack of Cys19 on Dha13, Scheme 2).37 Recent mechanistic studies on the ProcM/ProcA2.8 reaction demonstrated that both the dehydrations and the cyclizations occur in a C- to N-terminal direction (i.e., Ser13 is dehydrated before Ser9, and ring B is formed before ring A).13 Thus, directionality in the ProcM-catalyzed maturation of ProcA2.8 is opposite that of the HalM2-catalyzed maturation of HalA2. This result illustrates why making mechanistic generalizations for uncharacterized LanM enzymes is currently not possible and further underscores the need for quantitative kinetic analysis methods for these systems.
As with the HalM2-catalyzed reaction, a variety of ProcA2.8-derived intermediates were generated and consumed during the ProcM-catalyzed reaction (Figures 1 and SI S4, Table S2). Time courses for the major species in the reaction are shown in Figure 5, along with curves simulated for these species using the mechanism in Scheme 5 and the set of rate constants given in Table 2. The relevant ProcM/ProcA2.8 reaction species include the starting material (26), uncyclized species containing either one (35) or two Dha residues (34), the penultimate species in the biosynthetic pathway containing a single Lan ring and a single Dha residue (29), and the final product containing two Lan rings (6). The LC/ESI-MS spectra indicated that several of the major reaction species (6, 26, and 29) were partially oxidized (to species 27, 36, and 30, respectively) and that two of the dehydrated peptides (29 and 34) formed adducts with TCEP (species 40 and 41, respectively). For the kinetic analysis depicted in Figure 5 and Scheme 5, the fractional abundances of the oxidized species and TCEP adducts were summed with the fractional abundances of their corresponding parent peptides. Finally, several phosphorylated intermediates present at low abundance (33, 38, and 39) were observed but were excluded from the model in Scheme 5. A preliminary kinetic model that incorporates these phosphorylated species into the pathway for ProcA2.8 maturation is presented in the Supporting Information (Scheme S3 and Figure S19).
Figure 5.
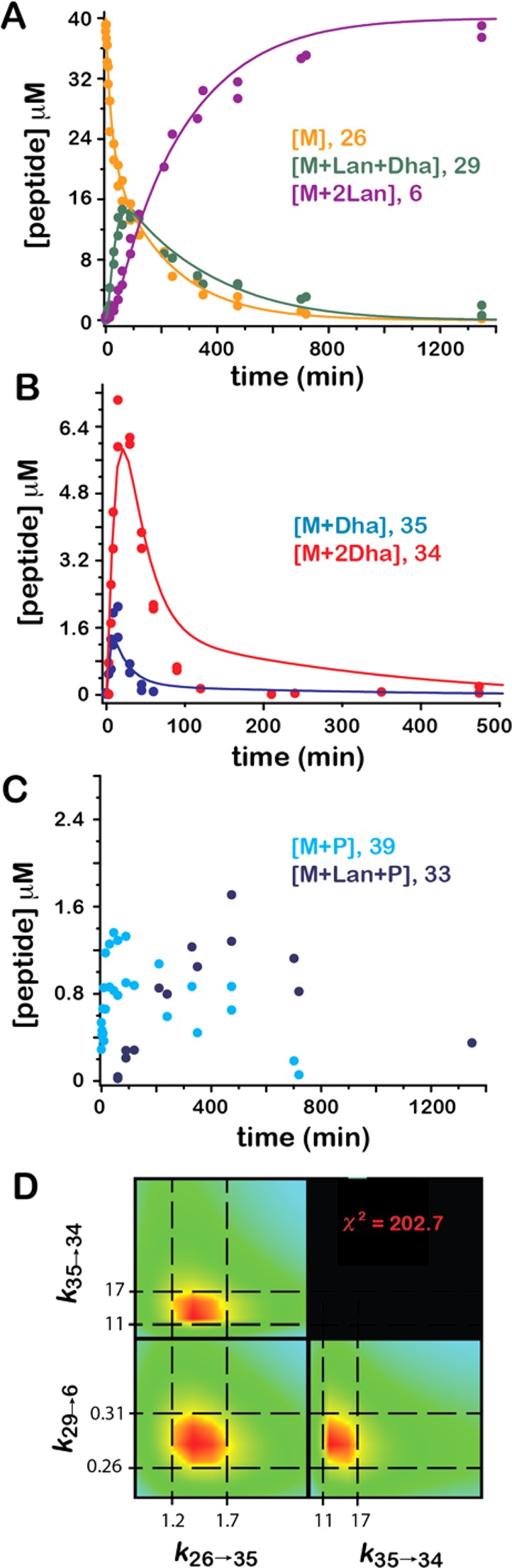
Time courses for peptide species involved in the ProcM/ProcA2.8 reaction. The progress curves overlying the data in panels A–C were generated by numerical simulation of the data with the kinetic model shown in Scheme 5 using the rate constants given in Table 2. The phosphorylated species (33 and 39, panel C) were not included in this model (see pages S31–S32 of the Supporting Information for a more detailed discussion). The confidence contours provided by FitSpace for the three variable parameters in the model (k26→35, k35→34, and k29→6) are shown in panel D along with the parameter boundaries (in units of min–1) reported in Table 2 and calculated at 1.14 times the χ2 minimum of 202.7.
Scheme 5. Kinetic Model for ProcM-Catalyzed Maturation of ProcA2.8.
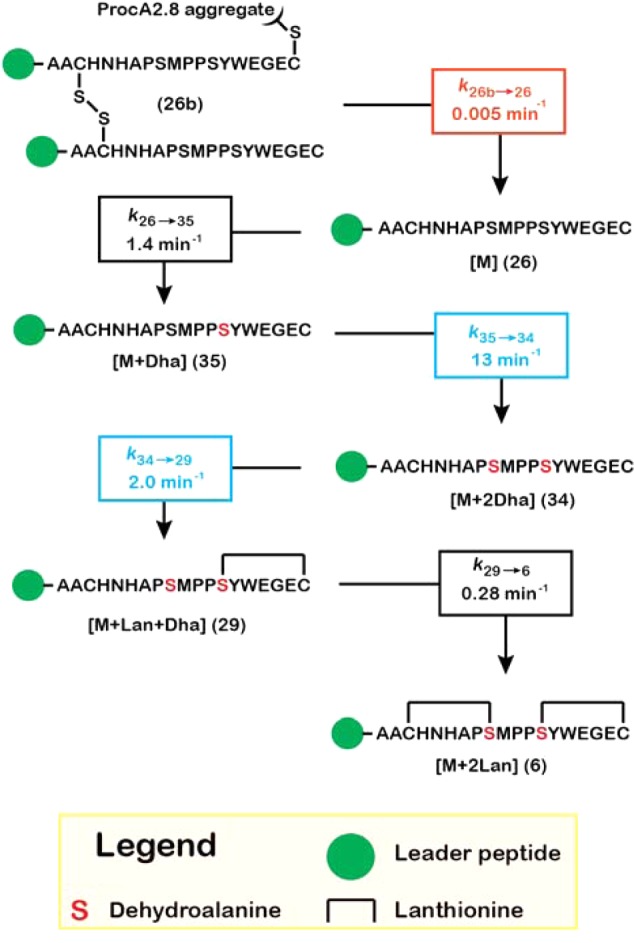
For clarity, peptide binding and dissociation steps from the enzyme are not shown. These steps were included in the model (Scheme 4) and were held constant for each species, as described in the text. Rate constants are color-coded: black, allowed to vary freely during nonlinear regression; red, held fixed at the indicated value; blue, held at a fixed ratio relative to each other. The progress curves simulated for each species in this mechanism are shown along with the experimental data in Figure 5. Compound numbering corresponds to the NEM-alkylated forms of the peptides that were observed in the mass spectra for the reaction (Figure 1D) and are assigned in the Supporting Information (Figure S4, Table S2). Evidence for the putative ProcA2.8 aggregate (26b) is discussed in more detail in the Supporting Information.
Table 2. Summary of Simulated Rate Constants for the ProcM/ProcA2.8 Reaction.
| kx | best fit ± SE (min–1) | FitSpace boundaries (min–1)** | knet (μM–1min–1)° | max concn (μM)′ | processivity factor″ |
|---|---|---|---|---|---|
| kon | 4.26* | held constant | |||
| koff | 20 | held constant | |||
| k26b→26 | 0.005 | held constant | |||
| k26→35 | 1.4 ± 0.1 | 1.2–1.7 | 0.27 | 0.07 | |
| k35→34 | 13 ± 2 | 11–17 | 1.7 | 2.4 | 0.65 |
| k34→29 | 2.0 | fixed relative to k35→34 | 0.39 | 7.2 | 0.1 |
| k29→6 | 0.28 ± 0.02 | 0.26–0.31 | 0.059 | 14 | 0.014 |
kon is in units of μM–1 min–1.
FitSpace boundaries are given as 1.14 times the χ2 minimum of the best fit.
knet = (konkx)/(koff + kx).
Refers to the maximum concentration of the substrate for the indicated reaction. The total peptide concentration was 40 μM.
Processivity factor is defined as kx/koff.
The maturation of ProcA2.8 could be modeled as a series of sequential reactions (26 → 35 → 34 → 29 → 6, Scheme 5) using the same assumptions that were made for the HalM2/His6P-HalA2 reaction. A binding constant for the unmodified precursor peptide with ProcM (Kd = 4.7 μM) was measured by fluorescence polarization (SI Figure S15). All ProcA2.8-derived peptide species involved in the reaction were assumed to have the same binding affinity, and the Kd was held fixed during nonlinear fitting of the data to the model in Scheme 5. Simulations indicated that only three of the four rate constants involved in the net conversion of starting material to compound 6 were well-constrained by the data and that holding the k35→34/k34→29 ratio fixed gave the best fit with the most well-defined FitSpace boundaries for the variable parameters (χ2/DoF = 1.635, Table 2). In a manner similar to the HalM2/His6P-HalA2 system, we also attempted to measure koff for the ProcM/ProcA2.8 complex using single molecule binding experiments, but these studies were not successful (data not shown). Nevertheless, simulations of the mechanism in Scheme 5 with different fixed values of koff indicated that similarly constrained values for the variable parameters in the model could be obtained over a relatively broad range of koff values (SI Table S6).
The most obvious kinetic feature of the ProcM/ProcA2.8 reaction is the relative timing of the dehydration and cyclization reactions. The two dehydration reactions (26 → 35 → 34) and the first cyclization (34 → 29) occur on a more rapid time scale, wheras the final cyclization (29 → 6) occurs more slowly. Following the first dehydration (k26→35 = 1.4 min–1), the reaction seems to partition strongly toward the second dehydration (k35→34 = 13 min–1) rather than toward the cyclization of the B ring to give an [M + Lan] species with a single Lan ring and no additional dehydration (an [M + Lan] intermediate containing only ring B was not detected in the mass spectra). The dehydrated intermediates 34 and 35 reach their peak concentrations at ∼15 min and are then completely consumed within 90 min as intermediate 29 (M + Lan + Dha) accumulates to its maximum value. Importantly, at its maximum, the partially cyclized intermediate 29 accounts for ∼35% of the total peptide in the reaction. The significant accumulation of 29 suggests that the first cyclization to form ring B (34 → 29) is significantly faster than the second cyclization to form ring A (29 → 6), a result that is supported by the simulated values for k34→29 and k29→6 (2.0 and 0.28 min–1, respectively). From these data, it is obvious that the second cyclization limits the net conversion of starting material to compound 6.
Finally, an additional step (26b → 26, Scheme 5) had to be included in the model to account for the biphasic consumption of starting material (26). In this model, there is an inactive form of the substrate (26b) that is slowly converted to an active form during the course of the reaction. The conversion of 26b to 26 is slow and limits overall turnover after the initial pool of 26 is consumed in the fast phase. Species 26b is likely an oxidized form of 26, perhaps involving (intermolecular) disulfide bonds. Evidence for this claim and further discussion of alternative kinetic models for the ProcM/ProcA2.8 reaction are presented in the Supporting Information.
Comparison of the ProcM- and HalM2-Catalyzed Reactions
Despite the technical obstacles encountered during the kinetic studies of the ProcM-catalyzed reaction, several features of the reaction stand in marked contrast to the HalM2-catalyzed reaction. The most obvious difference between the two enzymes is that the ProcM-catalyzed reaction takes much longer to reach completion than the HalM2-catalyzed reaction under nearly identical reaction conditions (1 μM enzyme, 40 μM peptide), and this difference is not caused by ProcM activity loss during the lengthy ProcM assay (SI Table S7). This observation comes despite the fact that HalM2 must catalyze more reactions than ProcM (18 vs 6) to fully modify the His6P-HalA2 precursor peptide. Using the 10 min reaction time points for comparison, the levels of the fully modified ProcA2.8 and His6P-HalA2 peptides (species 6 and 4, respectively) are roughly 2% and 35% of the total peptide. The sluggishness of the ProcM-catalyzed reaction is reflected in the simulated values for the rate constants (Tables 1 and 2). The rates of both dehydrations in the ProcM kinetic model (k26→35 = 1.4 min–1 and k35→34 13 min–1) are slower than each of the 11 dehydration rates included in the HalM2 model, which range from 16 to 81 min–1 (Scheme 3). Similarly, the rates for the two cyclization steps in the ProcM-catalyzed reaction (k34→29 = 2.0 min–1 and k29→6 0.28 min–1) are ∼10–100 fold slower, respectively, than the major cyclization steps in the HalM2-catalyzed reaction (k9→14 = 104 min–1, k18→21 = 20 min–1, k22→24 = 41 min–1, k24→4 > 41 min–1). Clearly, despite the similar chemistry mediated by the two enzymes, the kinetic barriers to ProcA2.8 maturation are more severe under these experimental conditions.
Another intriguing distinction between the two enzymes is that the rates of the cyclization reactions appear to get faster as His6P-HalA2 matures into the final product (i.e., k24→4 > k22→24 > k18→21, Scheme 3), whereas the opposite trend is observed in ProcA2.8 maturation, and the final cyclization event (k29→6) is the slowest step in the pathway. It should be noted that despite the extremely slow rate of ring A cyclization in ProcA2.8 (k29→6 = 0.28 min–1), this reaction is enzyme-catalyzed.13 Preliminary kinetic characterization of ProcM with several other ProcA substrates (ProcA2.11, ProcA3.3, and ProcA4.3) has revealed a similar pattern in cyclization kinetics: in each reaction, some thioether rings form quickly, while others require a much longer time (data not shown). Collectively, these data suggest that HalM2 may have evolved substrate specificity to efficiently catalyze a defined set of cyclization reactions toward a single product. The cyclization reactions occur more quickly as the reaction progresses and as the His6P-HalA2 intermediate structure more closely resembles the final product. Assuming similar intrinsic chemistry rates for each successive cyclization, this interpretation of the kinetic data implies evolved complementarity between the structure of the HalM2 cyclase and the structure of the maturing peptide intermediates. In contrast, the slow cyclization kinetics of ProcA2.8 maturation may reflect an evolutionary pressure on ProcM to maintain relaxed substrate specificity for the cyclization reaction. Thus, the ability of ProcM to assemble multiple different thioether ring architectures on the 29 different ProcA precursor peptides encoded in the Prochlorococcus MIT9313 genome appears to come at a substantial kinetic price.
A final notable difference between the two enzymes is in the directionality of the installed modifications6,13 and in the relative timing of dehydration and cyclization events for each reaction. In His6P-HalA2 maturation, dehydration and cyclization occur in parallel at each stage of the reaction, such that the two enzymatic activities are approximately kinetically comparable (Scheme 3). This observation implies a similar level of access to the core peptide by the dehydratase and cyclase domains of HalM2 or a similar binding affinity of the core peptide to each domain. The alternation of the two activities may be functionally important because it could assist HalM2 in guiding formation of the proper ring architecture found in mature His6P-HalA2. Similar alternating activities have been observed for the class III lanthipeptide synthetase involved in curvopeptin biosynthesis24 and have been proposed for class I nisin biosynthesis.48 In contrast, dehydration and cyclization in ProcA2.8 maturation seem to be largely uncoupled, with the dehydrations occurring first, and the cyclizations occurring last. A similar observation was recently made for the ProcM/ProcA3.3 reaction.13 Whereas the generality of this observation must await future studies on other ProcM/ProcA reactions, these initial data seem to suggest a greater access to or a greater affinity of the ProcA core peptide for the dehydratase active site of ProcM.
The differences in directionality for the two reactions (i.e., N- to C-terminal for HalA2 maturation and C- to N-terminal for ProcA2.8 maturation) may reflect different leader peptide binding modes of the two enzymes, which are not phylogenetically closely related.7 The class III lanthipeptide synthetases involved in biosynthesis of labyrinthopeptin A2 and catenulipeptin also display a C- to N-terminal processing mode,28,49 and a predominantly C-to-N terminal directionality has also been reported for the azole-installing synthetase involved in the biosynthesis of the RiPP plantazolicin.44 Different binding modes of the leader peptides could alter the relative spatial positioning of the core peptide to the enzyme active sites and, in the case of lanthipeptide synthetases, may influence access of the dehydratase and cyclase to the core peptide, as discussed above. Structural studies of a LanM enzyme in complex with a LanA substrate or leader peptide would undoubtedly help to illuminate some of these remaining questions.
Mechanistic Implications
The kinetic models for HalM2 and ProcM presented in this work provide estimates for the net rates of conversion of the major reaction intermediates observed in the LC/ESI-MS assay. These net rates are useful for determining the major pathways of intermediate flux and for determining the relative catalytic efficiencies of the various chemical transformations involved in the pathway. However, it should be emphasized that these net rates could reflect several microscopic steps that are simply not resolvable with the current assay. For example, the phosphorylation of Ser/Thr residues is known to be involved in the net dehydration of these residues, but phosphorylated species were not detected during His6P-HalA2 maturation. As such, the dehydration reactions in our kinetic models reflect at least two microscopic steps: phosphorylation and phosphate elimination. In this regard, it is intriguing that phosphorylated intermediates are observed in both the ProcM/ProcA2.8-catalyzed reaction and in the reactions catalyzed by the class III lanthipeptide synthetases.24,50 In the latter case, reaction intermediates containing up to three phosphate moieties have been observed.24 These observations suggest inherently different levels of kinetic coupling between the kinase and elimination activities of lanthipeptide synthetase enzymes.
A number of observations in this study suggest that conformational sampling of the LanM/LanA complex could make important contributions to the observed rates of intermediate conversion. First, the amino acid residues that have been implicated in the dehydration51 and cyclization52 reactions catalyzed by the well studied class II lacticin 481 synthetase (LctM) are all conserved in both HalM2 and ProcM. Thus, the differences in the observed kinetics between the HalM2 and ProcM-catalyzed reactions are unlikely to be due to drastic structural alterations in the active sites of these two enzymes. Second, the general decrease in dehydration rates at sites more distal from the leader peptide in the HalM2/His6P-HalA2 reaction (Scheme 3) is easiest to explain as a trend that reflects greater access of Ser/Thr residues on the N-terminus of the core peptide to the HalM2 dehydratase domain via conformational sampling, rather than as a trend that reflects intrinsically different chemical reactivity of the Ser/Thr residues toward dehydration. The similar kinetics and alternation of many of the dehydration and cyclization steps in the HalM2/His6P-HalA2 reaction also suggest that access of the dehydratase and cyclase active sites to the peptide is contributing to the observed rate, unless the intrinsic rates for cyclization and dehydration chemistry are coincidentally similar. The apparent kinetic uncoupling of dehydration and cyclization in the ProcM/ProcA2.8 reaction could simply reflect a higher probability of ProcA core peptide binding by the ProcM dehydratase domain.
Several additional features of the cyclization kinetics for the HalM2 and ProcM-catalyzed reactions support the hypothesis that conformational sampling contributes to rate limitation. According to our model for HalM2, the formation of ring B (k18→21 = 20 min–1) is the slowest cyclization step in His6P-HalA2 maturation. This is intriguing because ring B is the only lanthionine ring in Halβ (the other three thioether rings are methyllanthionines), and the less sterically hindered Cβ atom of the Dha residue should be intrinsically more reactive toward attack by a His6P-HalA2-derived thiolate nucleophile.53,54 This unexpected trend in the relative rates of Lan and MeLan ring formation can be rationalized if structural features of the His6P-HalA2 peptide (such as the presence of certain Dha/Dhb residues or thioether rings) help to stabilize docking interactions in the cyclase active site that gate the chemical transformation. For ProcM, the unusual Cys3 coordination sphere of the Zn2+ atom is typically associated with enhanced thiolate nucleophilicity in enzymes and model complexes that catalyze Zn2+-dependent thiol alkylation.55−59 The enhanced thiolate nucleophilicity in these Cys3 systems has been attributed to more facile dissociation of the substrate thiolate from the Zn, a result of the more negative net charge of the fully ligated Zn in this coordination geometry (the Cys4 ligated Zn carries a net 2– charge). The cyclization reactions in ProcM, however, are substantially slower than the cyclizations mediated by HalM2 (whose fully ligated Zn has Cys3His coordination and carries a net 1– charge). Furthermore, the rates of ring A and ring B formation during ProcA2.8 maturation are substantially different (Table 2). The slower than expected cyclization reactions (relative to HalM2/His6P-HalA2) and the different cyclization rates of ring A and ring B in the ProcM/ProcA2.8 reaction could once again be explained with a kinetic model characterized by different binding affinities of the ProcA2.8 core peptide in the ProcM cyclase active site, such that the cyclization steps are kinetically gated by binding steps.60,61
A model for LanM function that incorporates conformational changes is illustrated in Scheme 6. The key feature of the model is that following LanA leader peptide binding to form the LanM/LanA complex (Kd), a competition ensues between the dehydratase and cyclase domains for binding to the core peptide. The equilibrium constants for core peptide docking (Kdehyd and Kcyc) could be influenced by the relative spatial orientation of the dehydratase, cyclase, and core peptide. As such, the location of the LanA leader peptide binding site on the LanM could be important for determining which domain (dehydratase or cyclase) has greater access to the core peptide. This relative spatial orientation could also be an important factor in determining the directionality of post-translational modification. In addition to relative orientation, specific molecular interactions between the LanA core peptide and the LanM active sites could also alter Kdehyd and Kcyc such that certain peptide sequences or patterns of post-translational modification could shift the docking equilibrium. For example, the installation of the correct set of thioether rings may increase the value of Kcyc for subsequent cyclization events, as we suggest may be occurring during the late stages of His6P-HalA2 maturation (vide supra). The extent to which these conformational sampling steps (kdehyd and kcyc) limit turnover will determine the extent to which the rates for the intrinsic chemistry steps (Kdehyd and Kcyc) are reflected in the steady state rate constants measured in the current study. In this model, the role of the leader peptide appears to be that of a simple docking unit, but many experiments have indicated that the leader peptide in fact facilitates catalysis, possibly through allosteric activation.14,16,19 Regardless of the mechanism by which the leader peptide achieves overall activation, conformational sampling of the dehydration and cyclization active sites may be rate-limiting and may determine the timing of the various transformations.
Scheme 6. A Putative Model for LanM Function Involving Conformational Sampling of the LanM/LanA Michaelis Complex.

The model is drawn with a single leader peptide binding site, but separate leader peptide binding sites for the dehydratase and cyclase cannot be excluded.
Conclusions
In summary, we have developed the first semiquantitative kinetic assay capable of interrogating the complex multistep reactions mediated by the class II lanthipeptide synthetases. The assay is based on relative quantitation of LC/ESI-MS signals using a charge-state weighting procedure employed by Kelleher and co-workers to study the catalytic properties of polyketide and nonribosomal peptide synthases.30,31 The kinetic assay and control experiments described in this report should be readily applicable to study the kinetic properties of other lanthipeptide and RiPP biosynthetic enzymes. A comparative kinetic analysis of the class II synthetases, HalM2 and ProcM, under nearly identical reaction conditions revealed a number of stark differences in the kinetic properties of the reactions catalyzed by these enzymes. The most notable observation in these studies is the enhanced catalytic efficiencies of the HalM2-mediated reactions relative to the ProcM-mediated reactions. This difference in kinetic properties most likely reflects different evolutionary pressures placed on HalM2 and ProcM to maintain substrate selectivity and substrate flexibility, respectively. In addition, the kinetic assay and numerical simulation analysis revealed a number of other intriguing observations, including the possible involvement of rate-limiting LanM/LanA conformational sampling in many of the reactions. Clearly, as these initial kinetic studies of HalM2 and ProcM suggest, there are many interesting kinetic features of the LanM-catalyzed reaction that merit further investigation. Given the different types and distant evolutionary relationships of the lanthipeptide synthetases,7 it is likely that similar studies on other lanthipeptide biosynthetic systems will reveal additional catalytic and kinetic complexity. The stage is now set for a more detailed biochemical understanding of this versatile class of enzymes, which have garnered significant recent interest due their potential utility in the engineering of biologically active cyclic peptides.
Methods
Kinetic Assays
LanM/LanA reactions (1.5 mL total volume) contained 1 μM His6-tagged LanM enzyme, 40 μM LanA peptide (ProcA2.8 or His6P-HalA2), 5 mM ATP, 5 mM MgCl2, and 100 mM HEPES (pH 7.5). The TCEP concentration was 0.1 mM for the ProcM/ProcA2.8 reaction (1.25 Cys equiv) and 0.5 mM for the HalM2/His6P-HalA2 reaction (2.5 Cys equiv); higher concentrations resulted in more TCEP adduct formation to the dehydroamino acids during the assay. All components (minus enzyme) were mixed, and the pH was adjusted to 7.5 with several microliters of 5 M NaOH using a microtip electrode (Acumet). The reaction mixtures were incubated at 25 °C for 1 h prior to the addition of the appropriate LanM to initiate the reaction. Following the addition of enzyme, 100 μL reaction aliquots were removed at the desired time points and were quenched into 0.9 mL of quench buffer (111 mM citrate, 1.11 mM EDTA). After quenching, the pH of reaction aliquots was ∼3.5. The quenched aliquots were stored on ice (for HalM2 reactions) or at −80 °C (for ProcM reactions) until the last time point was quenched.
Derivitization of Kinetic Assay Time Points
After the final kinetic assay time point was quenched, the aliquots were warmed to 25 °C in a heat block, spiked with 100 μL of 100 mM TCEP, and incubated for an additional 10 min at 25 °C. The pH of each aliquot was then adjusted to 6.2–6.4 by addition of 35–40 μL of 5 M NaOH. To alkylate free Cys thiols, an 11 μL aliquot of 1 M NEM in EtOH was added to each quenched reaction aliquot to give ∼10 mM final NEM. The samples were then incubated for 10 min at 37 °C, at which point 11 μL of 100% TFA was added to acidify the reaction to pH ∼ 3. Samples were purified with a 1 mL Vydac BioSelect reversed phase C4 solid phase extraction (C4-SPE) column. The C4-SPE column was first equilibrated with 3 mL of 0.1% TFA in H2O. The peptide samples were loaded onto the column, washed with 3 mL of 0.1% TFA, and eluted with 2 mL of 0.1% TFA in 80% MeCN. Following elution, the samples were flash-frozen in liquid nitrogen and lyophilized to dryness. For LC/ESI-MS analysis, lyophilized samples were dissolved in 50 μL of H2O, and the concentration of peptide in each sample was then estimated by UV–visible absorption spectroscopy using extinction coefficients calculated by the ExPASy ProtParam tool (ε280 = 7115 M–1 cm–1 for ProcA2.8 and 5500 M–1 cm–1 for His6P-HalA2). Samples were diluted to final concentrations of 10 μM (ProcA2.8) or 20 μM (His6P-HalA2) total peptide in LC/ESI-MS injection solvent (50% MeCN in H2O, 0.1% formic acid) and were analyzed within 12 h at 8 °C to minimize solvent evaporation.
Liquid Chromatography Electrospray Ionization Mass Spectrometry
Peptide samples (5 μL) were injected into an Acquity UPLC BEH C8 1.7 μM column (1 × 100 mm) attached to a quadrupole/time-of-flight (Q/TOF) Synapt-G1 mass spectrometer (Waters). All kinetic and control experiments were performed in duplicate, all samples were run in a randomized injection order, and 10 μL H2O blanks were run between samples. The column, initially in 98% solvent A (0.1% formic acid in H2O), was eluted with a linear gradient of 2–100% solvent B (100% MeCN, 0.1% formic acid) over 20 min at a flow rate of 0.090 mL/min. All peptides analyzed in this study eluted as a broad peak between 7 and 9 min under these conditions. The Synapt instrument settings were as follows: positive ion mode, V optics, capillary voltage = 3.0 kV, cone gas = 20 L/h, desolvation gas = 500 L/h, source temperature = 110 °C, desolvation temperature = 150 °C. Data were collected in centroid mode with an extended linear range without precursor ion selection, the TOF detector was set to an m/z window of 800–1800 Da and a 1 s scan time. The instrument was externally calibrated with a 0.1% phosphoric acid standard. The m/z values for the ions of interest were determined by summing the mass spectra at full width at half-maximum (fwhm) of the main chromatographic peak observed in the total ion chromatogram (TIC) of each LC injection. Extracted ion chromatograms (EICs) were then generated by applying a mass window of variable size (1.5–2.5 Da, depending on the charge state and the intensity of the ion signal) around the center of the most intense isotope peak for each ion of interest. The EICs were integrated using MassLynx software (Waters) to generate peak areas. A correction of 50 peak area units was subtracted from each EIC peak area to account for background. The EIC peak areas were then used for calculation of the fractional abundances of the peptides of interest as described below.
Semiquantitative Analysis of Mass Spectrometry Data
All quantitation of relative peptide concentrations reported in this study was performed as follows: The extracted ion chromatograms were used to calculate the fractional abundance of each LanA species of interest using a procedure analogous to that reported by Kelleher and co-workers.30,31 For the kinetic assays, the species of interest included unmodified LanA starting materials, the final LanM-modified products, and any species detectable in the kinetic assay time points that could be assigned as a reaction intermediate on the basis of the known structures of fully modified prochlorosin 2.8 and haloduracin β.37,62 Complete lists of the LanA peptide species that were detected in the HalM2 and ProcM-catalyzed reactions are given in SI Tables S1 and S2, respectively. Using eq 1, the fractional abundance of peptide species X in charge state i (fX,i) was calculated as the ratio of the EIC peak area for species X in charge state i (AX,i) to the sum of the EIC peak areas for all the relevant peptides with charge state i (Ai).
| 1 |
The weight of charge state i (wi) is defined by eq 2 as the ratio of the sum of the EIC peak areas for all relevant peptides with charge state i (Ai) to the sum of the EIC peak areas for all of the charge states of all of the relevant peptide species observable in the mass spectrum (i.e., the total signal, Atotal).
| 2 |
Finally, the charge state-weighted fractional abundance of peptide species X (FX) was calculated with eq 3, where the sum is over all of the charge states (i) observed in the mass spectrum.
| 3 |
An identical relative quantitation approach was used to analyze the data for each of the control experiments reported in this work (with the peptides of interest changing depending on the experiment).
Numerical Simulation of Kinetic Data
The duplicate measurements for the time-dependent changes in the fractional abundance of each peptide species to be included in the kinetic model were combined into a single kinetic trace and were imported into KinTek Explorer for global numerical simulation.39,40 The standard deviation in the time-dependent fractional abundance measurements were then estimated for each species in the model by fitting each time course to exponential equations using the “aFit” module of KinTek Explorer. The standard deviations determined in this way for each kinetic trace were then used to normalize the residuals of the fit to the data points comprising that trace during the global nonlinear least-squares fitting process to determine the χ2 minimum of the fit. Goodness-of-fit was evaluated by the χ2/DoF statistic, where DoF (the degrees of freedom) is equal to the number of data points minus the number of variable parameters in the model. The quantity χ2/DoF should approach unity for a good fit. After a good fit was achieved, confidence contours were calculated for each variable parameter in the model using the FitSpace Explorer suite of KinTek Explorer. The confidence contours were calculated in an iterative fashion by holding each pairwise combination of variable parameters constant at different fixed ratios. The remaining parameters in the model are allowed to vary freely while the program searches for a new global χ2 minimum. As such, the confidence contours provide valuable information on what range of values a given parameter in the model can assume while still producing a good fit to the data. For a model that is well-constrained by the data, each variable parameter will be allowed to assume only a defined range of magnitudes. The confidence contours can then be used to calculate boundaries for the magnitude of each variable parameter using a user-defined threshold. The parameter boundaries for the variable rate constants in the HalM2 and ProcM kinetic models were calculated at 1.12 and 1.14 the χ2 minimum, respectively. These thresholds provide a larger boundary range for the parameter estimates than the thresholds suggested by FitSpace Explorer (1.06 and 1.07, respectively). Additional specific details regarding the numerical simulation of the HalM2 and ProcM kinetic data is given within the context of the discussion of these two enzymes in the main text and the Supporting Information.
Acknowledgments
The authors acknowledge funding from the Institute for Genomic Biology and the Center for the Physics of Living Cells at the University of Illinois, Urbana–Champaign (National Science Foundation Grant PHY 1430124 to T.H.) as well as National Institutes of Health Grants F32GM103291 (C.J.T.) and GM058822 (W.A.V.) and the Howard Hughes Medical Institute (W.A.V. and T.H.). We thank Dr. Gabrielle Thibodeaux for the determination of the Kd of HalA2 and HalM2.
Glossary
Abbreviations
- ATP
adenosine triphosphate
- Cys
cysteine
- Dhx
Dha or Dhb
- DoF
degrees of freedom
- EDTA
ethylenediaminetetraacetic acid
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- TCEP
tris(2-carboxyethyl)phosphine
- TFA
trifluoroacetic acid
Supporting Information Available
Descriptions of the reagents used in this study, supplemental methods, and additional results and discussion pertaining to the kinetic analysis of HalM2 and ProcM are provided in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Arnison P. G.; Bibb M. J.; Bierbaum G.; Bowers A. A.; Bugni T. S.; Bulaj G.; Camarero J. A.; Campopiano D. J.; Challis G. L.; Clardy J.; Cotter P. D.; Craik D. J.; Dawson M.; Dittmann E.; Donadio S.; Dorrestein P. C.; Entian K.-D.; Fischbach M. A.; Garavelli J. S.; Göransson U.; Gruber C. W.; Haft D. H.; Hemscheidt T. K.; Hertweck C.; Hill C.; Horswill A. R.; Jaspars M.; Kelly W. L.; Klinman J. P.; Kuipers O. P.; Link A. J.; Liu W.; Marahiel M. A.; Mitchell D. A.; Moll G. N.; Moore B. S.; Müller R.; Nair S. K.; Nes I. F.; Norris G. E.; Olivera B. M.; Onaka H.; Patchett M. L.; Piel J.; Reaney M. J. T.; Rebuffat S.; Ross R. P.; Sahl H.-G.; Schmidt E. W.; Selsted M. E.; Severinov K.; Shen B.; Sivonen K.; Smith L.; Stein T.; Süssmuth R. E.; Tagg J. R.; Tang G.-L.; Truman A. W.; Vederas J. C.; Walsh C. T.; Walton J. D.; Wenzel S. C.; Willey J. M.; van der Donk W. A. Nat. Prod. Rep. 2013, 30, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knerr P. J.; van der Donk W. A. Annu. Rev. Biochem. 2012, 81, 479. [DOI] [PubMed] [Google Scholar]
- Bierbaum G.; Sahl H. G. Curr. Pharm. Biotechnol. 2009, 10, 2. [DOI] [PubMed] [Google Scholar]
- Xie L.; Miller L. M.; Chatterjee C.; Averin O.; Kelleher N. L.; van der Donk W. A. Science 2004, 303, 679. [DOI] [PubMed] [Google Scholar]
- Chatterjee C.; Miller L. M.; Leung Y. L.; Xie L.; Yi M.; Kelleher N. L.; van der Donk W. A. J. Am. Chem. Soc. 2005, 127, 15332. [DOI] [PubMed] [Google Scholar]
- Lee M. V.; Ihnken L. A. F.; You Y. O.; McClerren A. L.; van der Donk W. A.; Kelleher N. L. J. Am. Chem. Soc. 2009, 131, 12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Yu Y.; Velásquez J. E.; van der Donk W. A. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 18361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierbaum G.; Szekat C.; Josten M.; Heidrich C.; Kempter C.; Jung G.; Sahl H. G. Appl. Environ. Microbiol. 1996, 62, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubelski J.; Overkamp W.; Kluskens L. D.; Moll G. N.; Kuipers O. P. Appl. Environ. Microbiol. 2008, 74, 4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee C.; Patton G. C.; Cooper L.; Paul M.; van der Donk W. A. Chem. Biol. 2006, 13, 1109. [DOI] [PubMed] [Google Scholar]
- Suda S.; Westerbeek A.; O’Connor P. M.; Ross R. P.; Hill C.; Cotter P. D. Chem. Biol. 2010, 17, 1151. [DOI] [PubMed] [Google Scholar]
- Islam M. R.; Shioya K.; Nagao J.; Nishie M.; Jikuya H.; Zendo T.; Nakayama J.; Sonomoto K. Mol. Microbiol. 2009, 72, 1438. [DOI] [PubMed] [Google Scholar]
- Mukherjee S.; van der Donk W. A. J. Am. Chem. Soc. 2014, 136, 10450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton G. C.; Paul M.; Cooper L. E.; Chatterjee C.; van der Donk W. A. Biochemistry 2008, 47, 7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oman T. J.; van der Donk W. A. Nat. Chem. Biol. 2010, 6, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levengood M. R.; Patton G. C.; van der Donk W. A. J. Am. Chem. Soc. 2007, 129, 10314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W.; van der Donk W. A. Nat. Chem. Biol. 2013, 9, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oman T. J.; Knerr P. J.; Bindman N. A.; Velásquez J. E.; van der Donk W. A. J. Am. Chem. Soc. 2012, 134, 6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; van der Donk W. A. Chem.—Eur. J. 2013, 19, 7662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levengood M. R.; Knerr P. J.; Oman T. J.; van der Donk W. A. J. Am. Chem. Soc. 2009, 131, 12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleyard A. N.; Choi S.; Read D. M.; Lightfoot A.; Boakes S.; Hoffmann A.; Chopra I.; Bierbaum G.; Rudd B. A.; Dawson M. J.; Cortés J. Chem. Biol. 2009, 16, 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter P. D.; Deegan L. H.; Lawton E. M.; Draper L. A.; O’Connor P. M.; Hill C.; Ross R. P. Mol. Microbiol. 2006, 62, 735. [DOI] [PubMed] [Google Scholar]
- Müller W. M.; Ensle P.; Krawczyk B.; Süssmuth R. D. Biochemistry 2011, 50, 8362. [DOI] [PubMed] [Google Scholar]
- Jungmann N. A.; Krawczyk B.; Tietzmann M.; Ensle P.; Süssmuth R. D. J. Am. Chem. Soc. 2014, 136, 15222. [DOI] [PubMed] [Google Scholar]
- Ortega M. A.; Hao Y.; Zhang Q.; Walker M. C.; van der Donk W. A.; Nair S. K. Nature 2014, 10.1038/nature13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y.; Li B.; Claesen J.; Shi Y.; Bibb M. J.; van der Donk W. A. PLoS Biol. 2010, 8, e1000339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H.; Gao Y.; Zhao F.; Wang J.; Teng K.; Zhang J.; Zhong J. Biochem. Biophys. Res. Commun. 2014, 450, 1126. [DOI] [PubMed] [Google Scholar]
- Krawczyk B.; Ensle P.; Müller W. M.; Süssmuth R. D. J. Am. Chem. Soc. 2012, 134, 9922. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Ortega M.; Shi Y.; Wang H.; Melby J. O.; Tang W.; Mitchell D. A.; van der Donk W. A. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks L. M.; O’Connor S. E.; Mazur M. T.; Walsh C. T.; Kelleher N. L. Chem. Biol. 2004, 11, 327. [DOI] [PubMed] [Google Scholar]
- Miller L. M.; Mazur M. T.; McLoughlin S. M.; Kelleher N. L. Protein Sci. 2005, 14, 2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrestein P. C.; Kelleher N. L. Nat. Prod. Rep. 2006, 23, 893. [DOI] [PubMed] [Google Scholar]
- Gatto G. J. Jr.; McLoughlin S. M.; Kelleher N. L.; Walsh C. T. Biochemistry 2005, 44, 5993. [DOI] [PubMed] [Google Scholar]
- McLoughlin S. M.; Kelleher N. L. J. Am. Chem. Soc. 2004, 126, 13265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns J. A.; Butler J. C.; Moran J.; Whitesides G. M. J. Org. Chem. 1991, 56, 2648. [Google Scholar]
- Hansen R. E.; Winther J. R. Anal. Biochem. 2009, 394, 147. [DOI] [PubMed] [Google Scholar]
- Li B.; Sher D.; Kelly L.; Shi Y.; Huang K.; Knerr P. J.; Joewono I.; Rusch D.; Chisholm S. W.; van der Donk W. A. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibodeaux G. N.; van der Donk W. A. Chem. Commun. 2012, 48, 10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K. A.; Simpson Z. B.; Blom T. Anal. Biochem. 2009, 387, 20. [DOI] [PubMed] [Google Scholar]
- Johnson K. A.; Simpson Z. B.; Blom T. Anal. Biochem. 2009, 387, 30. [DOI] [PubMed] [Google Scholar]
- Chatterjee C.; Paul M.; Xie L.; van der Donk W. A. Chem. Rev. 2005, 105, 633. [DOI] [PubMed] [Google Scholar]
- Koehnke J.; Bent A. F.; Zollman D.; Smith K.; Houssen W. E.; Zhu X.; Mann G.; Lebl T.; Scharff R.; Shirran S.; Botting C. H.; Jaspars M.; Schwarz-Linek U.; Naismith J. H. Angew. Chem., Int. Ed. 2013, 52, 13991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher N. L.; Belshaw P. J.; Walsh C. T. J. Am. Chem. Soc. 1998, 120, 9716. [Google Scholar]
- Melby J. O.; Dunbar K. L.; Trinh N. Q.; Mitchell D. A. J. Am. Chem. Soc. 2012, 134, 5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper L. E.; McClerren A. L.; Chary A.; van der Donk W. A. Chem. Biol. 2008, 15, 1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y.; Yang X.; Garg N.; van der Donk W. A. J. Am. Chem. Soc. 2011, 133, 2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W.; van der Donk W. A. Biochemistry 2012, 51, 4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubelski J.; Khusainov R.; Kuipers O. P. J. Biol. Chem. 2009, 284, 25962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; van der Donk W. A. ACS Chem. Biol. 2012, 7, 1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller W. M.; Schmiederer T.; Ensle P.; Süssmuth R. D. Angew. Chem., Int. Ed. 2010, 49, 2436. [DOI] [PubMed] [Google Scholar]
- You Y. O.; van der Donk W. A. Biochemistry 2007, 46, 5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul M.; Patton G. C.; van der Donk W. A. Biochemistry 2007, 46, 6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okeley N. M.; Zhu Y.; van der Donk W. A. Org. Lett. 2000, 2, 3603. [DOI] [PubMed] [Google Scholar]
- Zhou H.; van der Donk W. A. Org. Lett. 2002, 4, 1335. [DOI] [PubMed] [Google Scholar]
- Penner-Hahn J. Curr. Opin. Chem. Biol. 2007, 11, 166. [DOI] [PubMed] [Google Scholar]
- Wilker J. J.; Lippard S. J. Inorg. Chem. 1997, 36, 969. [DOI] [PubMed] [Google Scholar]
- Morlok M. M.; Janak K. E.; Zhu G.; Quarless D. A.; Parkin G. J. Am. Chem. Soc. 2005, 127, 14039. [DOI] [PubMed] [Google Scholar]
- Harris C. M.; Derdowski A. M.; Poulter C. D. Biochemistry 2002, 41, 10554. [DOI] [PubMed] [Google Scholar]
- Koutmos M.; Pejchal R.; Bomer T. M.; Matthews R. G.; Smith J. L.; Ludwig M. L. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCammon J. A.; Northrup S. H. Nature 1981, 293, 316. [DOI] [PubMed] [Google Scholar]
- Gora A.; Brezovsky J.; Damborsky J. Chem. Rev. 2013, 113, 5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClerren A. L.; Cooper L. E.; Quan C.; Thomas P. M.; Kelleher N. L.; van der Donk W. A. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 17243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.