

Abstract
Sepsis-induced acute lung injury is a common clinical disorder in critically ill patients that is associated with high mortality. In this study, we investigated the role of p120-catenin (p120), a constituent of endothelial adherens junctions, in regulating the innate immune function of lungs. In mice in which acute lung injury was induced by i.p. administration of LPS, we observed a rapid decrease in the expression of p120 in lungs. The p120 protein expression was correlated inversely with severity of inflammation. Suppression of p120 expression in lung endothelial cells in mice using small interfering RNA resulted in high sensitivity to endotoxin and greatly increased the mortality compared with controls. Knockdown of p120 also increased the expression of ICAM-1, neutrophil recruitment, production of cytokines TNF-α and IL-6, pulmonary transvascular protein permeability, and lung water content in response to LPS. We demonstrated that endothelial p120 modulates lung innate immune function by interfering with the association of TLR4 with its adaptor MyD88 to block TLR4 signaling and NF-κB activation in endothelial cells. In conclusion, these studies have uncovered a novel innate immune function of endothelial p120 in downregulating the lung inflammatory response to endotoxin through the suppression of TLR4 signaling.
Sepsis resulting from bacterial infection is the most common cause of acute lung injury (ALI) (1). Bacterial LPS (endotoxin) triggers a generalized inflammatory response that subsequently leads to multiple organ dysfunction syndrome (1, 2). The pulmonary vascular endothelium is a key target and a critical participant in the pathogenesis of sepsis-induced lung inflammation and injury. Binding of LPS to TLR4 results in loss of endothelial barrier and expression of cell-surface adhesion proteins such as ICAM-1 through MyD88-dependent and -independent signaling pathways (3). Recruitment of the adaptor protein MyD88 initiates early activation of NF-κB, whereas MyD88-independent pathway leads to delayed NF-κB activation (4) and rapid activation of IFN regulatory factor 3 (5, 6). Upon TLR4 activation, MyD88 induces the association with IL-1R–associated kinase-4 (IRAK-4) and IRAK-1 and recruitment of TNFR- associated factor 6 (TRAF6) to IRAK-1 (7–9). The IRAK-4/IRAK-1/TRAF6 complex dissociates from TLR4 and interacts with TGF-β–activated kinase 1 complex, which activates IkB kinases, leading to phosphorylation and degradation of IκB and release and translocation of NF-κB to the nucleus (10). The outcome of LPS/TLR4 signaling is the production of proinflamma-tory cytokines and upregulation of endothelial adhesion molecules, which, if unchecked, induces tissue injury (11).
p120-Catenin (p120) is a member of the catenin subfamily of armadillo repeat domain-containing proteins that associates with the juxtamembrane domain of multiple types of the cadherin family (12). p120 is widely expressed in all cells capable of adhering to other cells including endothelial and epithelial cells, fibroblasts, macrophages, cardiomyocytes, and neurons (12), but it is weakly expressed or absent in B- and T-lymphocytes (12). Alternative splicing gives rise to a number of p120 isoforms that are functionally modified upon serine/threonine and tyrosine phosphorylation (12–14). The 1A and 3A p120 isoforms are the most common and are typically found coexpressed in most cell types (12, 13). The p120 1A isoform predominates in mesenchymal and motile cells, whereas p120 3A is seen in sessile cells such as epithelial cells (12, 13). p120 is known to regulate cell–cell adhesion in part by controlling the amount of cadherin present at the cell surface and its availability for interaction with adjacent cells (15–17).
Although p120 is best known for its role in cell adhesion, recent observations showed that p120-null epidermal cells exhibited increased NF-κB activation, resulting in stimulation of proinflammatory NF-κB targets in vitro and in vivo (18, 19). These findings raise the intriguing possibility that p120 has a function in regulating innate immunity. In the current study, we addressed this function and demonstrated that p120 expressed in endothelial cells modulates endotoxin-induced lung inflammation through its ability to interfere with LPS receptor TLR4 signaling. We propose that p120-mediated inhibition of TLR4 signaling represents an important innate immune mechanism capable of regulating the lung host defense function. In this regard, p120 degradation, such as after endotoxemia, leads to amplification of TLR4-mediated NF-κB signaling and lung inflammation. These results suggest that blockade of p120 degradation in sepsis may be beneficial in preventing lung inflammation and ALI.
Materials and Methods
Animals and lung inflammatory injury
Seventy-four male C57BL/6J mice (25–30 g) were used in this study. Mice were housed in microisolator cages under specific pathogen-free conditions, fed with autoclaved food, and used in experiments at 8–12 wk of age. Animal protocols received institutional review and committee approval, and all studies were conducted under anesthesia using either inhaled isoflurane or i.p.-injected ketamine (60 mg/kg). ALI was induced by i.p. injection of LPS (10 mg/kg).
Endothelial cell culture and LPS challenge
Rat lung microvascular endothelial cells (RLMVECs) were obtained from Vec Technologies (Rensselaer, NY). For monolayer cultures, the cells were plated at a density of 1.6–1.8 × 104 cells/cm2 on fibronectin-coated dishes in MCDB-131 complete medium supplemented with 10% FBS, incubated (37°C) under a humidified atmosphere of 5% CO2–95% air, and used at passages 3–5. LPS was diluted with the appropriate basal culture media and added to cells deprived of serum for 2 h.
Virus packaging and infection
p120 1A cDNA was transfected into Phoenix 293 packaging cells with Lipofectamine 2000 to produce retrovirus. The RLMVECs were then infected with LZRS-MS-IRES-neo virus containing p120 1A cDNA or vector as described previously (15). The transfection rate and expression level were confirmed by Western blot analysis.
p120 small interfering RNA transfection in endothelial cells and mouse lungs
p120 small interfering RNA (siRNA), which is a pool of three target-specific 20–25 nt siRNAs (Santa Cruz Biotechnology) at a concentration of 50 nmol/l, was added to 50–70% confluent RLMVECs to deplete p120 using the protocol provided by the manufacturer. All experiments were performed 48 h posttransfection. In vivo cationic liposome–siRNA complexes were made as described (20, 21). The liposome–siRNA complex was prepared by addition of 1.0 mg/kg siRNA into 500 ml liposome suspensions. Successful transfection of p120 siRNA was confirmed by p120 Western blotting of lung homogenates.
Survival studies
Mice transfected with scrambled or p120 siRNAwere injected i.p. with LPS at a dose of 20 mg/kg in 0.9% saline (12 mice/group), monitored every 2 h, and sacrificed when moribund or after 96 h when the observations were terminated.
Lung tissue myeloperoxidase activity
Lung polymorphonuclear neutrophil (PMN) sequestration was determined by measuring myeloperoxidase (MPO) activity as described previously (22). At the end of experiment, lungs were immediately removed, frozen, and stored at −70°C until assayed. Lungs were homogenized in 5% hexadecyltrimethyl-ammonium bromide buffer, sonicated three times for 15 s on ice, and centrifuged at 16,100 × g for 30 min at 4°C. Protein concentrations were determined by bicinchoninic acid (BCA) protein assay. A 10 μl sample of the supernatant was loaded into a cuvette plate. o-Dianisidine dihydrochloride with 0.0005% hydrogen peroxide in phosphate buffer (190 μl) was then added to samples. Absorbance change was measured at 460 nm for 3 min. MPO activity was expressed as change in absorbance per minute per milligram of protein.
PMN counts in bronchoalveolar lavage fluid
Following LPS exposure, mice were anesthetized with an i.p. injection of ketamine (125 mg/kg) and xylazine (12.5 mg/kg) mixture. The trachea was cannulated, and 1 ml PBS was infused intratracheally and withdrawn. This procedure was repeated three times. The pooled bronchoalveolar lavage (BAL) fluid was centrifuged at 400 × g for 5 min, and cell pellets were suspended in PBS. The total cell number was determined after incubation in trypan blue with a hemocytometer. Cell suspensions were diluted to a final concentration of 1 × 105 cells/ml and a 200-μl volume of resus-pended cells cytospun onto slides at 300 rpm for 5 min with a cytocen-trifuge (Shandon, Southern Sewickley, PA). Slides were stained with Diff-Quick dye (Dade Behring, Newark, DE) and examined at a magnification of ×20 and ×40 by light microscopy. The percentage of PMNs and macrophages were determined after counting 300 cells in randomly selected fields.
Determination of lung vascular permeability and edema formation
An increase in BAL protein concentration was taken as a measure of increased permeability of the alveolar-capillary barriers (22, 23). Protein concentration was determined with BCA method. At the end of experiments, lungs were weighed, dried, and reweighed. Wet-to-dry lung weight ratio was used as an index of lung water content and edema.
Cytokine generation
Cytokine levels in the lung and plasma were determined using commercial ELISA kits for TNF-α and IL- 6 (Biolegend, San Diego, CA) according to the manufacturer's instructions. Each value represents the means of trip-licate determinations.
Lung histology
For untreated mice or mice treated with LPS, the trachea was cannulated, and the lungs were fixed by instillation of 4% paraformaldehyde (Sigma-Aldrich) in PBS and held for 2 h under 25 cm H2O pressure. The lungs were removed and preserved in 10% neutral buffered formalin overnight. Formalin-fixed tissue was washed with PBS and dehydrated in 70% ethanol before paraffin embedding. Four-micrometer-thick sections were stained with H&E and chloroacetate esterase (Leder stain; Sigma-Aldrich) for detection of PMNs and examined by light microscopy (22).
PMN isolation
Rat PMNs were isolated from peripheral blood by hetastarch exchange transfusion as described previously (20, 22) with minor modification. Briefly, blood was collected from the femoral vein with exchange performed by alternatively withdrawing blood and infusing hetastarch solution in 2-ml increments. The collected blood was maintained at room temperature to allow erythrocyte sedimentation, and then the leukocyte-rich plasma was collected and centrifuged at 500 × g for 10 min at 4°C. Contaminating erythrocytes in the pellet were removed with sterile distilled water and subsequent addition of 0.6 mol/l KCl. The leukocyte-rich suspension (2 ml) was then layered on top of 3 ml Ficoll-Paque and centrifuged at 800 × g at 4°C for 20 min. The purity of isolated PMNs was >98%, and viability was >95% as determined by trypan blue exclusion.
PMN adhesion assay
PMN adhesion was measured with Cytoselect Leukocyte-Endothelium Adhesion Assay kit (CBA-210; Cell Biolabs). Briefly, RLMVECs cultured in 96-well plates in 250 ml MCDB-131 complete medium supplemented with 10% FBS were transfected with either scrambled or p120 siRNA. At 48 h posttransfection, confluent monolayers were challenged with LPS for indicated times. Peripheral blood PMNs (5 × 105) labeled with leukoTracker were added to the monolayer and incubated for 30 min. After thorough washing, cells were lysed, and the fluorescence was measured at 480 nm excitation/520 nm emission. The number of adherent PMNs was calculated on the basis of a standard curve obtained with a known number of PMNs (22). All determinations were carried out in triplicate.
PMN transmigration assay
Transendothelial PMN migration was determined with Cytoselect Leukocyte Transmigration Assay kit (CBA-212; Cell Biolabs) (22). RLMVECs were plated onto 24-well format Transwell filter inserts in 200 μl 10% FBS containing MCDB-131 complete medium and transfected with either scrambled or p120 siRNA. At 48 h posttransfection, confluent cells were challenged with LPS for indicated times. Immediately before the addition of PMNs, the upper chambers were washed twice with serum-free MCDB-131, and medium in the lower chambers was replaced with 500 μl serum-free MCDB-131 or serum-free MCDB-131 with 1.0 μM fMLF. PMNs (5 × 105 cells) labeled with leukoTracker were added to the upper chamber and incubated for 3 h at 37°C in 5% CO2, and nonadherent cells in the upper chamber were then removed. PMNs that had migrated into the lower chamber were collected, lysed, and the fluorescence was measured at 480 nm excitation/520 nm emission. Absence of additional adherent PMNs was confirmed microscopically. All determinations were carried out in duplicate and repeated at least twice.
Western blot analysis and immunoprecipitation
Cells were lysed in RIPA buffer, and lung samples were homogenized in buffer containing 1.5% Triton X-100, 0.5% deoxycholic acid, 0.1% SDS, phosphatase inhibitor I and II (1:100), and protease inhibitor mixture. Protein concentration was determined with a BCA kit (Thermo Scientific). For immunoprecipitation, cells were lysed in buffer containing 50 mmol/l Tris-HCl (pH 8), 150 mmol/l NaCl, 1 mmol/l EDTA, 1% Triton X-100, 20 mmol/l NaF, 1 mmol/l PMSF, 1 mmol/l Na3VO4, and protease inhibitor mixture. Samples were precleared for 1 h at 4°C using 1 mg control IgG (normal rabbit IgG) together with protein A/G PLUS-agarose, and then incubated overnight at 4°C with primary Ab, followed by addition of 25 μl protein A/G PLUS- agarose and further incubation at 4°C for 2 h. Equal amounts of protein from the homogenates were electrophoresed on SDS-PAGE gels (10–12%) and subsequently transferred to 0.22-μm nitrocellulose membranes. The membranes were blocked with 5% nonfat milk and probed with the appropriate Abs (20, 21).
IRAK-4 activity assay
Lysates from RLMVECs were prepared in a lysis buffer consisting of 20 mmol/l HEPES (pH 7.4), 150 mmol/l NaCl, 1 mmol/l EGTA, 2 mmol/l DTT, 0.5% Triton X-100, 12.5 mmol/l β-glycerophosphate, 5 mmol/l pnitrophenylphosphate, 10 mmol/l NaF, 1 mmol/l PMSF, 1 mmol/l sodium orthovanadate, and protease inhibitors. IRAK-4 was immunoprecipitated using affinity-purified rabbit polyclonal Ab to full-length IRAK-4 and protein A-agarose beads. Immunoprecipitates pulled down with anti– IRAK-4 Ab were divided in half. One half was solubilized in protein loading buffer for Western blot analysis, and the other half was used for IRAK kinase assay. The immunoprecipitates were incubated with 30 μl kinase buffer containing 20 mmol/l HEPES (pH 7.4), 5 mmol/l MgCl2, 20 mmol/l β-glycerophosphate, 20 mmol/l p-nitrophenylphosphate, and 1 mmol/l sodium orthovanadate. IRAK-4 kinase activity was measured by γ[32P] incorporation using myelin basic protein (MBP) as substrate. The IRAK-4 kinase reactions were stopped by boiling, and samples were analyzed by SDS-PAGE. Dried gels were analyzed by phosphorimager. Enhanced phosphorylation of MBP indicated increased activity of IRAK-4 (24, 25).
RT-PCR
RNA was extracted from mouse lung tissues using a standard TRIzol (Invitrogen) protocol with additional genomic DNA digestion. Total RNA was reverse-transcribed using High-Capacity Reverse Transcription kits (Applied Biosystems) according to the manufacturer's protocol. PCR primer pairs employed were as follows: p120 forward: 5’-AGGAGCTTCGGAAGCCACTG-3’ and reverse: 5’-GCGAAGAAAGGAAAAAAATC-3’; and β-actin forward 5’-GTGGGGCGCCCAGGCACCAC-3’ and reverse: 5’-CTCCTTAATGTCACGCACGATTT-3’. The resultant PCR products were resolved on 1.5% agarose gels and stained with ethidium bromide (26–28).
EMSA
Nuclear protein extracts were made as described previously (28). EMSA was performed using a Gel Shift Assay System (catalog number E3300, Promega). Briefly, NF-κB consensus oligonucleotide end labeling was performed by T4 polynucleotide kinase in the presence of γ-[32P] ATP, respectively. Labeled oligonucleotides were then purified on a G-25 spin column. Nuclear extract (15 μg) was then incubated with the labeled consensus oligonucleotide in a binding buffer (10 mmol/l Tris-HCl [pH 7.5], 50 mmol/l NaCl, 0.5 mmol/l DTT, and 10% glycerol) for 20 min at room temperature. The DNA–protein complexes were resolved in nondenaturing 4% acrylamide gels in 0.5× TBE buffer. The gels were then dried and exposed to film at −20°C for 8–24 h. The sequences of the oligonucleotides were as follows: NF-κB, 5’-AGTTGAGGGGACTTTCCCAGGC-3’ and SP-1, 5’-ATTCGATCGGGGCGGGGCGAGC-3’ (nonspecific competitor). A negative control, a positive control, and two competition assays with HeLa cell extracts were conducted to confirm binding specificity.
Reagents and chemicals
LPS (Escherichia coli 0127:B8; purity >99%) was obtained from Sigma-Aldrich. p120 siRNA (r) and cDNA transfection reagent Lipofectamine 2000 were purchased from Invitrogen and Dharmacon, respectively. p120 siRNA (m) and control siRNA, ICAM-1, TLR4, and IκB-a Abs were obtained from Santa Cruz Biotechnology. p120 and b-catenin Abs were purchased from BD Biosciences. IRAK-4 and MyD88 Abs were obtained from Millipore and Abcam, respectively.
Statistical analysis
One-way ANOVA and Student's Newman-Keuls test for post hoc comparisons were used to determine differences between control and experimental groups. Student t test was performed for paired samples. Parameter changes between different groups over time were evaluated by a two-way ANOVA with repeated measures. Differences between survival curves were determined by the Mantel-Cox test. Data are expressed as mean ± SEM where applicable. Differences were considered significant when p < 0.05.
Results
Reduced p120 protein expression in mouse lungs and pulmonary endothelial cells following LPS challenge
We first determined the expression of p120 in mouse lungs by Western blot analysis following i.p. injection of LPS; a 120-kDa molecular mass protein was detected in mouse lung homogenates (Fig. 1A). p120 protein level was reduced in a time-dependent manner following LPS challenge (Fig. 1A, Supplemental Fig. 1A). The loss of lung p120 after LPS exposure (10 mg/kg) was coincident with appearance of a 70-kDa fragment. p120 expression in lungs gradually recovered 24 h after low-dose LPS exposure (10 mg/kg) but not upon challenge with a lethal LPS dose (20 mg/kg). We next examined the effects of LPS on p120 mRNA expression and observed no significant differences (Fig. 1B). These results suggest that the rapid loss of p120 after LPS challenge is due to its enhanced degradation. p120 protein loss following LPS treatment was also seen in RLMVECs (Fig. 1C, Supplemental Fig. 1B).
FIGURE 1.
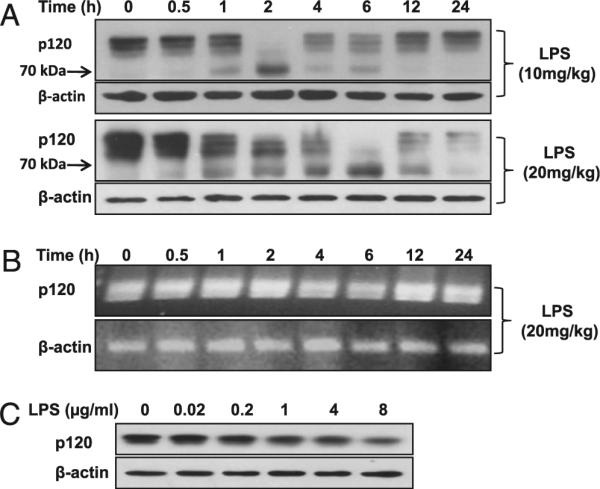
Reduction of p120 protein expression in mouse lung and pulmonary endothelial cells after LPS challenge. A, Time course of LPS-induced degradation of p120 protein in the lung. Mice were challenged by i.p. injection of LPS at different doses for the indicated times. p120 protein expression was determined by Western blot analysis. B, Time course of effect of LPS on p120 mRNA expression in the lung. p120 mRNA expression was determined by RT-PCR. C, Effect of different doses of LPS on p120 protein expression in RLMVECs. Cells were incubated with different doses of LPS for 24 h.
siRNA-induced depletion of p120 in mouse lung vessels increases sensitivity to LPS
To determine whether the observed decrease in p120 protein expression following LPS exposure is functionally relevant, we studied the effects of genetically manipulated p120 expression level in the pulmonary vasculature on lung inflammation. p120 expression was transiently reduced in pulmonary vascular endothelium using a liposome-based siRNA delivery method (20, 21, 29). p120 protein expression in lungs was reduced in a concentration- and time-dependent manner in the siRNA-treated mice (Supplemental Fig. 2A, 2B). The level of p120 protein expression started to decrease at 24 h after p120 siRNA injection and by 48 h expression was reduced by >80%. p120 depletion was maximal 72 h after siRNA treatment, and levels were fully recovered after 168 h (Supplemental Fig. 2A). The levels of vascular endothelial (VE)-cadherin and β-catenin also decreased following p120 knockdown (16, 30, 31). Scrambled siRNA had no effect on p120 expression in lungs (Supplemental Fig. 2B).
To address whether p120 has a role in the in vivo response to LPS, we carried out mortality studies with LPS using a dose previously shown to be lethal in mice. Intriguingly, the higher dosage of LPS (20 mg/kg, i.p.) resulted in a significantly greater mortality in mice with p120 depletion in lung endothelial cells compared with scrambled siRNA-treated mice (Fig. 2A). We observed that ~70% of these p120-depleted mice died within 96 h of LPS challenge in contrast to only 25% of the control mice dying during this period. Thus, loss of p120 increased the susceptibility to lethal LPS challenge.
FIGURE 2.
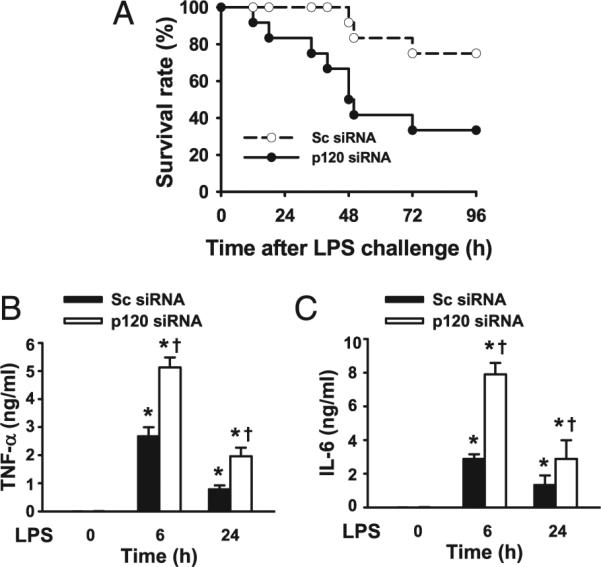
Increased mortality and cytokine production in endothelial p120-depleted mice following LPS challenge. A, Increased mortality in p120 depleted mice following LPS challenge. Mice were transfected with scrambled (Sc) or p120 siRNA as described above. At 48 h post siRNA transfection, mice were injected with LPS (20 mg/kg, i.p.) and housed under normal conditions. Differences in the survival between these two groups after LPS challenge was significant by Mantel-Cox test (n = 12 mice per group). Sera from mice transfected with Sc or p120 siRNA were collected at 6 and 24 h after PBS alone or LPS (10 mg/kg, i.p.) challenge, and TNF-α (B) and IL-6 (C) levels measured by ELISA (n = 3/group). *p < 0.05 versus control groups (without LPS), †p < 0.05 versus corresponding Sc siRNA groups.
Endotoxin mediates its inflammatory effects in part by increasing the production of proinflammatory cytokines, including TNF-α and IL-6. To determine whether p120 regulates LPS-induced cytokine release, mice transfected with scrambled or p120 siRNA were challenged with a sublethal dose of LPS (10 mg/kg), and serum was collected after 6 or 24 h. Serum TNF-α and IL-6 concentration were undetectable in the absence of LPS but expectedly increased markedly following LPS challenge. Depletion of p120, however, caused a further increase in serum concentration of TNF-α and IL-6 induced by LPS (Fig. 2B, 2C).
Suppression of p120 expression in endothelial cells increases severity of LPS-induced lung inflammation
To determine whether endothelial p120 plays a role in LPS-induced lung injury, we assessed the integrity of the alveolar–capillary barrier and pulmonary edema formation by measuring the concentration of total protein in BAL fluid (23, 32) and wet-to-dry lung weight ratio, respectively (Fig. 3A, 3B). LPS induced an increase in trans-alveolar protein permeability and lung edema formation in a time-dependent manner in mice treated with scrambled siRNA. However, these effects were significantly exaggerated in mice treated with p120 siRNA. As PMN adhesion to pulmonary vascular endothelial cells and migration into the air space are critical for induction of lung inflammation (33), we investigated the role of endothelial p120 expression in the mechanism of PMN infiltration in lungs. Lung MPO level (Fig. 3C) and PMN counts in BAL fluid (Fig. 3D) also increased in a time-dependent manner in the LPS-challenged scrambled siRNA-treated lungs. These responses were significantly enhanced in mice transfected with p120 siRNA at both 6 h and 24 h post LPS challenge (Fig. 3C). Importantly, p120 knockdown alone had no effect on pulmonary vascular permeability, edema formation, and PMN infiltration.
FIGURE 3.

Increased severity of LPS-induced lung injury in endothelial p120-depleted mice. Mice were injected with liposome–siRNA complexes. After 48 h, mice (n = 6/group) were challenged by i.p. injection of LPS (10 mg/kg) for 6 or 24 h and then sacrificed. BAL and lung homogenates were collected as described in Materials and Methods. A, PMN sequestration in lungs as determined by MPO activity. B, PMN recruitment into the alveolar space as determined by PMN counts in BAL fluid. C, Pulmonary vascular protein permeability as determined by protein concentration of BAL fluid. D, Pulmonary edema formation measured by wetto-dry (W/D) lung weight ratio. Levels of TNF-α (E) and IL-6 (F) in BAL fluid were determined by ELISA. *p < 0.05 versus control groups (without LPS), †p < 0.05 versus corresponding scrambled (Sc) siRNA groups (with LPS).
We next performed histological analysis of lungs 6 h after LPS challenge. As shown in Supplemental Fig. 3, in LPS-challenged mice, PMN infiltration increased in scrambled siRNA-treated mouse lungs. This effect was markedly enhanced in mouse lungs in which p120 was depleted. Furthermore, in response to LPS challenge, p120 knockdown lead to substantially greater BAL concentrations of TNF-α and IL-6 than in scrambled siRNA-treated lungs (Fig. 3E, 3F), whereas p120 knockdown alone had no effect on production of TNF-α and IL-6. These findings show that endothelial p120 is a crucial negative regulator of ALI during sepsis.
p120 dampens LPS-induced PMN adhesion and transmigration responses
We next measured PMN adhesion and transmigration across cultured pulmonary endothelial monolayers transfected with p120 siRNA or after overexpression of murine p120 by retroviral infection of p120 1A cDNA (11). Endothelial cells transduced with p120 siRNA showed a 90% decrease in p120 protein expression after 48 h. We observed that LPS-induced PMN adhesion to endothelial cells was 50% greater in p120-siRNA–transfected cells compared with control or scrambled siRNA-transfected cells (Fig. 4A). In the p120 overexpressing cells, LPS-induced PMN adhesion to endothelial cells was reduced by ~50% compared with control cells (Fig. 4B). PMN transmigration across LPS-stimulated endothelial monolayers was also increased compared with untreated endothelial cells, whereas knockdown of p120 further increased PMN transendothelial migration (Fig. 4C). In contrast, over-expression of p120 inhibited LPS-induced PMN transendothelial migration (Fig. 4D). These results show that p120 signaling negatively regulates LPS-induced PMN adhesion and transmigration in endothelial cells.
FIGURE 4.
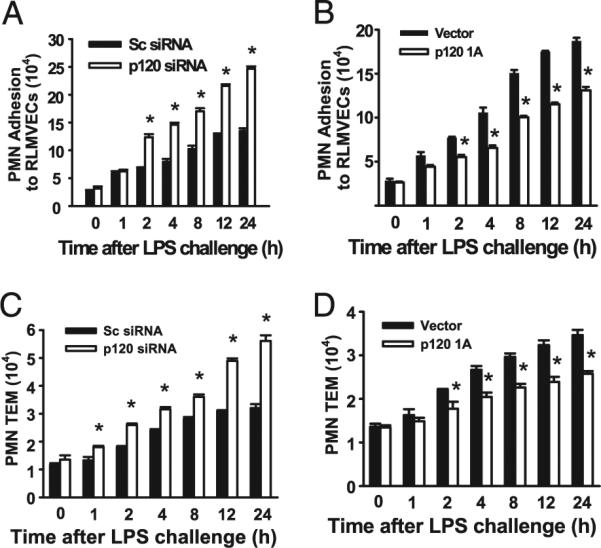
p120 attenuates LPS-induced PMN adhesion to pulmonary endothelial cells and PMN transendothelial migration. RLMVECs were transfected with scrambled (Sc) siRNA, p120 siRNA, empty vector, or retrovirus encoding p120 1A cDNA. Posttransfection, confluent mono-layers were formed, and then washed and stimulated with LPS (1.0 μg/ml) for the indicated times. PMN adhesion to RLMVECs and PMN transendothelial migration (TEM) were measured as described in Materials and Methods. Effects of p120 knockdown (A) and overexpression (B) on PMN adhesion to endothelial cells. Effects of p120 knockdown (C) and over-expression (D) on PMN transendothelial migration. n = 6/group. *p < 0.05 versus corresponding Sc siRNA groups.
p120 regulates LPS-induced ICAM-1 expression
ICAM-1 plays a major role in the recruitment of PMNs in lungs (34, 35). Therefore, we investigated the effects of endothelial p120 on ICAM-1 expression in response to LPS treatment. As shown in Fig. 5A (also Supplemental Fig. 4A), depletion of p120 in the lung vasculature increased ICAM-1 protein expression at 6 h and 24 h following LPS challenge compared with scrambled siRNA-treated lungs. In the presence of control siRNA, ICAM-1 expression in endothelial cells significantly increased in response to LPS stimulation in a time-dependent manner and depletion of p120 significantly enhanced this effect. However, p120 knockdown alone did not alter ICAM-1 expression in lungs or endothelial cells (Fig. 5A, 5B, Supplemental Fig. 4A, 4B). Fig. 5C shows expression levels of p120; expression of exogenous p120 did not affect basal ICAM-1 expression. In agreement with our observations in siRNA-treated cells demonstrating augmented expression of ICAM-1 following LPS exposure, we observed that overexpression of p120 in endothelial cells markedly attenuated LPS-induced increase in ICAM-1 expression (Fig. 5C, Supplemental Fig. 4C).
FIGURE 5.
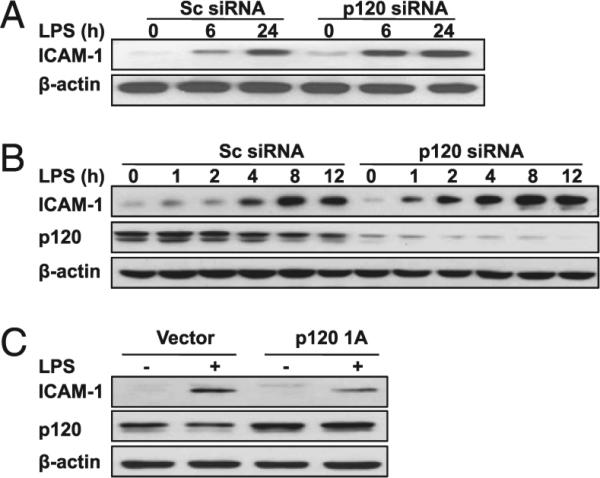
Effects of p120 expression level on LPS-induced ICAM-1 expression in mouse lungs and endothelial cells. A, ICAM-1 protein expression in lungs. Lungs from scrambled (Sc) and p120 siRNA-treated mice were collected at indicated time points after LPS injection (10 mg/kg i.p.). B, Time course of ICAM-1 and p120 protein expression following LPS stimulation in RLMVECs transfected with either Sc or p120 siRNA. RLMVECs grown in six-well plates were transfected with either scrambled or p120 siRNA. At 48 h posttransfection, cells were stimulated with LPS (1.0 μg/ml) for the indicated times. C, Overexpression of p120 1A inhibits LPS-induced ICAM-1 expression. RLMVECs grown in six-well plates were transfected with either vector or p120 1A cDNA. At 48 h posttransfection, cells were stimulated with LPS for 12 h. Data are representative of three independent experiments.
p120 suppresses LPS-induced NF-κB activation
LPS-induced increase in proinflammatory cytokine concentration and ICAM-1 expression is dependent on activation of NF-κB (36). Therefore, we determined the effects of endothelial p120 expression on LPS-induced activation of NF-κB. LPS induced an increase in activation of NF-κB in lungs (Fig. 6A) and endothelial cells (Fig. 6B) transduced with scrambled siRNA, whereas these effects were significantly enhanced in lungs and endothelial cells transduced with p120 siRNA. In contrast, p120-overexpressing cells treated with LPS showed a marked reduction in NF-κB activation (Fig. 6C). LPS-induced degradation of inhibitory IκB-a subunit was consistently increased in pulmonary endothelial cells transfected with p120 siRNA (Fig. 6D, Supplemental Fig. 5A). Addition of proteasome inhibitor MG-132 (25 μM) at the 60 min time point restored expression of IκB-α (Fig. 6E, Supplemental Fig. 5B), verifying IκB-α was degraded following LPS treatment. We also observed that overexpression of p120 prevented IκB-α degradation (Fig. 6F, Supplemental Fig. 5C). Our results show that endothelial p120 suppresses LPS-induced NF-κB activation.
FIGURE 6.

Effects of p120 expression level on LPS-induced activation of NK-κB in mouse lungs and cultured endothelial cells. Nuclear protein extracts from mouse lungs and endothelial cells were subjected to EMSA for measurement of NF-κB activity. Effect of p120 knockdown on NF-κB activity following LPS challenge in mouse lungs (A) and RLMVECs (B). Mouse lungs and RLMVECs were transfected with scrambled (Sc) siRNA or p120 siRNA. C, Effect of overexpression of p120 on NF-κB activity following LPS challenge in RLMVECs. Overexpression of p120 was performed by transfecting RLMVECs with LZRS retrovirus containing murine p120 1A cDNA or control vector. At 72 h posttransfection, cells were exposed to LPS (1.0 μg/ml) for 12 h. Cold oligonucleotides containing the transcription factor-binding site for NF-κB and SP1 were also added as competition controls. D, Effect of p120 knockdown on degradation of IκB-a following LPS challenge. E, Effect of proteasome inhibitor on IκB-a levels. Data are representative of three independent experiments. F, Effect of overexpression of p120 on degradation of IκB-α following LPS challenge. Endothelial cells were transfected with p120 1A cDNA or vector and then treated with LPS as described above (C). N, negative; NS, nonspecific; P, positive; S, specific.
p120 suppresses TLR4 signaling
To gain further mechanistic insights, we determined whether endothelial p120 has an effect on TLR4 signaling. We first investigated if p120 interfered with TLR4 interaction with MyD88, a required step in the activation of TLR4 signaling. We observed the expected pattern of association between MyD88 and TLR4; that is, a transient increase in the association between TLR4 and MyD88 upon stimulation with LPS (Fig. 7A, Supplemental Fig. 5D). Knockdown of p120 augmented LPS-induced association between TLR4 and MyD88, whereas p120 siRNA alone only slightly increased this interaction. In contrast, overexpression of p120 inhibited the TLR4–MyD88 interaction (Fig. 7B, Supplemental Fig. 5E). These findings indicate a crucial role of p120 in regulating association of MyD88 and TLR4 and thereby TLR4 signaling through the MyD88-dependent pathway. IRAK-4 activation following LPS stimulation has been demonstrated to augment TLR4 signaling and increase NF-κB activation (37, 38). We next determined the effect of endothelial p120 on LPS-induced IRAK-4 activation. As expected, a 30-min incubation period with LPS was sufficient to induce IRAK-4 phosphorylation (activation) in scrambled siRNA-treated endothelial cells (Fig. 7C, Supplemental Fig. 5F), whereas deletion of p120 with siRNA augmented LPS-induced IRAK-4 activation. These data show that p120 has an inhibitory effect on TLR4-induced IRAK-4 activation and that depletion of endothelial p120 amplifies TLR4 signaling, leading to intense NF-κB activation and proinflammatory cytokine production.
FIGURE 7.

p120 suppresses TLR4 signaling. RLMVECs grown to 50– 70% confluence were transfected with scrambled (Sc) and p120 siRNA. At 48 h posttransfection, cells were exposed to LPS (1.0 μg/ml) for the indicated times. A, Effect of p120 knockdown on the interaction of TLR4 and MyD88. Immunoprecipitation of TLR4 and immunoblotting with Ab against MyD88 were performed. The association of MyD88 and TLR4 was augmented in p120 knockdown endothelial cells. B, Effect of over-expression of p120 on the interaction of TLR4 and MyD88. Over-expression of p120 was performed by transfecting RLMVECs with LZRS retrovirus containing murine p120 1A cDNA or control vector. At 72 h posttransfection, cells were exposed to LPS (1.0 μg/ml) for the indicated times. C, Effect of p120 expression on IRAK-4 kinase activity in lung endothelial cells following LPS stimulation. RLMVECs were challenged with LPS for the indicated times and immunoprecipitation of IRAK-4 was performed. IRAK-4 kinase activity was measured by γ[32P] incorporation of MBP used as substrate (see Materials and Methods for details). Data are representative of three independent experiments.
Discussion
Our results have identified a heretofore-unknown function of the endothelial adherens junction protein p120 in downregulating the lung inflammatory response to LPS challenge. We observed that depletion of p120 in lung endothelia of mice with siRNA rendered the mice highly sensitive to endotoxin. We also demonstrated that this immunomodulatory function of p120 was secondary to suppression of NF-κB–dependent ICAM-1–mediated PMN infiltration in lungs and proinflammatory cytokine production. p120 was shown to act at the level of TLR4 signaling by preventing the association of TLR4 with the adaptor protein MyD88 to suppress LPS-mediated NF-κB activation.
We establish in this study the novel concept that endothelial p120 protein degradation, which we showed occurs during endotoxemia, results in amplification of lung host defense by enabling unfettered TLR4 signaling. This is therefore a likely factor responsible for the augmented lung inflammatory response as well as pulmonary vascular hyperpermeability and edema formation seen when lung endothelial p120 level was depleted using siRNA. Thus, the level of p120 protein expression was correlated inversely with the severity of lung inflammation. The mechanisms by which LPS challenge induces p120 degradation are not known; however, a tenable possibility is that LPS induces calpain m activation (39), which can activate proteolysis of p120 (40). In addition, there may be a direct role of p120 phosphorylation subsequent to LPS challenge in mediating p120 degradation (41).
PMN adhesion to pulmonary microvascular endothelial cells and migration into the air space are both critical for induction of inflammatory lung injury (33). We observed enhanced PMN sequestration in p120 siRNA-treated lungs after LPS challenge relative to scrambled siRNA-treated control lungs. In pulmonary microvascular endothelial cell monolayers, we also observed that depletion of p120 with siRNA amplified the LPS-induced PMN adhesion to endothelial cells and PMN transmigration across the endothelium. Further, overexpression of p120 in lung endothelia of mice reduced LPS-induced PMN adhesion and transmigration. These observations are all consistent with a key role of p120 in negatively regulating lung PMN sequestration induced by LPS.
LPS-induced PMN infiltration in lungs is known to depend on the interaction between PMNs and endothelial cells (3, 23, 32, 33). The adhesive protein ICAM-1 expressed on the endothelial plasma membrane functions as a key mechanism of recruitment of PMNs into the alveolar space in LPS-induced lung inflammation (34, 35, 42, 43). We found that the level of endothelial expression of p120 during sepsis has a major role in mediating PMN adhesion and transmigration in lungs. We observed a marked increase in ICAM-1 expression following LPS challenge in the p120-depleted lungs relative to scrambled siRNA-treated lungs. Also depletion of p120 enhanced LPS-induced increase in ICAM-1 expression in endothelial cells, whereas overexpression of p120 reduced the ICAM-1 expression. Because it is known that PMNs do not express p120 (12, 13), our results clearly indicate that p120 expressed in pulmonary vascular endothelium plays a critical role in the suppression of LPS-induced PMN infiltration into the lung.
Following LPS binding to TLR4, the inhibitory protein IκB-α is phosphorylated by activated IκB kinases α/β and degraded in the proteasomal complex (36). This releases NF-κB, which translocates to the nucleus and activates transcription of many genes encoding proinflammatory cytokines as well as adhesion molecules such as ICAM-1 (36). Using a genetic approach, we demonstrated that p120 inhibited IκB-α degradation and the subsequent activation of NF-κB induced by LPS in lungs and pulmonary microvascular endothelial cells in culture. NF-κB activation has also been previously seen in p120-null epidermal cells through stimulation of RhoA in the absence of inflammatory stimuli (18). Therefore, we cannot rule out the possibility that p120 affects NF-κB activation through the activation of Rho GTPase. We observed that p120 knockdown alone did not affect NF-κB activity, whereas it significantly increased LPS-induced NF-κB activation, suggesting an important role of endothelial p120 in modulating NF-κB signaling and hence ICAM-1 expression.
Endothelial TLR4 has been shown to be a key receptor molecule in LPS-induced neutrophil sequestration into lungs (32, 44), thus we surmised that interference with TLR4 signaling by endothelial p120 may be involved in the regulation of NF-кB activation and subsequent lung inflammation. Upon LPS-induced TLR4 activation, MyD88 is recruited to the membrane by interaction of its TIR domain with a homologous domain in TLR4. MyD88 binds to IRAK-4 and induces phosphorylation of IRAK-1 by IRAK-4 (45). IRAK-1 dissociates from the receptor complex and associates with TRAF6 to trigger downstream signaling pathways, including activation of NF-κB (46). The kinase activity of IRAK-4 is required for the recruitment of IRAK-1 to the receptor complex and activation of IRAK-1 (47). Our finding demonstrated that LPS induced the association of MyD88 and TLR4 as well as downstream activation of IRAK-4 in pulmonary endothelial cells, consistent with the described TLR4 signaling cascade (36–38, 45). Importantly, we observed that TLR4 signaling in endothelial cells was augmented by p120 depletion, whereas overexpression of p120 inhibited TLR4 signaling. Thus, although our study demonstrates that p120 functions as a negative regulator of TLR4-mediated NF-κB activation, the molecular mechanism of this intriguing effect of p120 is unknown. As there exists a pool of p120 that does not bind to VE-cadherin (14), it is possible that the cytosolic p120 has a role in directly interfering with TLR4 signaling.
p120 is known to regulate VE-cadherin expression at adherens junctions (17, 48). Therefore, p120 degradation may promote inflammation by weakening or disrupting the endothelial barrier (49). However, our data show that downregulation of p120 in the lung vasculature did not increase lung vascular protein permeability, even though levels of VE-cadherin and β-catenin expression were reduced. These results are consistent with studies in which barrier function was not altered in p120-null epidermal monolayers in vivo (18). A possible explanation for our finding is that siRNA-mediated depletion of endothelial p120 may not have been sufficient to cause endothelial barrier dysfunction.
In summary, the present results have identified a new function of endothelial p120 in regulating lung host defense in response to endotoxin. The observed decrease in endothelial p120 expression after the induction of endotoxemia was shown to be an important factor predisposing the lungs to inflammation and injury. p120 appears to function by preventing the interaction between TLR4 and MyD88 and thereby blocking TLR4 signaling, which modulates the severity of endotoxin-induced lung injury. In this context, normalizing the level of p120 expression in endothelial cells after endotoxemia may be a novel pharmacological approach to preventing sepsis-induced lung injury.
Supplementary Material
Acknowledgments
We thank Maricela Castellon (Departments of Pharmacology and Anesthesiology, University of Illinois College of Medicine) for technical assistance.
This work was supported by American Heart Association Scientist Development Grant 0730331N (to G.H.) and National Institutes of Health National Heart, Lung, and Blood Institute Grants HL104092 (to G.H.), HL071626 (to R.D.M.), and P01 HL060678 (to R.D.M. and A.B.M.).
Abbreviations used in this article
- ALI
acute lung injury
- BAL
bronchoalveolar lavage
- BCA
bicinchoninic acid
- IRAK
IL-1R–associated kinase
- MBP
myelin basic protein
- MPO
myeloperoxidase
- p120
p120-catenin
- PMN
polymorphonu-clear neutrophil
- RLMVEC
rat lung microvascular endothelial cell
- siRNA
small interfering RNA
- TRAF6
TNFR-associated factor 6
- VE
vascular endothelial
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, Lamy M, Legall JR, Morris A, Spragg R. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am. J. Respir. Crit. Care Med. 1994;149:818–824. doi: 10.1164/ajrccm.149.3.7509706. [DOI] [PubMed] [Google Scholar]
- 2.Bannerman DD, Goldblum SE. Mechanisms of bacterial lipopolysaccharide-induced endothelial apoptosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003;284:L899–L914. doi: 10.1152/ajplung.00338.2002. [DOI] [PubMed] [Google Scholar]
- 3.Dauphinee SM, Karsan A. Lipopolysaccharide signaling in endothelial cells. Lab. Invest. 2006;86:9–22. doi: 10.1038/labinvest.3700366. [DOI] [PubMed] [Google Scholar]
- 4.Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 5.Kawai T, Takeuchi O, Fujita T, Inoue J, Mühlradt PF, Sato S, Hoshino K, Akira S. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J. Immunol. 2001;167:5887–5894. doi: 10.4049/jimmunol.167.10.5887. [DOI] [PubMed] [Google Scholar]
- 6.Covert MW, Leung TH, Gaston JE, Baltimore D. Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science. 2005;309:1854–1857. doi: 10.1126/science.1112304. [DOI] [PubMed] [Google Scholar]
- 7.Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. TRAF6 is a signal transducer for interleukin-1. Nature. 1996;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- 8.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA., Jr. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol. Cell. 1998;2:253–258. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 9.Muzio M, Ni J, Feng P, Dixit VM. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science. 1997;278:1612–1615. doi: 10.1126/science.278.5343.1612. [DOI] [PubMed] [Google Scholar]
- 10.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat. Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 11.Uhlig S, Brasch F, Wollin L, Fehrenbach H, Richter J, Wendel A. Functional and fine structural changes in isolated rat lungs challenged with endotoxin ex vivo and in vitro. Am. J. Pathol. 1995;146:1235–1247. [PMC free article] [PubMed] [Google Scholar]
- 12.Mo YY, Reynolds AB. Identification of murine p120 isoforms and heterogeneous expression of p120cas isoforms in human tumor cell lines. Cancer Res. 1996;56:2633–2640. [PubMed] [Google Scholar]
- 13.Keirsebilck A, Bonné S, Staes K, van Hengel J, Nollet F, Reynolds AB, van Roy F. Molecular cloning of the human p120ctn catenin gene (CTNND1): expression of multiple alternatively spliced isoforms. Genomics. 1998;50:129–146. doi: 10.1006/geno.1998.5325. [DOI] [PubMed] [Google Scholar]
- 14.Anastasiadis PZ, Reynolds AB. The p120 catenin family: complex roles in adhesion, signaling and cancer. J. Cell Sci. 2000;113:1319–1334. doi: 10.1242/jcs.113.8.1319. [DOI] [PubMed] [Google Scholar]
- 15.Ireton RC, Davis MA, van Hengel J, Mariner DJ, Barnes K, Thoreson MA, Anastasiadis PZ, Matrisian L, Bundy LM, Sealy L, et al. A novel role for p120 catenin in E-cadherin function. J. Cell Biol. 2002;159:465–476. doi: 10.1083/jcb.200205115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davis MA, Ireton RC, Reynolds AB. A core function for p120-catenin in cadherin turnover. J. Cell Biol. 2003;163:525–534. doi: 10.1083/jcb.200307111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reynolds AB. p120-catenin: Past and present. Biochim. Biophys. Acta. 2007;1773:2–7. doi: 10.1016/j.bbamcr.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perez-Moreno M, Davis MA, Wong E, Pasolli HA, Reynolds AB, Fuchs E. p120-catenin mediates inflammatory responses in the skin. Cell. 2006;124:631–644. doi: 10.1016/j.cell.2005.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perez-Moreno M, Song W, Pasolli HA, Williams SE, Fuchs E. Loss of p120 catenin and links to mitotic alterations, inflammation, and skin cancer. Proc. Natl. Acad. Sci. USA. 2008;105:15399–15404. doi: 10.1073/pnas.0807301105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu G, Vogel SM, Schwartz DE, Malik AB, Minshall RD. Intercellular adhesion molecule-1-dependent neutrophil adhesion to endothelial cells induces caveolae-mediated pulmonary vascular hyperpermeability. Circ. Res. 2008;102:e120–e131. doi: 10.1161/CIRCRESAHA.107.167486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun Y, Hu G, Zhang X, Minshall RD. Phosphorylation of caveolin-1 regulates oxidant-induced pulmonary vascular permeability via paracellular and transcellular pathways. Circ. Res. 2009;105:676–685. 15, 685. doi: 10.1161/CIRCRESAHA.109.201673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu G, Ye RD, Dinauer MC, Malik AB, Minshall RD. Neutrophil caveolin-1 expression contributes to mechanism of lung inflammation and injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008;294:L178–L186. doi: 10.1152/ajplung.00263.2007. [DOI] [PubMed] [Google Scholar]
- 23.Hirano S. Migratory responses of PMN after intraperitoneal and intratracheal administration of lipopolysaccharide. Am. J. Physiol. 1996;270:L836–L845. doi: 10.1152/ajplung.1996.270.5.L836. [DOI] [PubMed] [Google Scholar]
- 24.Mamidipudi V, Lin C, Seibenhener ML, Wooten MW. Regulation of interleukin receptor-associated kinase (IRAK) phosphorylation and signaling by iota protein kinase C. J. Biol. Chem. 2004;279:4161–4165. doi: 10.1074/jbc.C300431200. [DOI] [PubMed] [Google Scholar]
- 25.Kim TW, Staschke K, Bulek K, Yao J, Peters K, Oh KH, Vandenburg Y, Xiao H, Qian W, Hamilton T, et al. A critical role for IRAK4 kinase activity in Toll-like receptor-mediated innate immunity. J. Exp. Med. 2007;204:1025–1036. doi: 10.1084/jem.20061825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sampson HW, Dearman AC, Akintola AD, Zimmer WE, Parrish AR. Immunohistochemical localization of cadherin and catenin adhesion molecules in the murine growth plate. J. Histochem. Cytochem. 2007;55:845–852. doi: 10.1369/jhc.7A7184.2007. [DOI] [PubMed] [Google Scholar]
- 27.van Hengel J, Vanhoenacker P, Staes K, van Roy F. Nuclear localization of the p120(ctn) Armadillo-like catenin is counteracted by a nuclear export signal and by E-cadherin expression. Proc. Natl. Acad. Sci. USA. 1999;96:7980–7985. doi: 10.1073/pnas.96.14.7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garrean S, Gao XP, Brovkovych V, Shimizu J, Zhao YY, Vogel SM, Malik AB. Caveolin-1 regulates NF-kappaB activation and lung inflammatory response to sepsis induced by lipopolysaccharide. J. Immunol. 2006;177:4853–4860. doi: 10.4049/jimmunol.177.7.4853. [DOI] [PubMed] [Google Scholar]
- 29.Zhou MY, Lo SK, Bergenfeldt M, Tiruppathi C, Jaffe A, Xu N, Malik AB. In vivo expression of neutrophil inhibitory factor via gene transfer prevents lipopolysaccharide-induced lung neutrophil infiltration and injury by a b2 integrin-dependent mechanism. J. Clin. Invest. 1998;101:2427–2437. doi: 10.1172/JCI407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao K, Allison DF, Buckley KM, Kottke MD, Vincent PA, Faundez V, Kowalczyk AP. Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J. Cell Biol. 2003;163:535–545. doi: 10.1083/jcb.200306001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davis MA, Reynolds AB. Blocked acinar development, E-cadherin reduction, and intraepithelial neoplasia upon ablation of p120-catenin in the mouse salivary gland. Dev. Cell. 2006;10:21–31. doi: 10.1016/j.devcel.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 32.Hu G, Malik AB, Minshall RD. Toll-like receptor 4 mediates neutrophil sequestration and lung injury induced by endotoxin and hyperinflation. Crit. Care Med. 2010;38:194–201. doi: 10.1097/CCM.0b013e3181bc7c17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abraham E, Carmody A, Shenkar R, Arcaroli J. Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000;279:L1137–L1145. doi: 10.1152/ajplung.2000.279.6.L1137. [DOI] [PubMed] [Google Scholar]
- 34.Basit A, Reutershan J, Morris MA, Solga M, Rose CE, Jr., Ley K. ICAM-1 and LFA-1 play critical roles in LPS-induced neutrophil recruitment into the alveolar space. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006;291:L200–L207. doi: 10.1152/ajplung.00346.2005. [DOI] [PubMed] [Google Scholar]
- 35.Moreland JG, Fuhrman RM, Pruessner JA, Schwartz DA. CD11b and intercellular adhesion molecule-1 are involved in pulmonary neutrophil recruitment in lipopolysaccharide-induced airway disease. Am. J. Respir. Cell Mol. Biol. 2002;27:474–480. doi: 10.1165/rcmb.4694. [DOI] [PubMed] [Google Scholar]
- 36.Banerjee A, Gerondakis S. Coordinating TLR-activated signaling pathways in cells of the immune system. Immunol. Cell Biol. 2007;85:420–424. doi: 10.1038/sj.icb.7100098. [DOI] [PubMed] [Google Scholar]
- 37.Li S, Strelow A, Fontana EJ, Wesche H. IRAK-4: a novel member of the IRAK family with the properties of an IRAK-kinase. Proc. Natl. Acad. Sci. USA. 2002;99:5567–5572. doi: 10.1073/pnas.082100399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Janssens S, Beyaert R. Functional diversity and regulation of different interleukin-1 receptor-associated kinase (IRAK) family members. Mol. Cell. 2003;11:293–302. doi: 10.1016/s1097-2765(03)00053-4. [DOI] [PubMed] [Google Scholar]
- 39.Li X, Li Y, Shan L, Shen E, Chen R, Peng T. Over-expression of calpastatin inhibits calpain activation and attenuates myocardial dysfunction during endotoxaemia. Cardiovasc. Res. 2009;83:72–79. doi: 10.1093/cvr/cvp100. [DOI] [PubMed] [Google Scholar]
- 40.Ohno H, Uemura K, Shintani-Ishida K, Nakamura M, Inomata M, Yoshida K. Ischemia promotes calpain-mediated degradation of p120-catenin in SH-SY5Y cells. Biochem. Biophys. Res. Commun. 2007;353:547–552. doi: 10.1016/j.bbrc.2006.12.061. [DOI] [PubMed] [Google Scholar]
- 41.Gong P, Angelini DJ, Yang S, Xia G, Cross AS, Mann D, Bannerman DD, Vogel SN, Goldblum SE. TLR4 signaling is coupled to SRC family kinase activation, tyrosine phosphorylation of zonula adherens proteins, and opening of the paracellular pathway in human lung microvascular endothelia. J. Biol. Chem. 2008;283:13437–13449. doi: 10.1074/jbc.M707986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumasaka T, Quinlan WM, Doyle NA, Condon TP, Sligh J, Takei F, Beaudet AL, Bennett CF, Doerschuk CM. Role of the intercellular adhesion molecule-1(ICAM-1) in endotoxin-induced pneumonia evaluated using ICAM-1 antisense oligonucleotides, anti-ICAM-1 monoclonal antibodies, and ICAM-1 mutant mice. J. Clin. Invest. 1996;97:2362–2369. doi: 10.1172/JCI118679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doerschuk CM, Quinlan WM, Doyle NA, Bullard DC, Vestweber D, Jones ML, Takei F, Ward PA, Beaudet AL. The role of P-selectin and ICAM-1 in acute lung injury as determined using blocking antibodies and mutant mice. J. Immunol. 1996;157:4609–4614. [PubMed] [Google Scholar]
- 44.Andonegui G, Bonder CS, Green F, Mullaly SC, Zbytnuik L, Raharjo E, Kubes P. Endothelium-derived Toll-like receptor-4 is the key molecule in LPS-induced neutrophil sequestration into lungs. J. Clin. Invest. 2003;111:1011–1020. doi: 10.1172/JCI16510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Akira S, Takeda K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 46.Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, Hurley P, Chien M, Chai S, Hitotsumatsu O, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat. Immunol. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 47.Lye E, Mirtsos C, Suzuki N, Suzuki S, Yeh WC. The role of interleukin 1 receptor-associated kinase-4 (IRAK-4) kinase activity in IRAK-4-mediated signaling. J. Biol. Chem. 2004;279:40653–40658. doi: 10.1074/jbc.M402666200. [DOI] [PubMed] [Google Scholar]
- 48.Iyer S, Ferreri DM, DeCocco NC, Minnear FL, Vincent PA. VE-cadherin-p120 interaction is required for maintenance of endothelial barrier function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004;286:L1143–L1153. doi: 10.1152/ajplung.00305.2003. [DOI] [PubMed] [Google Scholar]
- 49.Alcaide P, Newton G, Auerbach S, Sehrawat S, Mayadas TN, Golan DE, Yacono P, Vincent P, Kowalczyk A, Luscinskas FW. p120-Catenin regulates leukocyte transmigration through an effect on VE-cadherin phosphorylation. Blood. 2008;112:2770–2779. doi: 10.1182/blood-2008-03-147181. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.