

Abstract
Objectives
Inflammatory bowel disease (IBD) is heritable, but a total of 163 variants commonly implicated in IBD pathogenesis account for only 25% of the heritability. Rare, highly penetrant genetic variants may also explain mendelian forms of IBD and some of the missing heritability. To test the hypothesis that rare loss-of-function mutations can be causative, we performed whole exome sequencing (WES) on 5 members of a 2-generation family of European ancestry presenting with an early-onset and atypical form of IBD.
Methods
WES was performed for all of the 5 family members; the mother and 3 male offspring were affected, whereas the father was unaffected. Mapping, annotation, and filtering criteria were used to reduce candidate variants. For functional testing we performed forkhead box P3 (FOXP3) staining and a T-cell suppression assay.
Results
We identified a novel missense variant in exon 6 of the X-linked FOXP3 gene. The c.694A>C substitution in FOXP3 results in a cysteine-toglycine change at the protein position 232 that is completely conserved among all vertebrates. This variant (heterozygous in the mother and hemizygous in all 3 affected sons) did not impair FOXP3 protein expression, but significantly reduced the ability of the host's T regulatory cells to suppress an inappropriate autoimmune response. The variant results in a milder immune dysregulation, polyendocrinopathy, enteropathy, and X-linked phenotype with early-onset IBD.
Conclusions
Our study illustrates the successful application of WES for making a definitive molecular diagnosis in a case of multiply affected families, with atypical IBD-like phenotype. Our results also have important implications for disease biology and disease-directed therapeutic development.
Keywords: atypical, enteropathy, exome, immune dysregulation, inflammatory bowel disease, polyendocrinopathy, X-linked
Inflammatory bowel diseases (IBDs), comprising ulcerative colitis (UC) and Crohn disease (CD), are heritable disorders of intestinal inflammation that usually manifest in the second or third decade of life. Extremely early-onset IBDs, presenting in infancy and young children, have unique characteristics and include primary immunodeficiencies that may be difficult to distinguish from chronic IBD. Recent genome-wide association studies identified >163 genetic loci for IBD (1,2), with low effect sizes that explain only 25% of the heritability in these diseases. These loci fail to explain highly penetrant, extremely early-onset IBD and some of its mendelian forms. An alternative hypothesis suggests that rare, highly penetrant genetic variants may play a major role in some forms of IBD. Recent targeted sequencing discovered rare interleukin (IL)-10/IL-10R loss-of-function variants and serves as proof-of-principle that extremely early-onset IBD cohorts are fertile targets for further gene discovery efforts (3). Whole exome sequencing (WES) has proved useful in helping clinicians make the proper diagnosis and has successfully demonstrated discordance between clinical diagnosis and molecular findings (4,5).
We ascertained a 2-generation family of 5 members, 4 of whom (the mother and her 3 sons) are affected with atypical IBD phenotypes. The youngest male member of the family (the proband) was referred to our IBD clinic and presented with atypical clinical features and phenotypes of IBD that were more diverse and severe than his siblings or his mother. For 10 years, he was given an equivocal diagnosis despite intensive and expensive investigation. In this study, we performed WES to determine whether a definitive genetic diagnosis could be made for this atypical clinical presentation.
METHODS
Patients and Clinical Evaluation
We recruited a 5-member family with the mother and 3 sons affected and the father unaffected (Fig. 1). Parents are of European ancestry and unrelated. All of the participants provided written consent, and the institutional review boards of Children's Hospital and Emory University approved all of the studies. Patient II.1 is the oldest, experiencing diarrhea in infancy, and has eczema and hypogammaglobulinemia with chronic lung disease and cutaneous candidiasis. He presented with sinusitis, food allergy, and allergic rhinitis, whereas his immunoglobulin (Ig) E and T/B cells were normal. Patient II.2, the second child, developed eczematous dermatitis, chronic candidiasis, recurrent otitis media, and pneumonia in infancy. He developed type 1 diabetes mellitus at age 3. He also developed arthritis and had intermittent diarrhea, attributed to food allergies. At age 13, he had poor specific antibody responses and did not produce IgG to ϕX174, despite normal total IgG levels. Serum IgE was normal. His symptoms of arthritis and recurrent infections improved on monthly γ-globulin therapy. Patient II.3 (the proband) is the youngest; he was a full-term baby delivered after an uneventful pregnancy. He developed severe eczematous dermatitis (Fig. 2) in infancy, with recurrent bacterial skin infections. He experienced intermittent diarrhea and poor weight gain, attributed to food allergy. He had bacterial sepsis at age 2 and recurrent candidiasis, onychomycosis, and recurrent pneumonia. He developed bilateral deep venous thrombosis (DVT) at age 2 and had a recurrence of a right DVT extending into the inferior vena cava at age 5. He has unilateral right hearing loss and has not developed any endocrinopathies or autoimmune disease. His mitogen response was normal, as were his serum IgG, IgM, and IgA. Serum IgE was elevated. Colonoscopy showed evidence of multiple deep mucosal ulcerations in his colon (Fig. 3). All of the affected male members of the family shared similar phenotypes of diarrhea from food allergy and eczematous dermatitis. The patients’ mother, patient I.2, has psoriasis and had a subtotal colectomy for UC. Her brother also had psoriasis and CD, and required a bone marrow transplant for aplastic anemia. The father, patient I.1, carries a diagnosis of multiple sclerosis. He has minimal symptoms, no physical limitation, and is not receiving any therapy.
FIGURE 1.
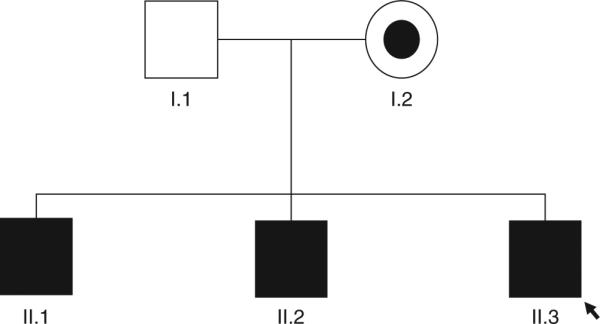
Five individuals of European descent were recruited for exome sequencing. Square symbols are male members and the circle is the female member of the family. Filled symbols are affected individuals; partially filled symbol is the carrier individual; empty symbol is the unaffected individual; arrow indicates the proband (patient II.3).
FIGURE 2.
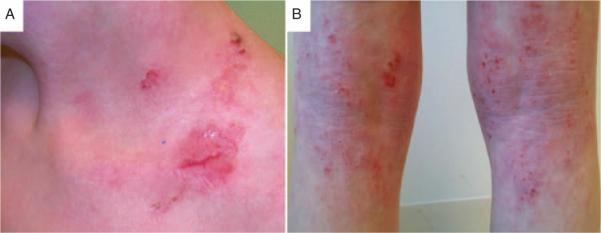
Severe eczematous dermatitis of patient II.3 (the proband) in the supraclavicular (A) and the popliteal region (B).
FIGURE 3.

Patient II.3 colonoscopy showing multiple mucosal deep ulcerations and severe inflammation consistent with the diagnosis of inflammatory bowel disease.
Genomic DNA Isolation
Genomic DNA was extracted from patients’ whole blood with a Puregene Blood Kit (Qiagen, Valencia, CA). The quantity and quality of the DNAwere determined on the NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Waltham, MA), and the integrity was assessed on 1% agarose gel stained with ethidium bromide.
Array-Based Genotyping and Copy Number Variant Detection
Genotyping and copy number variant (CNV) detection were performed in all of the patients using the Genome-Wide Human SNP Array 6.0 kit (Affymetrix, Santa Clara, CA). Two aliquots of 250 ng DNA were digested with StyI and NspI (New England Biolabs, Ipswich, MA), ligated with respective oligonucleotide adaptors (Affymetrix), and amplified by polymerase chain reaction (PCR) (Clontech Laboratories, Mountain View, CA). Forty-five microliters of the Agencourt AMPure-purified (Beckman Coulter, Brea, CA) DNA was fragmented and labeled (Affymetrix). The labeled DNAwas hybridized to the single nucleotide polymorphism (SNP) arrays for 16 hours, then washed, stained (Fluidics Station 450, Affymetrix), and scanned (Scanner, Affymetrix). Positive and negative controls were included for quality control. Arrays with <88% call rate, <0.4 contrast QC, or mismatched sex concordance were excluded from the analysis. Genotyping was performed using the Birdseed (version 2; Affymetrix) algorithm, as implemented in the Affymetrix Power Tool software (version 1.12.0), and used to calculate the pairwise identity by descent (PLINKv1.07 (6)). For CNV analysis, log(2) ratio calculation was performed using the Affymetrix Power Tool. To make the CNV calls and minimize false-positives, we used 3 algorithms: GLAD (7), GADA (8), and BEAST (G.A. Satten, unpublished data). We required each CNV to have a BEAST cutoff score of 5.0 and the corresponding call to be made by at least 2 of the 3 algorithms for further analysis.
Exome Capture Library Preparation and Quality Control
Three micrograms of genomic DNA was fragmented by sonication (Covaris Inc, Woburn, MA) to yield 300-bp fragments. The fragmented DNA was purified using Agencourt's AMPure XP (Beckman Coulter), followed by polishing of the DNA ends using T4 DNA polymerase and Klenow fragment (New England Biolabs). A single “A” base overhang was added to the 3’ end of the polished DNA fragments (using Klenow fragment), followed by ligation of Illumina (San Diego, CA) paired-end adaptors with T4 DNA ligase. The ligated samples were purified using SPRI beads and amplified by 6 cycles of PCR. After size assessment (Agilent Bioanalyzer, Agilent Technologies, Santa Clara, CA) and quantitation (PicoGreen, Invitrogen, Carlsbad, CA), 1 μg of the purified library was denatured and hybridized onto the Human Exome Library version 2.0 (Roche NimbleGen, Madison, WI) by incubation at 47°C for 68 hours. Streptavidin-coated magnetic beads were used to purify the hybridized library. The hybridized library was washed, amplified during an 8-cycle PCR reaction, and then repurified for size assessment. Final quantitation of the library was performed (Kapa Biosystems Real-Time PCR assay, Wilmington, MA), and the appropriate amount was loaded onto the Illumina HiSeq2000 for paired-end (PE) sequencing.
WES
Sequencing was performed using standard Illumina protocols as described in Bentley et al (9). The exome capture libraries were diluted to 10 nmol/L and used to generate a PE cluster density of 800,000 to 900,000/mm2. Fifty-base-pair PE sequencing was performed on the Illumina HiSeq2000 sequencer (v3 reagents).
Sequencing Data Analysis
The raw sequence reads were mapped relative to a ~30.8-Mb human exome reference sequence (NCBI37/hg19) using the PEMapper software tool (Emory University, Atlanta, GA) to identify single nucleotide variants and insertions and deletions (indels). A custom Perl script (blat_snp.pl) was used to identify the subset of variants that mapped to unique genomic regions. These unique variants were functionally annotated using SeqAnt (10), which reports the variant's type, functional classification (nonsense, replacement, silent, 5’ or 3’ untranslated region, intronic, inter-genic), presence in databases such as dbSNP, and measures of its evolutionary conservation (PhyloP, PhastCons). Data on the sequence reads, quality, and mapping have been deposited in the National Center for Biotechnology Information Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra; accession number, SRA071565).
Identification and Validation of Novel Functional Candidate Variants
Common variants catalogued in the dbSNP137 database (dbSNP137 also contains data from the 1000 Genomes Project) were excluded, and novel variants shared only by the affected family members (the mother and all 3 sons) were retained. Final novel coding variants consisted of those predicted to be functionally damaging by SIFT (11) and PolyPhen-2 (12). Sanger sequencing validated novel functional variants in each affected patient. Primers used for validation (Table 1) were designed using Primer3Plus (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) or EmPrime (http://primer.genetics.emory.edu) to generate PCR amplicons encompassing each coding variant. The amplicons were purified using Centrifugal units (Millipore, St Charles, MO) or Wizard gel and PCR cleanup (Promega, Madison, WI). The Sanger sequencing results were analyzed using SeqMan pro (version 10.1.1; DNASTAR, Madison, WI).
TABLE 1.
Sequence of primers used for Sanger sequencing validation experiment (5′-3′ orientation)
| Genes | Forward primer | Reverse primer |
|---|---|---|
| FOXP3 | CTTGGTCAGTGCCATTTTCC | CTGAGGGGCATGTGTTAAGG |
| TUFM | GGAAACAGCCAAGTTCAACG | AGCTCTGCCTCTAGCACTGG |
Cell Isolation, Flow Cytometry, and Forkhead Box P3 (FOXP3) Staining
Human T regulatory cells (Tregs) were isolated from patients or healthy donors by Ficoll separation and magnetic bead sorting (CD4+CD25+ Tregs isolation kit, 130–091–301; Miltenyi Biotec, San Diego, CA), with the addition of human CD127 antibody microbead (130–094–945) to select CD4+, CD25 bright, and CD127dim T regulatory cells. FOXP3 enrichment was determined by flow cytometry in an aliquot of the final product upon cell separation. Only cells of the highest CD25 expression (CD252+) were selected with a limited quantity of anti-CD25 beads, as described in Beyer et al (13). Nonregulatory CD25− cells were selected by collecting flow-through from saturating amounts of anti-CD25 Ab (20 μL of anti-CD25 beads/107 cells). CD4+CD252+ CD127dim cells are >90% FOXP3 positive, and CD4+CD25− cells are 2% positive for FOXP3 upon intracellular staining and flow cytometry. CD4 positivity was >98% by flow cytometry, which was performed using the FACSCalibur (BD Biosciences, San Jose, CA) and 4-color staining. Commercially available antibodies for FOXP3, CD4, and CD25 (BioLegend, San Diego, CA) were used.
T-Regulatory Cell Suppression Assays
Antigen-presenting cells, CD4+CD25− T responder cells (Tresponders), and CD4+CD25+ Tregs were isolated from 2 healthy independent control patients, patient II.3, and his mother (patient I.2). Mixed lymphocyte reactions were performed using 5 × 103 CD4+CD25− Tresponders and 5 × 104 irradiated (at 3300 rad) T-cell–depleted antigen-presenting cells isolated from healthy blood donors. Tregs were added to cell culture at titrations of 1:32 to 1:1. The culture medium was cRPMI supplemented with 10% human serum and 2.5 μg/mL of soluble anti-CD3 (UCHT1) and anti-CD28 (BD Biosciences). Proliferation was read (in a scintillation counter) at day 7 upon addition of 1 μCi tritiated thymidine (Perkin Elmer, Waltham, MA) for the last 18 hours of culture. The suppressive phenotype of CD252+ cells was confirmed through in vitro suppression assays. Using Tregs from healthy donors, suppression of Tresponder proliferation at a 1:1 ratio is typically >75%.
RESULTS
Genotyping and CNV Analysis
All DNA samples were genotyped as described in the Methods section. Allele sharing was confirmed to be 50% between parents and offspring after QC filtering. We identified 2 CNVs (Table 2) that were both maternally inherited: a duplication of ~42 kb shared by all of the affected family members but containing no known gene, and a deletion of ~224 kb overlapping the TMPRSS11E gene, shared by the mother (I.2) and 1 affected son (II.2). Neither of the CNVs identified is known to be pathogenic or likely to explain the phenotypes seen in this family.
TABLE 2.
CNVs detected in the case family
| Chr:start | Size of CNV, kb | Type of CNV | Genes within the CNV | Mode of inheritance | Individual |
|---|---|---|---|---|---|
| chr1:72768904 | 42.23 | Duplication | — | Maternal | All affected |
| chr4:69374929 | 224.60 | Deletion | TMPRSS11E | Maternal | Patient I.2 (mother) and II.2 (2nd son) |
chr = chromosome; CNV = copy number variation.
WES Revealed a Novel Missense Variant in the FOXP3 Gene
For each individual, sequencing generated 5.2 to 6.6 Gb of genomic sequence, of which ~3.9 Gb was mapped relative to the human exome reference. The mapped sequence has ~129× average depth of coverage per base (Table 3). On average, 93.8% of the targeted exome sequences had >8× coverage, a threshold set to confidently call variant sites. A total of 21,180 variants (single nucleotide variants and indels) that mapped to a single unique genomic location were discovered in the 5 exomes sequenced. Variants were annotated using SeqAnt (Table 4). To identify pathogenic mutations, we assumed that variants should be novel and affect protein structure or function. Priority was given to novel exonic replacement variants, small indels, and untranslated region variants called in splice acceptor and/or donor sites because they are likely the most pathogenic. We also required that variants be shared by all of the affected family members and variants be expected to damage protein by the prediction algorithms SIFT (11) and Poly-Phen-2 (12).
TABLE 3.
Summary sequencing statistics for 5 exomes from a 2-generation family
| II.2 | II.3 | I.2 | I.1 | Average | ||
|---|---|---|---|---|---|---|
| % Reads that uniquely map to target | 37.75 | 36.58 | 38.27 | 38.03 | 38.15 | 37.76 |
| % Bases with at least 1 read | 97.09 | 97.23 | 97.27 | 96.96 | 97.55 | 97.22 |
| % Bases with at least 8× coverage | 92.83 | 93.60 | 93.91 | 93.09 | 94.95 | 93.77 |
| Average depth of coverage | 116.65 | 121.37 | 144.62 | 112.38 | 153.22 | 129.65 |
| % Exons with at least 1 read | 98.30 | 98.41 | 98.41 | 98.30 | 98.31 | 98.35 |
TABLE 4.
Classification of genomic variants detected from exome sequencing
| In dbSNP137 |
Novel |
||||
|---|---|---|---|---|---|
| Total | Count | % of total | Count | % of total | |
| Replacement SNPs | 9033 | 8574 | 94.92 | 459 | 5.08 |
| Silent SNPs | 10785 | 10492 | 97.28 | 293 | 2.72 |
| Exonic indels | 72 | 40 | 55.56 | 32 | 44.44 |
| UTR SNPs | 1237 | 1186 | 95.88 | 51 | 4.12 |
| UTR indels | 9 | 6 | 66.67 | 3 | 33.33 |
| Intronic SNPs | 33 | 29 | 87.88 | 4 | 12.12 |
| Intronic indels | 3 | 1 | 33.33 | 2 | 66.67 |
| Intergenic SNPs | 8 | 8 | 100.00 | 0 | 0 |
| Intergenic indels | 0 | 0 | 0 | 0 | 0 |
indels = insertions and deletions; SNP = single nucleotide polymorphism; UTR = untranslated region.
After imposing these criteria, a single missense variant in the FOXP3 gene emerged as a possible candidate. The variant was successfully validated in all of the affected family members. The mother was heterozygous, whereas all of the male offspring were hemizygous (Fig. 4). The unaffected father carried the wild-type reference nucleotide.
FIGURE 4.

Sequencing chromatogram of the FOXP3 variant in each family member. DNA: A>C on (−) strand (or T>G on (+) strand). Protein: cysteine (C) to glycine (G).
The novel A-to-C missense variant (c.694A>C) is in exon 6 of FOXP3, is predicted to be damaging by SIFT (score of 0.01) and PolyPhen-2 (score of 3.168), and replaces a cysteine (C) with a glycine (G) at position 232 (p.C232G) of the FOXP3 protein. The cysteine 232 is completely conserved among all vertebrates (Table 5). As described above, this variant was maternally inherited in all of the affected hemizygous sons, and the inheritance scenario clearly supports an X-linked recessive pattern.
TABLE 5.
Comparison of FOXP3 in vertebrates
| Human | L | L | D | E | K | G | R | A | Q | C | L | L | Q | R | E | M | V | Q | S |
| Rhesus | L | L | D | E | K | G | R | A | Q | C | L | L | Q | R | E | M | V | Q | S |
| Mouse | L | L | D | E | K | G | K | A | Q | C | L | L | Q | R | E | V | V | Q | S |
| Dog | L | L | D | E | K | G | R | A | Q | C | L | L | Q | R | E | V | V | Q | S |
| Elephant | L | L | D | E | K | G | R | A | Q | C | L | L | Q | K | E | M | V | Q | S |
| Opossum | R | L | D | E | K | G | K | A | Q | C | L | I | Q | K | E | V | V | Q | N |
| Xenopus tropicalis | H | L | D | D | K | S | T | V | Q | C | L | L | Q | T | E | V | V | H | R |
| Zebrafish | A | L | D | D | K | S | T | A | Q | C | R | V | Q | M | E | V | V | Q | Q |
| Bos taurus | L | L | D | E | K | G | R | A | Q | C | L | L | Q | R | E | V | V | Q | S |
The cysteine 232 is completely conserved among all vertebrates.
Effect of Mutation on Protein Expression: Staining of Intracellular FOXP3 Protein
To determine the functional impact of the novel FOXP3 missense variant, we assessed the expression of the FOXP3 protein in peripheral blood mononuclear cells. The percentage of CD4+, CD25+ FOXP3+ Tregs, and the mean fluorescence intensity of FOXP3 were determined by flow cytometry in the affected proband (patient II.3) and his mother (patient I.2). Neither the absolute number nor the percent enrichment of FOXP3+ cells within the CD4+CD25+ cell subset was significantly different in the proband (90.8% FOXP3+, green, Fig. 5A) compared with the mother (84.5%, brown, Fig. 5A) or a healthy control (78.8%, blue, Fig. 5A). Thus, the normal expression of FOXP3 in the affected proband and his mother suggests that the c.694A>C mutation does not impair transcription and translation.
FIGURE 5.
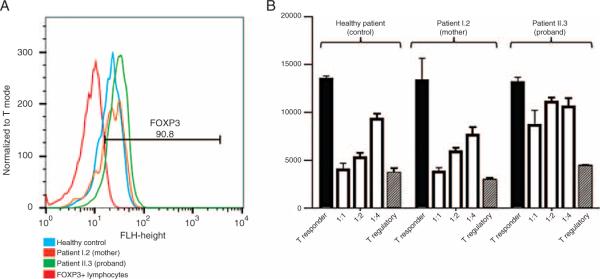
A, Profile of flow cytometry analysis for FOXP3 (FL1 channel) staining in CD4+CD25+IL7Rlo peripheral lymphocytes isolated from healthy control (blue), proband's mother (brown), and proband (green) demonstrating >90% enrichment for FOXP3+ lymphocytes (red line, isotype control). B, CD4+CD25+IL7Rlo Tregs are cocultured with increasing titer of CD4+CD25− T responders (open histograms) from the same healthy control. Healthy control and patient I.2 demonstrate normal suppressive function with increasing titer. Patient II.3 suppresses poorly, even at a 1:1 ratio.
Cell Isolation and Suppressive Function of Tregs
The FOXP3 protein is considered critical for the function of CD4+CD25+ Tregs. Because the c.694A>C change did not affect protein expression, we investigated the correlation between normal FOXP3 expression and its activity (suppressive function of CD4+CD25+ Tregs).
Using irradiated CD4− cells as stimulation and CD4+CD25− as Tresponders, Tregs from an independent healthy control, patient II.3, and patient I.2 were tested for regulatory function via in vitro suppression assay. As demonstrated in Figure 5B, Tregs isolated from patient II.3 (proband) could not efficiently suppress proliferation of Tresponders at 1:4, 1:2, or 1:1 ratios (Tregs:Tresponders). At 1:1 ratio, Tregs from the proband suppressed Tresponder proliferation by only 34.2% (thymidine counts 8627 ± 1583 vs 13,107 ± 539.9, mean/SE), whereas both patient I.2 (mother) and the healthy control suppressed Tresponder proliferation by >70% (mother 3780 ± 442.3 vs 13,275 ± 2365; control 4005 ± 686.5 vs 13,434 ± 342.7, Fig. 5B). These data demonstrate the functional relevance of the c.694A>C mutation.
DISCUSSION
We performed a genetic investigation of a nonconsanguineous family of 5, 4 of whom (the mother and her 3 sons) presented with an atypical chronic gastroenteritis. The 3 sons were affected with similar phenotypes of diarrhea from food allergy and eczematous dermatitis. In addition, each sibling presented with distinct clinical features (see Methods section). We performed WES on all of the members of the family, and with a pedigree consistent with a pattern of X-linkage, we focused on novel variants predicted to be damaging and that were shared by all of the affected individuals. We identified 1 novel missense variant, c.694A>C in exon 6 of the FOXP3 gene on chromosome X, which replaces a cysteine (C) with a glycine (G) at position 232 (p.C232G) of the FOXP3 protein. All of the affected males were hemizygous for the variant, and their mother was heterozygous. Thus, the FOXP3 c.694A>C variant is putatively causative of this atypical IBD-like phenotype.
The FOXP3 gene provides instructions for the production of the FOXP3 protein, the master transcription factor of regulatory T cells mediating both cell development and suppressive function (14). FOXP3 was initially discovered as the gene altered in the Scurfy mouse, leading to a fatal lymphoproliferative disorder (15,16). The FOXP3 gene was later found to cause the immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) in humans. IPEX is phenotypically similar to Scurfy and is characterized by systemic autoimmunity, typically beginning in the first year of life. All of the affected males have a common clinical triad of enteropathy, dermatitis, and endocrinopathy (17–19). Most die within the first year or 2 of life from metabolic derangements or sepsis (20), although a few patients with a milder phenotype have survived into the second or third decade of life (21–23). Therefore, a later age of onset cannot rule out an IPEX diagnosis.
To date, 40 different mutations within FOXP3 have been described and reported in the literature (17,20,24–38) (Fig. 6). It is now clear that variable manifestations of IPEX relate directly to the precise site of the FOXP3 mutation. Indeed, even food allergy has been attributed to FOXP3 mutation (a deletion of a 3’ splice donor site in exon1 of the FOX3 gene (24)). Our data demonstrate the c.694A>C mutation significantly abrogates the function of FOXP3, but not expression. This scenario is reminiscent of the Wiskott-Aldrich syndrome (WAS), in which mutations in the WAS gene do not affect protein expression, but lead to a milder phenotype of WAS, termed X-linked thrombocytopenia (39).
FIGURE 6.

List of all FOXP3 gene published mutations. The novel mutation identified (represented by the big open arrow) is located between the zinc finger and the leucine zipper domain.
Studies of the functional relevance of IPEX-inducing FOXP3 mutations have uncovered key FOXP3 domains, including the nuclear localization signal, the protein dimerization domain, and the DNA-binding domains. The majority of mutations identified in FOXP3 that lead to Tregs dysfunction and typical IPEX cluster in the proline-rich (protein-protein interaction domain), leucine zipper (dimerization domain), or the forkhead domain (DNA-binding domain) of the FOXP3 protein (40). The novel c.694A>C FOXP3 mutation affects the cysteine 232, immediately upstream (3 amino acids) of the leucine zipper domain known to specifically mediate homodimerization of FOXP3 (40–42). Furthermore, the cysteine 232 has been identified as 1 of 7 core residues (heptad repeats) of the FOXP3 coiled-coil domain, removal of which disrupts the dimerization (43) required for its function as a transcriptional regulator (40–42). These data argue strongly that the c.694A>C (p.C232G) mutation plays a causal role in this atypical extremely early-onset IBD phenotype.
To the best of our knowledge, this is the first report of a family in whom a novel c.694A>C mutation in the FOXP3 gene leads to an atypical IBD-like phenotype representing a milder form of IPEX. This novel mutation in FOXP3 abrogates the suppressive function of T regulatory cells. Supporting the utility of WES in familial clusters of atypical IBD, this approach led to a definitive diagnosis in this case, resulting in a justifiable treatment strategy of allogeneic bone marrow transplantation, the treatment of choice for IPEX (44).
Acknowledgment
The authors thank the family for its participation in the study.
This work was supported by National Institutes of Health grants DK087694 (S.K.), AI089714 (W.A.F.), and DK078683 (L.A.D.).
Footnotes
The authors report no conflicts of interest.
REFERENCES
- 1.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–24. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Imielinski M, Baldassano RN, Griffiths A, et al. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat Genet. 2009;41:1335–40. doi: 10.1038/ng.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moran CJ, Walters TD, Guo CH, et al. IL-10R polymorphisms are associated with very-early-onset ulcerative colitis. Inflamm Bowel Dis. 2012;19:115–23. doi: 10.1002/ibd.22974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009;106:19096–101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13:255–62. doi: 10.1097/GIM.0b013e3182088158. [DOI] [PubMed] [Google Scholar]
- 6.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Human Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hupe P, Stransky N, Thiery JP, et al. Analysis of array CGH data: from signal ratio to gain and loss of DNA regions. Bioinformatics. 2004;20:3413–22. doi: 10.1093/bioinformatics/bth418. [DOI] [PubMed] [Google Scholar]
- 8.Pique-Regi R, Monso-Varona J, Ortega A, et al. Sparse representation and Bayesian detection of genome copy number alterations from microarray data. Bioinformatics. 2008;24:309–18. doi: 10.1093/bioinformatics/btm601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–9. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shetty AC, Athri P, Mondal K, et al. SeqAnt: a web service to rapidly identify and annotate DNA sequence variations. BMC Bioinformatics. 2010;11:471. doi: 10.1186/1471-2105-11-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–74. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beyer M, Kochanek M, Darabi K, et al. Reduced frequencies and suppressive function of CD4+CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood. 2005;106:2018–25. doi: 10.1182/blood-2005-02-0642. [DOI] [PubMed] [Google Scholar]
- 14.Sakaguchi S, Ono M, Setoguchi R, et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 15.Clark LB, Appleby MW, Brunkow ME, et al. Cellular and molecular characterization of the scurfy mouse mutant. J Immunol. 1999;162:2546–54. [PubMed] [Google Scholar]
- 16.Brunkow ME, Jeffery EW, Hjerrild KA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 17.Chatila TA, Blaeser F, Ho N, et al. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J Clin Invest. 2000;106:R75–81. doi: 10.1172/JCI11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bennett CL, Christie J, Ramsdell F, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–1. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 19.Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: a model of immune dysregulation. Curr Opin Allergy Clin Immunol. 2002;2:481–7. doi: 10.1097/00130832-200212000-00002. [DOI] [PubMed] [Google Scholar]
- 20.Gambineri E, Perroni L, Passerini L, et al. Clinical and molecular profile of a new series of patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: inconsistent correlation between forkhead box protein 3 expression and disease severity. J Allergy Clin Immunol. 2008;122:1105.e1–12.e1. doi: 10.1016/j.jaci.2008.09.027. [DOI] [PubMed] [Google Scholar]
- 21.Powell BR, Buist NR, Stenzel P. An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. J Pediatr. 1982;100:731–7. doi: 10.1016/s0022-3476(82)80573-8. [DOI] [PubMed] [Google Scholar]
- 22.Gambineri E, Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical regulator of T-cell homeostasis. Curr Opin Rheumatol. 2003;15:430–5. doi: 10.1097/00002281-200307000-00010. [DOI] [PubMed] [Google Scholar]
- 23.Bennett CL, Brunkow ME, Ramsdell F, et al. A rare polyadenylation signal mutation of the FOXP3 gene (AAUAAA–>AAUGAA) leads to the IPEX syndrome. Immunogenetics. 2001;53:435–9. doi: 10.1007/s002510100358. [DOI] [PubMed] [Google Scholar]
- 24.Torgerson TR, Linane A, Moes N, et al. Severe food allergy as a variant of IPEX syndrome caused by a deletion in a noncoding region of the FOXP3 gene. Gastroenterology. 2007;132:1705–17. doi: 10.1053/j.gastro.2007.02.044. [DOI] [PubMed] [Google Scholar]
- 25.Myers AK, Perroni L, Costigan C, et al. Clinical and molecular findings in IPEX syndrome. Arch Dis Child. 2006;91:63–4. doi: 10.1136/adc.2005.078287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bacchetta R, Passerini L, Gambineri E, et al. Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest. 2006;116:1713–22. doi: 10.1172/JCI25112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gavin MA, Torgerson TR, Houston E, et al. Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc Natl Acad Sci U S A. 2006;103:6659–64. doi: 10.1073/pnas.0509484103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kobayashi I, Shiari R, Yamada M, et al. Novel mutations of FOXP3 in two Japanese patients with immune dysregulation, polyendocrinopathy, enteropathy, X linked syndrome (IPEX). J Med Genet. 2001;38:874–6. doi: 10.1136/jmg.38.12.874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moudgil A, Perriello P, Loechelt B, et al. Immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome: an unusual cause of proteinuria in infancy. Pediatr Nephrol. 2007;22:1799–802. doi: 10.1007/s00467-007-0532-0. [DOI] [PubMed] [Google Scholar]
- 30.De Benedetti F, Insalaco A, Diamanti A, et al. Mechanistic associations of a mild phenotype of immunodysregulation, polyendocrinopathy, enteropathy, x-linked syndrome. Clin Gastroenterol Hepatol. 2006;4:653–9. doi: 10.1016/j.cgh.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 31.Lee SM, Gao B, Dahl M, et al. Decreased FoxP3 gene expression in the nasal secretions from patients with allergic rhinitis. Otolaryngol Head Neck Surg. 2009;140:197–201. doi: 10.1016/j.otohns.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 32.Halabi-Tawil M, Ruemmele FM, Fraitag S, et al. Cutaneous manifestations of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Br J Dermatol. 2009;160:645–51. doi: 10.1111/j.1365-2133.2008.08835.x. [DOI] [PubMed] [Google Scholar]
- 33.Hashimura Y, Nozu K, Kanegane H, et al. Minimal change nephrotic syndrome associated with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Pediatr Nephrol. 2009;24:1181–6. doi: 10.1007/s00467-009-1119-8. [DOI] [PubMed] [Google Scholar]
- 34.Harbuz R, Lespinasse J, Boulet S, et al. Identification of new FOXP3 mutations and prenatal diagnosis of IPEX syndrome. Prenat Diagn. 2010;30:1072–8. doi: 10.1002/pd.2613. [DOI] [PubMed] [Google Scholar]
- 35.Rubio-Cabezas O, Minton JA, Caswell R, et al. Clinical heterogeneity in patients with FOXP3 mutations presenting with permanent neonatal diabetes. Diabetes Care. 2009;32:111–6. doi: 10.2337/dc08-1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.An YF, Xu F, Wang M, et al. Clinical and molecular characteristics of immunodysregulation, polyendocrinopathy, enteropathy, X-linked syndrome in China. Scand J Immunol. 2011;74:304–9. doi: 10.1111/j.1365-3083.2011.02574.x. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki S, Makita Y, Mukai T, et al. Molecular basis of neonatal diabetes in Japanese patients. J Clin Endocrinol Metab. 2007;92:3979–85. doi: 10.1210/jc.2007-0486. [DOI] [PubMed] [Google Scholar]
- 38.Wildin RS, Ramsdell F, Peake J, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27:18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- 39.Albert MH, Bittner TC, Nonoyama S, et al. X-linked thrombocytopenia (XLT) due to WAS mutations: clinical characteristics, long-term outcome, and treatment options. Blood. 2010;115:3231–8. doi: 10.1182/blood-2009-09-239087. [DOI] [PubMed] [Google Scholar]
- 40.Lopes JE, Torgerson TR, Schubert LA, et al. Analysis of FOXP3 reveals multiple domains required for its function as a transcriptional repressor. J Immunol. 2006;177:3133–42. doi: 10.4049/jimmunol.177.5.3133. [DOI] [PubMed] [Google Scholar]
- 41.Chae WJ, Henegariu O, Lee SK, et al. The mutant leucine-zipper domain impairs both dimerization and suppressive function of Foxp3 in T cells. Proc Natl Acad Sci U S A. 2006;103:9631–6. doi: 10.1073/pnas.0600225103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li B, Samanta A, Song X, et al. FOXP3 is a homo-oligomer and a component of a supramolecular regulatory complex disabled in the human XLAAD/IPEX autoimmune disease. Int Immunol. 2007;19:825–35. doi: 10.1093/intimm/dxm043. [DOI] [PubMed] [Google Scholar]
- 43.Song X, Li B, Xiao Y, et al. Structural and biological features of FOXP3 dimerization relevant to regulatory T cell function. Cell Rep. 2012;1:665–75. doi: 10.1016/j.celrep.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rao A, Kamani N, Filipovich A, et al. Successful bone marrow transplantation for IPEX syndrome after reduced-intensity conditioning. Blood. 2007;109:383–5. doi: 10.1182/blood-2006-05-025072. [DOI] [PubMed] [Google Scholar]