

Abstract
Overexpression of hypoxia-inducible factor-1α (HIF-1α) in human tumors is associated with poor prognosis and poor outcome to radiation therapy. Inhibition of HIF-1α is considered as a promising approach in cancer therapy. The purpose of this study was to test the efficacy of a novel HIF-1α inhibitor PX-478 as a radiosensitizer under normoxic and hypoxic conditions in vitro. PC3 and DU 145 prostate carcinoma cells were treated with PX-478 for 20 hr, and HIF-1α protein level and clonogenic cell survival were determined under normoxia and hypoxia. Effects of PX-478 on cell cycle distribution and phosphorylation of H2AX histone were evaluated. PX-478 decreased HIF-1α protein in PC3 and DU 145 cells. PX-478 produced cytotoxicity in both cell lines with enhanced toxicity under hypoxia for DU-145. PX-478 (20 μmol/L) enhanced the radiosensitivity of PC3 cells irradiated under normoxic and hypoxic condition with enhancement factor (EF) 1.4 and 1.56, respectively. The drug was less effective in inhibiting HIF-1α and enhancing radiosensitivity of DU 145 cells compared to PC3 cells with EF 1.13 (normoxia) and 1.25 (hypoxia) at 50 μmol/L concentration. PX-478 induced S/G2M arrest in PC3 but not in DU 145 cells. Treatment of PC3 and DU 145 cells with the drug resulted in phosphorylation of H2AX histone and prolongation of γH2AX expression in the irradiated cells. PX-478 is now undergoing Phase I clinical trials as an oral agent. Although the precise mechanism of enhancement of radiosensitivity remains to be identified, this study suggests a potential role for PX-478 as a clinical radiation enhancer.
Keywords: PX-478, hypoxia, normoxia, HIF-1α, radiosensitivity
Radiation therapy is a widely used treatment modality for local control of solid tumors. However, solid tumors often contain heterogeneous hypoxic areas. Clinical studies have shown that tumor hypoxia is not only a poor prognostic marker but also a major dose-limiting factor for radiation therapy.1-5 Hypoxic cells are 2- to 3-fold more resistant to radiation than well-oxygenated cells because the biological effect of radiation is greatly influenced by the presence or absence of molecular oxygen at the time of irradiation. 6,7 Some of the approaches to circumvent the hypoxiainduced therapeutic resistance include improving tumor oxygenation using oxygen-mimetic radiosensitizers, using drugs that are preferentially toxic to hypoxic cells, or devising radiation sources and regimens that are less affected by hypoxia.8
The hypoxic microenvironment in solid tumors also leads to several biochemical changes in tumor cells, mostly to adapt to the low oxygen tension. At molecular level, many of these changes are mediated by upregulation of hypoxia-inducible transcription factor HIF-1α, which activates target genes involved in erythropoiesis, glycolysis and angiogenesis.9 In addition, HIF-1α regulates the expression of genes coding for growth factors/receptors, apoptotic pathway, cell cycle regulators, invasiveness and metastasis. 10 Importantly, in some cancers, HIF-1α can be activated by nonhypoxic mechanisms.9 HIF-1α is overexpressed in the majority of human tumors and their metastasis as compared with the surrounding normal tissues.11,12 Therefore, HIF-1α is recognized as an important target for cancer therapy. Strategies to inhibit HIF pathways include pharmacological intervention of HIF-1α,13-18 genetic disruption of HIF-1α19-24 or blockade of hypoxia-inducible transcription.25 The precise role of HIF-1α in radiation response is yet unclear. Inhibition of HIF by chetomin, a small molecule inhibitor of transcriptional coactivation of HIF pathway is shown to radiosensitize HT 1080 fibrosarcoma cells under hypoxia in vitro.26 It has been suggested that inhibition of HIF could alter pleiotropic cellular processes initiated by hypoxia and radiation, and thereby enhance the radiosensitivity of tumors.21
PX-478 (S-2-amino-3-[4′-N,N,-bis (2-chloroethyl) amino]-phenyl propionic acid N-oxide dihydrochloride) is an experimental HIF-1α inhibitor that has shown antitumor activity against several aggressive human tumor xenografts.13,27 A recent study demonstrated that PX-478 inhibited HIF-1α protein at multiple levels and was cytotoxic to a variety of cancer cell lines under normoxia and hypoxia in vitro with an IC50 ~20–30 μM.28 The aim of our study was to investigate the efficacy of PX-478 as a radiosensitizer. In addition to producing cytotoxicity, PX-478 enhanced the radiosensitivity of prostate carcinoma cells irradiated under normoxic and hypoxic conditions in vitro. Treatment of cells with PX-478 resulted in the phosphorylation of H2AX histone and cell type-dependent cell cycle perturbations. PX-478 prolonged γH2AX expression in the irradiated cells.
Material and methods
Materials
PC3 and DU 145 human prostate carcinoma cells were obtained from American Type Culture Collection (Rockville, MD). Cells were maintained as monolayer cultures in RPMI 1640 media supplemented with 10%FBS, glutamine and antibiotics. All the tissue culture reagents were purchased from Invitrogen (Carlsbad, CA). Anti-HIF-1α monoclonal antibody was purchased from Transduction Labs (BD Biosciences, San Jose, CA); anti-mouse IgG and anti-topoisomerase-1 polyclonal antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). 5-BrdU was from Calbiochem (La Jolla, CA), Anti-BrdU antibody was from Becton Dickinson (San Jose, CA) and anti-mouse-FITC was from Sigma-Aldrich (St. Louis, MO). Anti-γH2AX (clone JBW301) and anti-Histone H1o/H5 (clone 3H9) antibodies were from Upstate Biotechnology (Lake Placid, NY). FITC-conjugated Anti-mouse IgG was from Jackson ImmunoResearch Labs (West Grove, PA). PX-478 was generously provided by ProlX Pharmaceuticals, AZ. PX-478 was prepared as a 10 mmol/L stock in distilled water and used immediately.
Irradiation
Cells were irradiated at room temperature in a PANTAK highfrequency X-ray generator (East Haven, CT), operated at 300 kV and 10 mA using 0.10-mm Cu and 2.50-mm Al filter. The dose rate was 1.6 Gy/min.
Clonogenic cell survival assay under normoxia
To determine the effect of PX-478 in combination with radiation, cells were treated with the drug for 24 hr under normoxic condition, irradiated and plated after 1 hr. Colonies were stained with crystal violet after 12 days and the colonies of >50 cells were counted. For combination treatments, net survival was calculated by correcting the toxicity of PX-478 alone. Enhancement factor (EF) was calculated by dividing the dose of radiation required to reduce plating efficiency to 10% when cells were treated with radiation alone by the dose of radiation required to reduce plating efficiency to 10% when cells were treated with PX-478 and radiation.
Hypoxia treatment
Cells were plated in 70-cm2 glass flasks (for Western blotting) or small glass flasks (for clonogenic assay). Next day, media was removed and fresh media with or without PX-478 was added to the flasks. After incubating with the drug for 18–20 hr under normoxia, flasks were tightly sealed with rubber stopper. Two 19-gauge needles were inserted in the rubber stoppers to introduce hypoxic gas mixture and to interconnect flasks. Hypoxia was induced by gassing the flasks with a mixture of 95% N2 and 5% CO2 for 1 hr in a warm room.15,29 Needles were removed at the end of 1 hr gassing and the rubber-stopper sealed flasks were incubated for desired duration. Previous measurements with a Thermox probe (Ametek, Pittsburgh, PA) indicated that the oxygen tension in flask was <10 ppm (0.02%) producing radiobiologic hypoxia.29
After 1 hr of gassing, the cells were harvested by scraping for Western blot analysis or trypsinized and plated for clonogenic survival assay. For combination treatment, cells were pretreated with the drug for 20 hr under normoxia and subjected to 1 hr gassing as described. At the end of 1-hr hypoxic, gassing cells were irradiated under hypoxic condition and plated for clonogenic assay within 1 hr. To investigate the effect of prolonged treatment with PX-478 during hypoxia, in some experiments PX-478 was added to cells just before hypoxic gassing and cells were maintained under hypoxic condition with the drug for 18 hr. For clonogenic assays, cells were plated in drug-free media.
Western blot analysis
Cell extracts were prepared in lysis buffer containing 50 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 1% Igepal, 0.1% SDS, 1 mM EDTA, 600 μmol/L sodium orthovanadate, 50 mmol/L sodium fluoride, 1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L DTT and “complete” protease inhibitor cocktail (Roche Diagnostics, Cat. No. 1836153).15 After incubating on ice for 20 min, cell extracts were centrifuged at 20,000g for 10 min and supernatants were collected. Protein concentrations were determined by Bio-Rad Dc protein assay. Sixty microgram of protein from normoxic samples and 15 μg protein from hypoxic samples were separated on 6% gels and transferred to PVDF membrane for HIF-1α analysis. Membranes were stripped and reprobed with topoisomerase-1 (TOPO) to ascertain uniform loading. Protein bands were developed with enhanced chemiluminescence detection kit (Santa Cruz, CA) and visualized by autoradiography. After scanning the autoradiographs, the band intensities were quantitated using ImageQuant (5.2 version). Fold change in HIF-1α protein (ΔHIF) was calculated by dividing the HIF-1α band density with the TOPO density (loading control), and then normalized to the control.30
Cell cycle analysis
Effect of PX-478 on cell cycle distribution was analyzed by flow cytometry by propidium iodide staining after treating cells with the drug for 24 hr. For BrdU staining, cells were incubated with 10 μmol/L BrdU for the last 1 hr of incubation and processed as described.31 Briefly, cells were trypsinized, washed with PBS and fixed in 70% ethanol overnight. Cells were pelleted and nuclei were isolated by pepsin/HCl digestion followed by treatment with 10 mmol/L borate (pH 8.6) to neutralize the acid. Cells were then incubated with anti-BrdU antibody as described in the manufacturer’s protocol followed by incubation with FITC-labeled antimouse IgG and PI staining. Cell cycle data were collected on BD FACSCalibur Flow Cytometer (San Jose, CA) and analyzed using CellQuest/MOD-Fit software (Verity Software House, Topsham, ME).
Immunoflourescent staining for γH2AX
PC3 cells were plated in 4-well chamber slides (20,000 cells/ml/well) and treated with PX-478. At desired time interval, PX-478 was removed by aspirating the drug media and cells were irradiated and further incubated in drug-free media. At 6- and 24-hr phosphorylated histone, H2AX (γH2AX) foci were analyzed by immunoflourescent staining as described.32 Briefly, cells were fixed in 4% paraformaldehyde, permeablized with 0.1% NP-40 and blocked with 5% Goat serum in 1% BSA. Cells were covered with antiphospho-histone H2AX primary antibody (1:2,000) and incubated overnight at 4°C. After washing with 1%BSA, cells were treated with FITC Goat anti-rabbit secondary antibody (1:100) for 1 hr followed by 30 min DAPI (1 μg/mL) staining in the dark. Coverslips were mounted with an antifade solution (DAKO, Carpinteria, CA). Slides were examined on a Leica DMRXA fluorescent microscope (Leica, Wetzlar, Germany). Images were captured by a photometrics Sensys CCD camera (Roper Scientific, Tucson, AZ) and imported into IP Labs image analysis software package (Scanalytics, Fairfax, VA) running on a Macintosh G3 computer (Apple, Cupertino, CA). For each condition, ~70–100 cells from 2 to 3 separate experiments were analyzed to determine the number of γH2AX foci per cell.
Immunoblot analysis for γH2AX
Cells were lysed in 20 mmol/L Tris-HCl, pH 7.4 containing 150 mmol/L NaCl, 1 mmol/L EDTA, 1% NP-40 and “complete” protease inhibitor cocktail. Histones from the nuclear pellet were extracted in 0.2 mol/L sulfuric acid by incubating samples on ice for 4–6 hr. After centrifugation, acid-soluble histones were transferred to fresh tubes and 9 volumes of acetone were added. Histones were precipitated at −20°C overnight and were pelleted by centrifugation at 20,000g for 10 min at 4°C. Supernatant was discarded and pellets were air-dried. Histones were solubilized in 4 mol/L urea and protein concentration was determined by BioRad Dc protein assay. Histones were separated on 18% gel by loading 15 μg samples and transferred to nitrocellulose membrane. Membranes were incubated overnight at 4°C with anti-γH2AX antibody (1:1,000), washed 3 times with PBS-T and incubated with HRP-conjugated anti-mouse antibody. γH2AX was visualized by ECL detection kit using Fuji LAS 3000 CCD imaging camera device. Membranes were stripped and reprobed with anti-H1o/H5 antibody to ascertain uniform loading. Signal intensities were normalized to their loading control H1o/H5 and expressed as fold change compared to controls.
Data analysis
Each data point represents average ± SEM of 3 experiments. Differences between the groups were statistically evaluated by 2- tailed paired t-test. A p value less than 0.05 was considered statistically significant.
Results
PX-478 inhibited HIF-1α protein in PC3 and DU 145 cells
PC3 and DU 145 cells express HIF-1α protein under normoxic condition. Figure 1 shows the Western blot analysis of dose response of HIF-1α protein in PC3 and DU 145 treated with PX-478 for 20 hr under normoxia (Fig. 1a). PC3 cells were more sensitive to PX-478 as compared with DU 145 cells. Densitometric analysis showed that the IC50 for HIF-1α inhibition for PC3 cells under normoxic condition was 20–25 μmol/L (ΔHIF: 0.56 ± 0.08, n = 5), whereas the IC50 for HIF-1α inhibition for the DU 145 cells was ~40–50 μmol/L (ΔHIF: 0.47 ± 0.08, n = 6).
Figure 1.
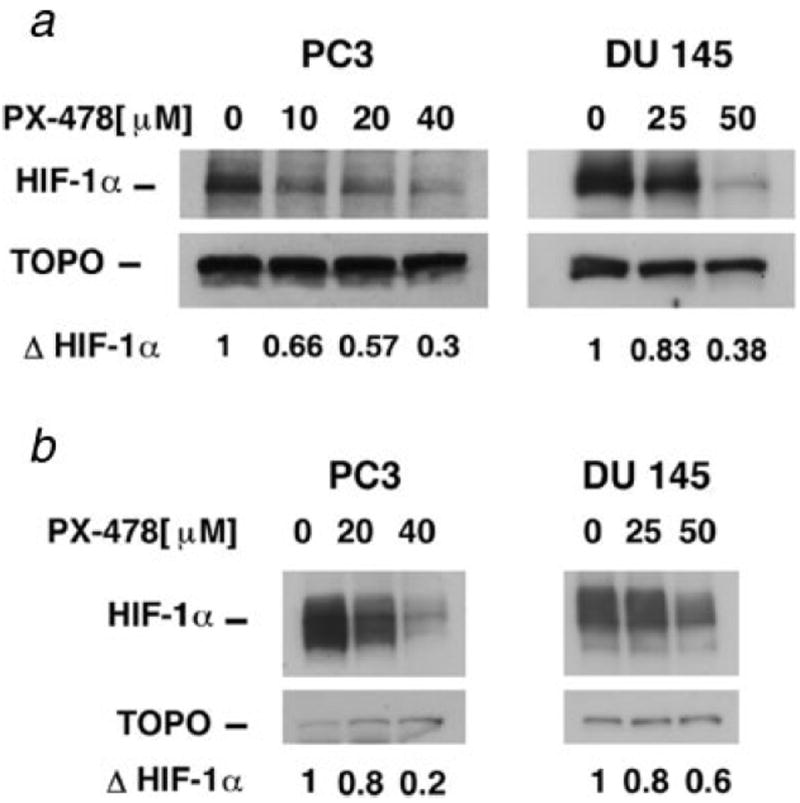
Western blot analysis of HIF-1α in PC3 and DU 145 cells. (a) Cells were treated with PX-478 for 20 hr under normoxic condition. (b) Cells were treated with PX-478 for 20 hr under normoxic condition, exposed to 1-hr hypoxic gassing and analyzed. ΔHIF-1α: fold change in HIF-1α compared to the control. For normoxic samples, 60 μg protein, and for hypoxic samples, 15 μg protein was analyzed.
HIF-1α protein was significantly upregulated by 1-hr hypoxia in both PC3 and DU 145 cells. When cells were pretreated with PX-478 for 20 hr under normoxia and then subjected to 1-hr hypoxia, hypoxia-induced accumulation of HIF-1α was attenuated (Fig. 1b). Pretreatment of PC3 cells with 20 μmol/L PX-478 resulted in 40% inhibition of HIF-1α compared to the cells treated with hypoxia alone (ΔHIF: 0.60 ± 0.08, n = 3). Pretreatment of DU 145 cells with 50 μmol/L PX-478 resulted in 35% inhibition of HIF-1α compared to the cells treated with hypoxia alone (ΔHIF: 0.65 ± 0.05, n = 3). Thus, preincubation with PX-478 inhibited hypoxia-induced HIF-1α protein accumulation at 1 hr after hypoxia.
Clonogenic survival of cells treated with PX-478 under normoxic and hypoxic condition
PC3 and DU 145 cells were treated with different concentrations of PX-478 for 18–20 hr under normoxia or hypoxia, and the effect of drug on cell survival was analyzed by clonogenic assay (Fig. 2). Under normoxia, PC3 cells were more sensitive to PX-478 than DU 145 cells. Figure 2 shows that IC50 for clonogenic survival (n = 3) was 17 μmol/L for PC3 cells and 35 μmol/L for DU 145 cells. When cells were treated with the drug under hypoxic condition for 18 hr, the IC50 was 16 μmol/L for PC3 cells and 22 μmol/L for DU 145 cells. Thus DU 145 cells were more sensitive to PX-478 under hypoxic condition.
Figure 2.
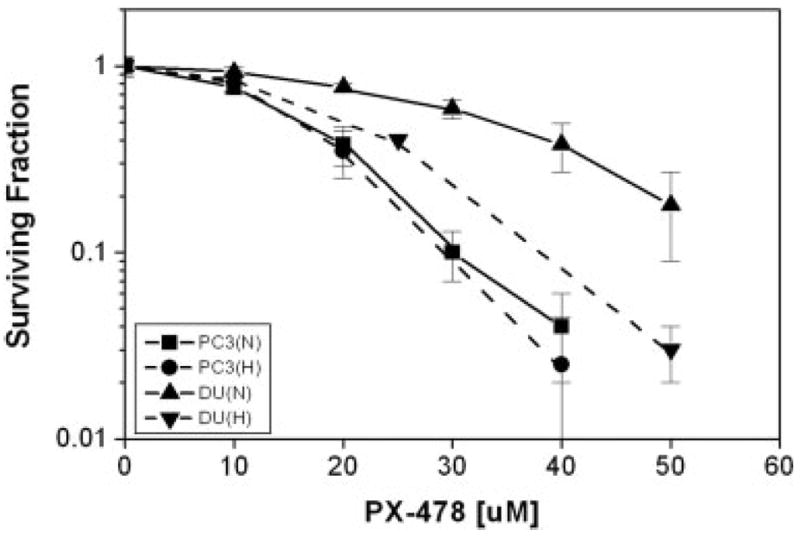
Clonogenic cell survival of PC3 and DU 145 cells treated with PX-478 for 20 hr under normoxic (N) and 18 hr under hypoxic (H) condition. Each data point represents Av ± SEM of 3 separate experiments. The IC50 for PC3 cells was 17 μM (N) and 16 μM (H). The IC50 for DU 145 cells was 35 μM (N) and 22 μM (H).
Based on the aforementioned data on HIF-1α inhibition and clonogenic survival, we treated PC3 cells with 0–30 μmol/L concentrations of PX-478 and DU 145 cells with 0–60 μmol/L concentrations of PX-478 for further analysis of radiosensitivity, cell cycle alterations and H2AX phosphorylation. The radiation survival curves in combination with PX-478 were done at concentrations at which treatment with PX-478 alone resulted in comparable reduction in surviving fraction (SF) of PC3 and DU 145 cells.
Effect of PX-478 on radiation survival of PC3 cells irradiated under normoxic or hypoxic condition
The SF of PC3 cells treated with 15 and 20 μmol/L PX-478 for 24 hr in normoxic condition was reduced to 0.56 ± 0.06 and 0.3 ± 0.03 (n = 3), respectively. Figure 3a shows that at 0.1 survival level, PX-478 enhanced the radiosensitivity of PC3 cells by an EF of 1.19 and 1.44 for 15 and 20 μmol/L concentrations, respectively (Fig. 3a).
Figure 3.
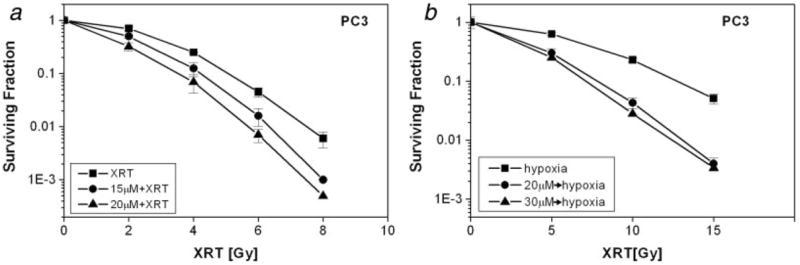
Radiosensitivity of PC3 cells treated with PX-478. (a) PC3 cells were treated with PX-478 under normoxia for 24 hr, irradiated under normoxia and plated after 1 hr. (b) PC3 cells were treated with PX-478 under normoxic condition for 20 hr, subjected to 1-hr hypoxia, irradiated under hypoxic condition and plated after 1 hr. Each data point represents Av ± SEM of 3 separate experiments. Survival curve with 30 μM PX-478 is from a single experiment.
Figure 3b shows the clonogenic survival of PC3 treated with PX-478 for 20 hr under normoxic condition and then irradiated under hypoxic condition. With this protocol, the SF of PC3 cells was 0.36 ± 0.08 (n = 3) and 0.17 at 20 and 30 μmol/L PX-478. At 0.1 survival level, preincubation of PC3 cells with 20 and 30 μmol/L PX-478 enhanced the radiosensitivity by an EF of 1.56 and 1.78, respectively, when the cells were irradiated under hypoxia (Fig. 3b).
Effect of PX-478 on radiation survival of DU 145 cells irradiated under normoxic or hypoxic condition
The SF of DU 145 cells treated with 40, 50 and 60 μmol/L PX-478 for 24 hr in normoxic condition was reduced to 0.43 ± 0.02, 0.24 ± 0.02 and 0.15 ± 0.07, respectively (n = 3). The same concentrations enhanced the radiosensitivity of DU 145 cells by an EF of 1.09, 1.13 and 1.28, respectively (Fig. 4a).
Figure 4.
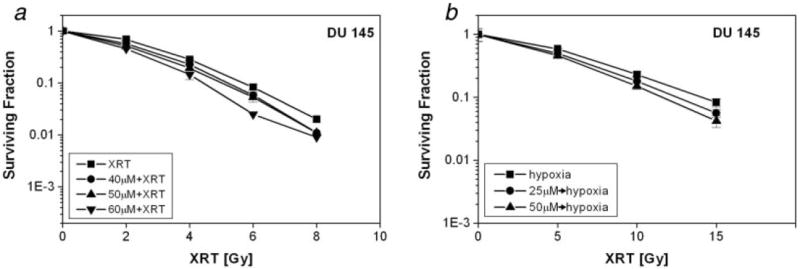
Radiosensitivity of DU 145 cells treated with PX-478. (a) DU 145 cells were treated with PX-478 under normoxia for 24 hr irradiated under normoxia and plated after 1 hr. (b) DU 145 cells were treated with PX-478 20 hr under normoxic condition for 20 hr, subjected to 1- hr hypoxia, irradiated under hypoxic condition and plated after 1 hr. Each data point represents Av ± SEM of 3 separate experiments.
Figure 4b shows the clonogenic survival DU 145 cells treated with PX-478 for 20 hr and then irradiated under hypoxic condition. With this protocol, the SF of DU 145 cells was 0.8 ± 0.2 (n = 3) and 0.18 ± 0.04 (n = 3) at 25 and 50 μmol/L PX-478. At 0.1 survival level, preincubation of DU 145 cells with 25 and 50 μmol/L PX-478 enhanced the radiosensitivity by an EF of 1.12 and 1.25, respectively, when the cells were irradiated under hypoxia (Fig. 4b).
Thus pretreatment with PX-478 enhanced the radiosensitivity of PC3 and DU 145 cells irradiated under normoxic or hypoxic condition. However, even at higher drug concentrations the EFs of DU 145 cells were smaller compared to the PC3 cells. Because DU cells appeared to be more sensitive to the drug under prolonged hypoxic condition (Fig. 2), we examined the radiosensitivity of DU 145 cells that were irradiated after treatment with 25 μmol/L PX-478 under hypoxia for 18 hr. The SF of cells treated with 25 μmol/L PX-478 alone was 0.28 + 0.07 (n = 3). Incubation with PX-478 for 18 hr under hypoxic condition increased the radiosensitivity of DU 145 cells by an EF of 1.48 at 0.1 survival level (Fig. 5b). There was no major difference in HIF-1α protein level at the end of 18 hr-hypoxia in cells treated with hypoxia alone or with PX-478 and hypoxia together (Fig. 5a).
Figure 5.
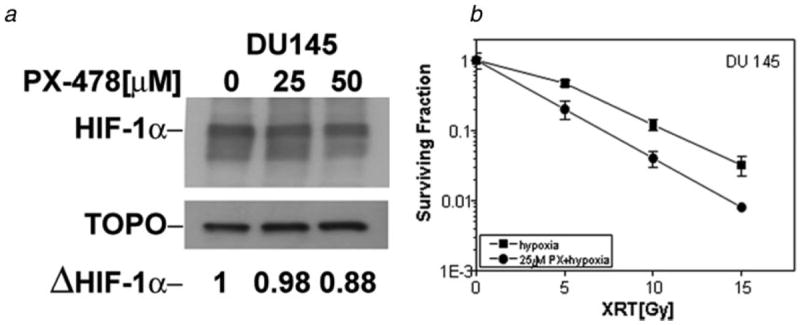
HIF-1α protein and radiosensitivity of DU 145 cells treated with PX-478 for 18 hr under hypoxic condition. (a) Cells were treated with PX-478 under hypoxic condition for 18 hr, and HIF-1α protein was analyzed by Western blotting. (b) Cells were treated with PX-478 for 18 hr under hypoxic condition and irradiated under hypoxia. Each data point represents Av ± SEM of 3 separate experiments.
Effect of PX-478 on cell cycle
To examine if PX-478 enhances radiosensitivity by accumulating cells in radiosensitive cell cycle phase, cells were treated with different concentrations of PX-478 for 24 hr and analyzed by flow cytometry. Treatment of PC3 cells with the drug significantly reduced the percentage of cells in G1, and resulted in an accumulation of cells at the S/G2M boundary (Table I). BrdU staining revealed a significant increase in cells in S and G2M compartments. However, treatment of DU 145 cells with PX-478 did not alter the cell cycle distribution significantly (Table I).
TABLE I.
EFFECT OF PX-478 ON CELL CYCLE DISTRIBUTION
| PX-478 (μM) | %G1 | %S | %G2/M |
|---|---|---|---|
| PC3 | |||
| 0 | 39.98 ± 0.72 | 41.54 ± 1.71 | 17.76 ± 2.01 |
| 10 | 27.06 ± 1.63** | 48.11 ± 2.09** | 23.81 ± 2.95* |
| 20 | 20.45 ± 0.97** | 51.71 ± 4.00* | 26.02 ± 3.41* |
| 30 | 15.03 ± 0.97** | 60.40 ± 1.53** | 23.33 ± 1.53** |
| DU 145 | |||
| 0 | 48.49 ± 4.11 | 29.07 ± 2.21 | 21.94 ± 3.44 |
| 15 | 42.85 ± 1.94 | 30.40 ± 0.45 | 26.75 ± 1.65 |
| 40 | 39.73 ± 2.42 | 34.02 ± 1.68 | 26.26 ± 2.01 |
| 50 | 41.50 ± 4.50 | 31.15 ± 2.83 | 27.35 ± 4.47 |
PC3 and DU 145 cells were treated with different concentrations of PX-478 for 24 hr and the effect of PX-478 on cell cycle distribution was analyzed by flow cytometry. Each data point represents Av ± SEM of 4 separate experiments.
p < 0.05,
p ≤ 0.001.
Effect of PX-478 on phosphorylation of histone H2AX
To examine if PX-478 causes DNA double-strand breaks (DSB) or prolongs radiation-induced DSB, phosphorylation of histone H2AX (γH2AX) indicative of DSB was analyzed by counting immunoreactive foci and by Western blot analysis. PC3 cells were treated with 20 μmol/L PX-478 for 18 hr, irradiated and after 1-hr media were replaced with drug-free media. Figure 6a shows the number of γH2AX foci in PC3 cells at 24 hr after radiation. Treatment with PX-478 alone significantly increased the number of foci compared to the control. The number of foci was significantly higher in cells treated with combination of PX-478 and 2Gy as compared with 2Gy radiation alone. Figure 6b shows immunoblot analysis of γH2AX in PC3 and DU 145 cells treated with PX-478 alone and 4Gy radiation with or without PX-478. For these experiments, cells were treated with the drug for 18 hr, irradiated and then incubated in drug-free media. Cells were analyzed at 4 hr (lanes 1–4) and 24 hr (lanes 5–8) after irradiation. Treatment with PX-478 for 18 hr resulted in an increase in γH2AX (lane 2). The increase in γH2AX persisted at 24 hr, although the cells were in drug-free media (Fig. 6b, lane 6). Radiation alone increased the expression of γH2AX at 4 hr, which subsided partially in PC3 cells and completely in DU 145 cells by 24 hr (Fig. 6b, compare lanes 3 and 7). At 24 hr, the expression of γH2AX in PC3 cells treated with PX-478 and radiation was higher compared to cells treated with PX-478 alone. In DU 145 cells, the γH2AX expression after combined treatment was comparable to PX-478 alone.
Figure 6.
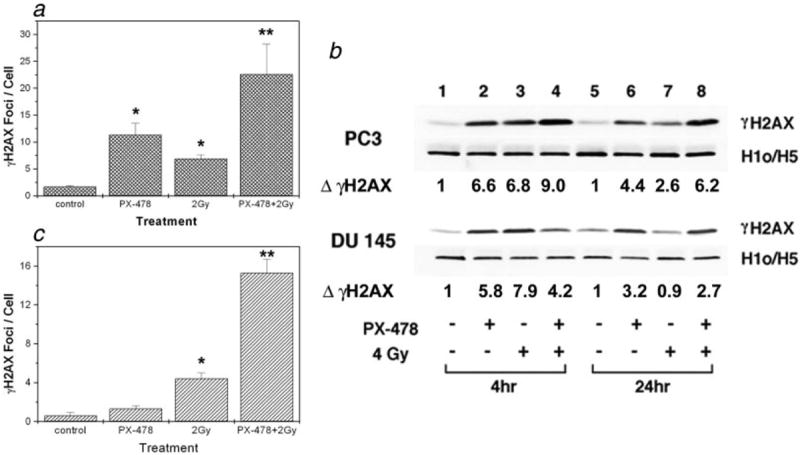
Analysis of γH2AX following treatment with PX-478 and radiation. (a) γH2AX foci per cell in PC3 cells. Cells were treated with 20 μmol/l PX-478 for 18 hr, irradiated and after 1 hr changed to drug-free media. Cells were incubated for another 24 hr in drug-free media and γH2AX foci were counted in ~70 cells. Each data point represents Av ± SEM of 3 separate experiments. *Compared to the control, **compared to the 2Gy. (b) Western blot analysis of γH2AX in PC3 (top) and DU 145 (bottom) cells. Cells were treated as above and γH2AX was analyzed at 4 hr and 24 hr after radiation. Histone H1o/H5 was used as a loading control. ΔγH2AX: fold change compared to the control. Data shown are representative of 3 separate experiments. (c) Effect of short exposure to PX-478 on H2AX phosphorylation in PC3 cells. Cells were treated with PX-478 for 30min and changed to drug-free media. After 1h cells were irradiated and H2AX foci/cell were counted at 4h (data not shown) and at 24h. *Compared to the control, **compared to the 2Gy.
In some experiments, PC3 cells were treated with PX-478 for 30 min and the effect of the short exposure on the H2AX phoshorylation was evaluated (Fig. 6c). After 30-min treatment with 20 μmol/L PX-478, cells were incubated in drug-free media for 1 hr and irradiated. At 24 hr, the number of foci in the irradiated cells pretreated with PX-478 was significantly higher than the number of foci in cells treated with radiation alone. Thus, even a short exposure to PX-478 resulted in the phosphorylation of H2AX and prolongation of γH2AX expression in the irradiated PC3 cells.
Discussion
The overexpression of HIF-1α in human cancers is correlated with poor patient prognosis and poor response to radiotherapy. 1,2,4,33 HIF-1α is therefore considered to be a potential therapeutic target and HIF-1α blockade is pursued as a promising strategy for cancer therapy in combination with radiation. Our study showed that HIF-1α inhibitor PX-478 enhanced the radiosensitivity of prostate carcinoma cells irradiated under normoxia. Furthermore, pretreatment with PX-478 under normoxic condition enhanced the radiosensitivity of cells irradiated under hypoxic condition. In addition to the inhibition of HIF-1α, treatment with PX-478 resulted in cell type-dependent alterations in cell cycle distribution and prolonged γH2AX expression in the irradiated cells.
Compared to PC3 cells, DU 145 cells were less sensitive to HIF-1α inhibition and radiation enhancing effect of PX-478 under normoxic condition. However, incubation with PX-478 for 18 hr under hypoxic condition enhanced the cytotoxicity as well as the radiosensitivity of DU 145 cells. A recent study reported that PX-478 inhibited HIF-1α at multiple levels by inhibiting HIF transcription, translation as well as deubiquitination.28 It was observed that although PX-478 decreased global protein translation in normoxia and hypoxia, HIF-1α was the major protein inhibited by PX-478 in hypoxia.28 Therefore, the enhancement of the radiosensitivity of DU 145 cells could be attributed to the inhibition of cellular targets of HIF-1α and changes in cellular biochemistry by PX-478 during hypoxic incubation.
Inhibition of tumor growth in vivo by inhibition of HIF-1α is generally attributed to the subsequent changes in tumor microenvironment and inhibition of angiogenesis. In vitro inhibition of HIF-1α under normoxic condition does not necessarily result in the inhibition of HIF-regulated proteins. Our previous study demonstrated that complete inhibition of HIF-1α protein by ibuprofen under normoxic condition did not inhibit VEGF or Glut-1 in prostate cancer cells.15 Other studies have shown that HIF inhibitors, including PX-478, are more potent in inhibition of HIF-dependent transcriptional activity under hypoxia than under normoxia.17,28,34 Therefore, the enhancement of radiosensitivity by PX-478 under normoxic condition is unlikely due to the inhibition of HIF-1α and HIF-regulated downstream proteins.
The precise mechanism of the radiation enhancement in cells preincubated with PX-478 under normoxia and irradiated in hypoxic condition is not clear. Preincubation with PX-478 under normoxia attenuated hypoxic induction of HIF-1α protein in prostate cancer cells, consistent with a recent observation that pretreatment of MCF-7 cells with 25 μmol/L PX-478 under normoxia inhibited the rate of HIF-1α synthesis upon adding hypoxia-equilibrated medium to the cells.28 It is more likely that the PX-478- induced cellular changes during normoxic incubation were responsible for the enhancement of radiosensitivity of cells irradiated under hypoxic condition. These results suggest that in addition to HIF-1α, PX-478 has other cellular targets.
Pharmacological agents with diverse mechanisms of action reduce the expression of HIF-1α protein and/or HIF-1α activity. Interestingly, many of the anticancer drugs, though not developed as HIF inhibitors, are now reported to inhibit HIF-1α.27 Agents that are shown to inhibit HIF-1α include inhibitors of PI-3/AKT-kinase and MAPK kinase pathway,12 Hsp90 antagonists18,35 topoisomerase-I and II inhibitors,17,36 thioredoxin inhibitors16 and anti-microtubule agents.14,37 HIF-1α is also inhibited by nonsteroidal anti-inflammatory drugs (NSAIDs), which affect multiple cellular targets.15,30,38,39 Thus most of the reported HIF-1α inhibitors have other cellular targets as well. These agents are also cytotoxic and separate studies indicate that many of them also enhance radiosensitivity.
In addition to HIF-1α, a recent study examined the effect of PX-478 on several other proteins including HSP-90, AKT, Src, Raf-1, cyclin B1, P53 and histone H1 under normoxia and hypoxia. 28 Although there was no major change in these protein levels, PX-478 increased P53 and lowered Raf-1 to a smaller extent under normoxia. Also, the transcription of SPAK and SART1 and UTR-driven translation of c-Myc were lowered by PX-478 in normoxia. Therefore, although selective in lowering HIF-1α transcription and translation, PX-478 is not completely specific for HIF-1α.28 The in vivo antitumor activity of PX-478 has been attributed to the inhibition of VEGF and glucose metabolism.13 Because HIF-1α may have a limited role in the radiosensitization in vitro, we looked for potential targets of PX-478 relevant in radiosensitization. To determine if the enhancement of radiosensitivity was associated with accumulation of cells in radiosensitive phases of cell cycle, we investigated the effect of PX-478 on cell cycle alterations. In PC3 cells, PX-478 significantly reduced the percentage of cells in G1 with an accumulation of cells in S/G2M. Cells in G2M are most sensitive, which may partially account for the increase in the radiosensitivity of PC3 cells treated with PX-478. Although cells in S phase are mostly radioresistant, many cytotoxic agents are known to accumulate cells in S phase. Interestingly, cell cycle alterations were not seen in DU 145 cells that also showed smaller radiation enhancement with PX-478. Thus, PX-478-induced cell cycle modulation appears to be cell type dependent. DU 145 and PC3 cells differ in pRb, p16INK4A and p21WAFI proteins, which are involved in cell cycle control.40
Ionizing radiation and some chemotherapeutic drugs produce DNA double-strand breaks, and phosphorylation of histone H2AX on serine residue (Ser139) forming foci of γH2AX immunoreactivity at sites flanking these breaks is identified as an early event following DNA damage.41,42 Cellular radiosensitivity is largely determined by the ability of cells to repair the DNA damage. Dephosphorylation of γH2AX and dispersal of γH2AX foci in irradiated cells correlates with the repair of DNA double-strand breaks.43 To determine if the enhancement of radiosensitivity by PX-478 is associated with changes in H2AX phoshorylation, we examined γH2AX expression in cells treated with PX-478. H2AX phosphorylation, as evaluated by increase in the number of γH2AX foci or Western blot analysis, was increased and persisted even after removing PX-478 from media suggesting that the drug itself caused DNA damage and the effect was persistent. Furthermore, at 24 hr, γH2AX levels in cells that were pretreated with PX-478 and irradiated were higher as compared with the cells that were treated with radiation alone, suggesting that the drug may inhibit DNA damage repair in the irradiated cells. Although radiation increased γH2AX expression in PC3 and DU 145 cells, clearance of the radiation-induced γH2AX appeared to be slower in PC3 cells compared to the DU 145 cells. Our data on γH2AX foci in cells treated with PX-478 suggests that the cytotoxicity and radiation enhancement by PX-478 may be partially associated with the drug-induced DNA lesions.
PX-478 has been studied in human tumor xenografts,13 and this in vivo study demonstrated the antitumor activity of the drug against established (0.15–0.40 cm3) or even large (0.83 cm3) aggressive human tumor xenografts. Tumor regression occurred typically 3–10 days after treatment and was characterized by extensive apoptosis. The antitumor response to PX-478 was found to be positively correlated with tumor HIF-1α levels. Treatment with PX-478 decreased VEGF expression by 38% and Glut-1 expression by 76% in HT-29 tumors. Although VEGF levels recovered rapidly, reduction in Glut-1 persisted for longer duration suggesting that inhibition of glycolysis rather than inhibition of angiogenesis was the primary mode of PX-478 anti-cancer activity in vivo.13,44
Solid tumors often contain heterogeneous hypoxic area. In addition to diffusion-limited chronic hypoxia in cells that are 100–150 mm away from blood vessels, some tumor cells may also experience perfusion-limited intermittent hypoxia because of intermittent blood supply caused by abnormal tumor vasculature. Our data showed that PX-478, a novel HIF-1α inhibitor, reduced cell survival and enhanced the radiosensitivity of prostate carcinoma cells irradiated under normoxic and hypoxic condition. Furthermore, the increase in radiosensitivity of cells treated with PX-478 under normoxia and then irradiated after 1-hr hypoxia suggests that cells experiencing intermittent hypoxia can also respond to radiation if they were pre-exposed to PX-478. In addition to inhibiting HIF-1α, PX-478 altered cell cycle progression and prolonged expression of γH2AX in irradiated cells. Our data showing enhancement of radiosensitivity of cells irradiated under normoxic as well as under hypoxic condition suggests that PX-478 is a promising radiation modifier. PX-478-induced phosphorylation of histone H2AX suggests that the drug may cause DNA damage indicative of drug toxicity. The major acute toxicity of PX-478 given daily for 5 days to nonimmunodeficient C57BL/6 mice was neutropenia, as described in a preclinical study.13 This drug is now in Phase I clinical trial (PX-478-001 NCT00522652, http://clinicaltrials.gov) making it a potential new agent for approaching hypoxic cells as part of radiation therapy treatment.
Acknowledgments
Grant sponsor: Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
References
- 1.Bachtiary B, Schindl M, Potter R, Dreier B, Knocke TH, Hainfellner JA, Horvat R, Birner P. Overexpression of hypoxia-inducible factor 1α indicates diminished response to radiotherapy and unfavorable prognosis in patients receiving radical radiotherapy for cervical cancer. Clin Cancer Res. 2003;9:2234–40. [PubMed] [Google Scholar]
- 2.Aebersold DM, Burri P, Beer KT, Laissue J, Djonov V, Greiner RH, Semenza GL. Expression of hypoxia-inducible factor-1α: a novel predictive and prognostic parameter in the radiotherapy of oropharyngeal cancer. Cancer Res. 2001;61:2911–16. [PubMed] [Google Scholar]
- 3.Fyles AW, Milosevic M, Wong R, Kavanagh MC, Pintilie M, Sun A, Chapman W, Levin W, Manchul L, Keane TJ, Hill RP. Oxygenation predicts radiation response and survival in patients with cervix cancer. Radiother Oncol. 1998;48:149–56. doi: 10.1016/s0167-8140(98)00044-9. [DOI] [PubMed] [Google Scholar]
- 4.Brizel DM, Sibley GS, Prosnitz LR, Scher RL, Dewhirst MW. Tumor hypoxia adversely affects the prognosis of carcinoma of the head and neck. Int J Radiat Oncol Biol Phys. 1997;38:285–9. doi: 10.1016/s0360-3016(97)00101-6. [DOI] [PubMed] [Google Scholar]
- 5.Nordsmark M, Overgaard M, Overgaard J. Pretreatment oxygenation predicts radiation response in advanced squamous cell carcinoma of the head and neck. Radiother Oncol. 1996;41:31–9. doi: 10.1016/s0167-8140(96)91811-3. [DOI] [PubMed] [Google Scholar]
- 6.Hall EJ. Radiobiology for the radiologist. 5. Philadelphia, PA: Lippincott Williams and Wilkins; 2000. The oxygen effects and reoxygenation; pp. 91–111. [Google Scholar]
- 7.Gray LH, Conger AD, Ebert M, Hornsey S, Scott OC. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br J Radiol. 1953;26:638–48. doi: 10.1259/0007-1285-26-312-638. [DOI] [PubMed] [Google Scholar]
- 8.Overgaard J. Hypoxic radiosensitization: adored and ignored. J Clin Oncol. 2007;25:4066–74. doi: 10.1200/JCO.2007.12.7878. [DOI] [PubMed] [Google Scholar]
- 9.Semenza GL. Regulation of mammalian O2 homeostasis by hypoxiainducible factor 1. Annu Rev Cell Dev Biol. 1999;15:551–78. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- 10.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 11.Harris AL. Hypoxia—a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 12.Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, Simons JW, Semenza GL. Modulation of hypoxia-inducible factor 1α expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60:1541–5. [PubMed] [Google Scholar]
- 13.Welsh S, Williams R, Kirkpatrick L, Paine-Murrieta G, Powis G. Antitumor activity and pharmacodynamic properties of PX-478, an inhibitor of hypoxia-inducible factor-1α. Mol Cancer Ther. 2004;3:233–44. [PubMed] [Google Scholar]
- 14.Ricker JL, Chen Z, Yang XP, Pribluda VS, Swartz GM, Van Waes C. 2-Methoxyestradiol inhibits hypoxia-inducible factor 1α, tumor growth, and angiogenesis and augments paclitaxel efficacy in head and neck squamous cell carcinoma. Clin Cancer Res. 2004;10:8665–73. doi: 10.1158/1078-0432.CCR-04-1393. [DOI] [PubMed] [Google Scholar]
- 15.Palayoor ST, Tofilon PJ, Coleman CN. Ibuprofen-mediated reduction of hypoxia-inducible factors HIF-1α and HIF-2α in prostate cancer cells. Clin Cancer Res. 2003;9:3150–7. [PubMed] [Google Scholar]
- 16.Welsh SJ, Williams RR, Birmingham A, Newman DJ, Kirkpatrick DL, Powis G. The thioredoxin redox inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin inhibit hypoxia-induced factor 1α and vascular endothelial growth factor formation. Mol Cancer Ther. 2003;2:235–43. [PubMed] [Google Scholar]
- 17.Rapisarda A, Uranchimeg B, Scudiero DA, Selby M, Sausville EA, Shoemaker RH, Melillo G. Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res. 2002;62:4316–24. [PubMed] [Google Scholar]
- 18.Isaacs JS, Jung YJ, Mimnaugh EG, Martinez A, Cuttitta F, Neckers LM. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1α-degradative pathway. J Biol Chem. 2002;277:29936–44. doi: 10.1074/jbc.M204733200. [DOI] [PubMed] [Google Scholar]
- 19.Mizuno T, Nagao M, Yamada Y, Narikiyo M, Ueno M, Miyagishi M, Taira K, Nakajima Y. Small interfering RNA expression vector targeting hypoxia-inducible factor 1α inhibits tumor growth in hepatobiliary and pancreatic cancers. Cancer Gene Ther. 2006;13:131–40. doi: 10.1038/sj.cgt.7700871. [DOI] [PubMed] [Google Scholar]
- 20.Li L, Lin X, Staver M, Shoemaker A, Semizarov D, Fesik SW, Shen Y. Evaluating hypoxia-inducible factor-1α as a cancer therapeutic target via inducible RNA interference in vivo. Cancer Res. 2005;65:7249–58. doi: 10.1158/0008-5472.CAN-04-4426. [DOI] [PubMed] [Google Scholar]
- 21.Moeller BJ, Dreher MR, Rabbani ZN, Schroeder T, Cao Y, Li CY, Dewhirst MW. Pleiotropic effects of HIF-1 blockade on tumor radiosensitivity. Cancer Cell. 2005;8:99–110. doi: 10.1016/j.ccr.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Q, Zhang ZF, Rao JY, Sato JD, Brown J, Messadi DV, Le AD. Treatment with siRNA and antisense oligonucleotides targeted to HIF-1α induced apoptosis in human tongue squamous cell carcinomas. Int J Cancer. 2004;111:849–57. doi: 10.1002/ijc.20334. [DOI] [PubMed] [Google Scholar]
- 23.Zhang X, Kon T, Wang H, Li F, Huang Q, Rabbani ZN, Kirkpatrick JP, Vujaskovic Z, Dewhirst MW, Li CY. Enhancement of hypoxia-induced tumor cell death in vitro and radiation therapy in vivo by use of small interfering RNA targeted to hypoxia-inducible factor-1α. Cancer Res. 2004;64:8139–42. doi: 10.1158/0008-5472.CAN-03-2301. [DOI] [PubMed] [Google Scholar]
- 24.Sun X, Kanwar JR, Leung E, Vale M, Krissansen GW. Regression of solid tumors by engineered overexpression of von Hippel-Lindau tumor suppressor protein and antisense hypoxia-inducible factor-1α. Gene Ther. 2003;10:2081–9. doi: 10.1038/sj.gt.3302118. [DOI] [PubMed] [Google Scholar]
- 25.Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A, Cornell-Kennon S, Lee J, Wang B, Wang J, Memmert K, Naegeli HU, et al. Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell. 2004;6:33–43. doi: 10.1016/j.ccr.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 26.Staab A, Loeffler J, Said HM, Diehlmann D, Katzer A, Beyer M, Fleischer M, Schwab F, Baier K, Einsele H, Flentje M, Vordermark D. Effects of HIF-1 inhibition by chetomin on hypoxia-related transcription and radiosensitivity in HT 1080 human fibrosarcoma cells. BMC Cancer. 2007;7:213. doi: 10.1186/1471-2407-7-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Powis G, Kirkpatrick L. Hypoxia inducible factor-1α as a cancer drug target. Mol Cancer Ther. 2004;3:647–54. [PubMed] [Google Scholar]
- 28.Koh MY, Spivak-Kroizman T, Venturini S, Welsh S, Williams RR, Kirkpatrick DL, Powis G. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1α. Mol Cancer Ther. 2008;7:90–100. doi: 10.1158/1535-7163.MCT-07-0463. [DOI] [PubMed] [Google Scholar]
- 29.Russo A, Mitchell JB, Finkelstein E, DeGraff WG, Spiro IJ, Gamson J. The effects of cellular glutathione elevation on the oxygen enhancement ratio. Radiat Res. 1985;103:232–9. [PubMed] [Google Scholar]
- 30.Palayoor ST, Burgos MA, Shoaibi A, Tofilon PJ, Coleman CN. Effect of radiation and ibuprofen on normoxic renal carcinoma cells overexpressing hypoxia-inducible factors by loss of von Hippel-Lindau tumor suppressor gene function. Clin Cancer Res. 2004;10:4158–64. doi: 10.1158/1078-0432.CCR-04-0005. [DOI] [PubMed] [Google Scholar]
- 31.Cook JA, Glass J, Lebovics R, Bobo H, Pass H, DeLaney TF, Oldfield EH, Mitchell JB, Glatstein E, Goffman TE. Measurement of thymidine replacement in patients with high grade gliomas, head and neck tumors, and high grade sarcomas after continuous intravenous infusions of 5-iododeoxyuridine. Cancer Res. 1992;52:719–25. [PubMed] [Google Scholar]
- 32.Dote H, Cerna D, Burgan WE, Carter DJ, Cerra MA, Hollingshead MG, Camphausen K, Tofilon PJ. Enhancement of in vitro and in vivo tumor cell radiosensitivity by the DNA methylation inhibitor zebularine. Clin Cancer Res. 2005;11:4571–9. doi: 10.1158/1078-0432.CCR-05-0050. [DOI] [PubMed] [Google Scholar]
- 33.Koukourakis MI, Bentzen SM, Giatromanolaki A, Wilson GD, Daley FM, Saunders MI, Dische S, Sivridis E, Harris AL. Endogenous markers of two separate hypoxia response pathways (hypoxia inducible factor 2α and carbonic anhydrase 9) are associated with radiotherapy failure in head and neck cancer patients recruited in the CHART randomized trial. J Clin Oncol. 2006;24:727–35. doi: 10.1200/JCO.2005.02.7474. [DOI] [PubMed] [Google Scholar]
- 34.Zhong H, Willard M, Simons J. NS398 reduces hypoxia-inducible factor (HIF)-1α and HIF-1 activity: multiple-level effects involving cyclooxygenase-2 dependent and independent mechanisms. Int J Cancer. 2004;112:585–95. doi: 10.1002/ijc.20438. [DOI] [PubMed] [Google Scholar]
- 35.Hur E, Kim HH, Choi SM, Kim JH, Yim S, Kwon HJ, Choi Y, Kim DK, Lee MO, Park H. Reduction of hypoxia-induced transcription through the repression of hypoxia-inducible factor-1α/aryl hydrocarbon receptor nuclear translocator DNA binding by the 90-kDa heatshock protein inhibitor radicicol. Mol Pharmacol. 2002;62:975–82. doi: 10.1124/mol.62.5.975. [DOI] [PubMed] [Google Scholar]
- 36.Chang H, Shyu KG, Lee CC, Tsai SC, Wang BW, Hsien Lee Y, Lin S. GL331 inhibits HIF-1α expression in a lung cancer model. Biochem Biophys Res Commun. 2003;302:95–100. doi: 10.1016/s0006-291x(03)00111-6. [DOI] [PubMed] [Google Scholar]
- 37.Mabjeesh NJ, Escuin D, LaVallee TM, Pribluda VS, Swartz GM, Johnson MS, Willard MT, Zhong H, Simons JW, Giannakakou P. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell. 2003;3:363–75. doi: 10.1016/s1535-6108(03)00077-1. [DOI] [PubMed] [Google Scholar]
- 38.Jones MK, Szabo IL, Kawanaka H, Husain SS, Tarnawski AS. von Hippel Lindau tumor suppressor and HIF-1α: new targets of NSAIDs inhibition of hypoxia-induced angiogenesis. FASEB J. 2002;16:264–6. doi: 10.1096/fj.01-0589fje. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Q, Tang X, Lu QY, Zhang ZF, Brown J, Le AD. Resveratrol inhibits hypoxia-induced accumulation of hypoxia-inducible factor-1α and VEGF expression in human tongue squamous cell carcinoma and hepatoma cells. Mol Cancer Ther. 2005;4:1465–74. doi: 10.1158/1535-7163.MCT-05-0198. [DOI] [PubMed] [Google Scholar]
- 40.Lanzi C, Cassinelli G, Cuccuru G, Supino R, Zuco V, Ferlini C, Scambia G, Zunino F. Cell cycle checkpoint efficiency and cellular response to paclitaxel in prostate cancer cells. Prostate. 2001;48:254–64. doi: 10.1002/pros.1105. [DOI] [PubMed] [Google Scholar]
- 41.Banath JP, Olive PL. Expression of phosphorylated histone H2AX as a surrogate of cell killing by drugs that create DNA double-strand breaks. Cancer Res. 2003;63:4347–50. [PubMed] [Google Scholar]
- 42.MacPhail SH, Banath JP, Yu TY, Chu EH, Lambur H, Olive PL. Expression of phosphorylated histone H2AX in cultured cell lines following exposure to X-rays. Int J Radiat Biol. 2003;79:351–8. doi: 10.1080/0955300032000093128. [DOI] [PubMed] [Google Scholar]
- 43.Nazarov IB, Smirnova AN, Krutilina RI, Svetlova MP, Solovjeva LV, Nikiforov AA, Oei SL, Zalenskaya IA, Yau PM, Bradbury EM, Tomilin NV. Dephosphorylation of histone γ-H2AX during repair of DNA double-strand breaks in mammalian cells and its inhibition by calyculin A. Radiat Res. 2003:160–309. doi: 10.1667/rr3043. [DOI] [PubMed] [Google Scholar]
- 44.Macpherson GR, Figg WD. Small molecule-mediated anti-cancer therapy via hypoxia-inducible factor-1 blockade. Cancer Biol Ther. 2004;3:503–4. doi: 10.4161/cbt.3.6.961. [DOI] [PubMed] [Google Scholar]