

Abstract
Background
Cardiac myocyte hypertrophy is regulated by an extensive intracellular signal transduction network. In vitro evidence suggests that the scaffold protein muscle A-kinase anchoring protein β (mAKAPβ) serves as a nodal organizer of hypertrophic signaling. However, the relevance of mAKAPβ signalosomes to pathological remodeling and heart failure in vivo remains unknown.
Methods and Results
Using conditional, cardiac myocyte–specific gene deletion, we now demonstrate that mAKAPβ expression in mice is important for the cardiac hypertrophy induced by pressure overload and catecholamine toxicity. mAKAPβ targeting prevented the development of heart failure associated with long-term transverse aortic constriction, conferring a survival benefit. In contrast to 29% of control mice (n=24), only 6% of mAKAPβ knockout mice (n=31) died in the 16 weeks of pressure overload (P=0.02). Accordingly, mAKAPβ knockout inhibited myocardial apoptosis and the development of interstitial fibrosis, left atrial hypertrophy, and pulmonary edema. This improvement in cardiac status correlated with the attenuated activation of signaling pathways coordinated by the mAKAPβ scaffold, including the decreased phosphorylation of protein kinase D1 and histone deacetylase 4 that we reveal to participate in a new mAKAP signaling module. Furthermore, mAKAPβ knockout inhibited pathological gene expression directed by myocyte-enhancer factor-2 and nuclear factor of activated T-cell transcription factors that associate with the scaffold.
Conclusions
mAKAPβ orchestrates signaling that regulates pathological cardiac remodeling in mice. Targeting of the underlying physical architecture of signaling networks, including mAKAPβ signalosome formation, may constitute an effective therapeutic strategy for the prevention and treatment of pathological remodeling and heart failure.
Keywords: heart failure, hypertrophy, signal transduction
Congestive heart failure (CHF), the end stage of many heart diseases, is a syndrome affecting >5 million people in the United States alone.1 Current pharmacological treatment for CHF includes angiotensin-converting enzyme inhibitors and antagonists for β-adrenergic, angiotensin II, and aldosterone receptors, as well as diuretics, calcium channel blockers, and other vasodilators. Nevertheless, 5-year mortality for CHF remains 50%, providing impetus for novel therapeutic approaches.
Clinical Perspective on p 672
Regardless of the pathogenesis, the common response of the heart to stress is cardiac myocyte hypertrophy.1 Hypertrophy can be compensatory for increased wall stress (Laplace law). However, in disease, hypertrophy is typically concomitant with impaired contractility, myocyte death, and myocardial interstitial fibrosis, resulting in progressive systolic and diastolic dysfunction. Consequently, left ventricular (LV) hypertrophy is a leading CHF risk factor. Within the myocyte, pathological remodeling is controlled by a network of signaling pathways composed of plasmalemmal receptors, second messengers, protein kinases and phosphatases, regulators of gene transcription, and other signaling molecules.2 These pathways are organized by scaffold proteins that physically colocalize signaling enzymes with their upstream activators and substrate effectors.3 Formation of signalosomes by scaffolds increases the efficiency of signal transduction by affording additional specificity to enzyme catalysis, accelerating pathway kinetics, amplifying responses, and providing the infrastructure for feed-forward and feedback loops. Scaffold proteins that organize nodes in the hypertrophic signaling network may provide alternative therapeutic targets specific to the disease process.
Muscle A-kinase anchoring protein β (mAKAPβ; AKAP6) is a 227 kDa scaffold located at the nuclear envelope in striated myocytes.4 Studies using cultured neonatal myocytes have implicated mAKAPβ signalosomes in G-protein–coupled receptor, cytokine receptor, and hypoxia-regulated signaling, including the activation of gene expression by nuclear factor of activated T-cells (NFATc), myocyte enhancer factor-2 (MEF2), and hypoxia-inducible factor-1α (HIF-1α) transcription factors.5–11 Despite extensive biochemical characterization of mAKAPβ complexes, the relevance of the scaffold to cardiac disease has not been investigated in vivo. We now show that mAKAPβ is required for cardiac remodeling in response to diverse stressors. Remarkably, in mice subjected to long-term pressure overload, mAKAPβ knockout conferred a survival benefit. The results presented below define mAKAPβ as a key scaffold in the myocyte hypertrophic signaling network and provide a rationale for the targeting of these signalosomes in CHF prevention.
Methods
All methods and reagents are described in detail in the Data Supplement.
Animal Studies
All experiments involving animals were approved by the Institutional Animal Care and Use Committee at the University of Miami. All mice for this project were in the C57BL/76 background. The mAKAPfl mouse was generated by the University of Cincinnati Gene Targeted Mouse Service using a vector designed to delete exon 9 of the mAKAP gene conditionally (Figure I in the Data Supplement). Tg(Myh6-cre/ Esr1*) mice (MerCreMer mice [MCM], The Jackson Laboratory) express a tamoxifen (Tam)-inducible, cre-estrogen receptor fusion protein under the control cardiac myocyte–specific, α-myosin heavy chain promoter.12 To induce mAKAP conditional knock-out (mAKAP CKO), 8-week-old mAKAPfl/fl;MCM mice were fed Tam-containing chow for 1 week (≈ 20 mg Tam/kg body weight). Control cohorts included MCM+Tam and mAKAPfl/fl;MCM mice that control for effects because of gene targeting and Tam administration, respectively. Mice were used for experiments after restoration of normal chow for 1 week.
Transverse aortic constriction (TAC) and infusion of isoproterenol (Iso) by osmotic pump (60 mg/kg per day for 2 weeks) were used to induce pathological hypertrophy by pressure overload and chronic catecholamine excess, respectively. Physiological hypertrophy was induced by a 5-week swimming regimen. Mice were studied by echocardiography under isoflurane anesthesia and postmortem by cardiac histology, including wheat germ agglutinin staining for myocyte cross section area, picrosirius red and Masson’s Trichrome staining for fibrosis, and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining for apoptosis. Additional analyses included isolation of adult myocytes for morphometry, assay of gene expression by Nanostring, and immunoblotting for detection of protein complexes and the activation of key regulatory molecules.
Statistics
For all experiments, n refers to the number of individual mice studied. All data are expressed as mean±SEM. For scatter plots, red bar indicates sample mean. P values were calculated using 2-tailed Student t tests, paired or unpaired as appropriate. Symbols represent uncorrected P values of different orders of magnitude: *P≤0.05, **P≤0.005, ***P≤0.0005. For multiple comparisons, 1- or 2-way ANOVA followed by Bonferroni multiple comparison testing was used as appropriate. Kaplan–Meier Survival curves were generated using GraphPad Prism 5; significance was determined by log-rank (Mantel–Cox) test. Gene expression data were analyzed using non-parametric testing using GraphPad Prism 5.
Results
Development of a Conditional mAKAP Knockout Mouse
Germline mAKAP knockout acquired by mating the mAKAPfl mouse (Figure I in the Data Supplement) to a cre transgenic under the transcriptional control of a human cytomegalovirus minimal promoter resulted in morphologically normal mice unable to survive more than a few hours after birth (data not shown). This neonatal lethality was not as a result of a defect in cardiac development because Nkx2-5–directed cre expression did not result in an overt phenotype by 6 months of age (data not shown).13 How mAKAP was required for survival was not investigated further.
Tam administration to adult mAKAPfl/fl;MCM mice inhibited mAKAPβ cardiac expression >80% but did not induce cardiomyopathy in unstressed mice (see below). In control heart tissue and cells, mAKAPβ was detectable only in cardiac myocytes and only at the nuclear envelope (Figure 1A). There was no apparent difference in mAKAPβ localization between neonatal and adult myocytes (Figure II in the Data Supplement) or in hypertrophied or failing hearts (data not shown).
Figure 1.

Muscle A-kinase anchoring protein β (mAKAPβ) is important for pressure overload–induced hypertrophy. A, Frozen left ventricular (LV) tissue sections from MCM+tamoxifen (Tam) control (top left 2 panels) and mAKAP CKO (bottom left 2 panels) mice were stained with mAKAP FL100 antibody (gray scale panels and green), Hoechst nuclear stain (blue), and wheat germ agglutinin (red, shown in enlarged control image only). Bar, 20 μm (n=3). B–I, Mice were subjected to 2-week transverse aortic constriction (TAC) or sham operation. mAKAP CKO mice were mAKAPfl/fl;MCM+Tam. MCM+Tam and mAKAPfl/fl;MCM were controls. B and C, M-mode echocardiography for LV posterior wall thickness in diastole (LVPW;d) and ejection fraction; cf Table I in the Data Supplement. D, Biventricular weight indexed to body weight; cf Table II in the Data Supplement. E and F, Myocyte cross section area was determined by wheat germ agglutinin staining. Bar, 50 μm (n=4–6). G–I, Isolated adult myocytes were measured for width and length for the mAKAP CKO and MCM+Tam cohorts. Bar=20 μmol/L (n=3–5 mice); >100 myocytes assayed per mouse. *Vs sham-operated for same genotype; †vs MCM+Tam; ‡vs mAKAPfl/fl;MCM.
mAKAPβ Requirement for Pressure Overload–Induced Hypertrophy
To test whether mAKAPβ was important for the induction of compensated cardiac hypertrophy in vivo, mAKAP CKO and control mice (both MCM+Tam mice and mAKAPfl/fl;MCM mice never exposed to Tam) were subjected to 2 weeks of pressure overload by TAC. By echocardiography (Figure III and Table I in the Data Supplement), TAC induced in control mice a concentric cardiac hypertrophy without significant ventricular dilatation or change in contractility (ejection fraction). Both control TAC groups had larger LV wall thicknesses and calculated LV masses compared with sham-operated mice. The difference in wall thickness from sham-operated was less for mAKAP CKO mice (difference in left ventricular posterior wall thickness in diastole 60% and 64% less than MCM+Tam and mAKAPfl/fl;MCM, respectively; Figure 1B), whereas ejection fraction was preserved (Figure 1C). The echocardiographic data correlated with gravimetric heart measurements (Table II in the Data Supplement). Biventricular weight was 57% and 36% greater than sham for the MCM+Tam and mAKAPfl/fl;MCM TAC cohorts, respectively, but only 23% for mAKAP CKO mice (Figure 1D).
Concentric cardiac hypertrophy is usually because of increased cardiac myocyte width.14 LV myocyte cross section area was 48% and 49% larger than sham for MCM+Tam and mAKAPfl/fl;MCM, respectively, but only 17% larger for mAKAP CKO TAC mice as shown by wheat germ agglutinin histochemistry (Figure 1E and 1F). Morphometry of isolated adult myocytes revealed that control MCM+Tam TAC myocytes were 21% greater in width than sham, without an increase in length (Figure 1G–1I). In contrast, mAKAP CKO TAC myocytes were not wider than sham. Together, these data show that mAKAPβ is important for the induction of concentric cardiac hypertrophy by pressure overload.
mAKAPβ Requirement for Cardiac Hypertrophy in Other Stress Models
Iso infusion mimics the excessive neurohumoral stimulation associated with CHF.15 By echocardiography, Iso induced a prominent enlargement in LV wall thickness in control mice, but not in mAKAP CKO mice, without cardiac decompensation (Figure 2A; Figure IV and Table III in the Data Supplement; 40%, 53%, and 14% greater left ventricular posterior wall thickness in diastole for MCM+Tam, mAKAPfl/fl;MCM, and mAKAP CKO Iso cohorts, respectively, compared with saline-infused mice). Accordingly, indexed biventricular weight was 25%, 33%, and 13% greater for MCM+Tam, mAKAPfl/fl;MCM, and mAKAP CKO Iso cohorts than saline-infused mice, respectively (Table IV in the Data Supplement and Figure 2B). Wheat germ agglutinin histochemistry corroborated these results (Figure 2C and 2D). LV myocyte cross section area was 33%, 29%, and 9% greater for the MCM+Tam, mAKAPfl/fl;MCM, and mAKAP CKO Iso cohorts than saline-infused mice, respectively. Likewise, the width of isolated adult myocytes was significantly greater only for the control MCM+Tam cohort (10%) and not the mAKAP CKO cohort (Figure 2E). Unlike pressure overload, Iso often increases myocyte length.14 Myocyte length was similar for control and knockout mice (both saline-infused and Iso-infused mice; Figure 2F), albeit the increase in length for the control cohort did not reach statistical significance. C57BL/6 mouse strains can be resistant to Iso toxicity,14 and there was no increase in cell death (TUNEL staining) for any of the cohorts (Figure IV in the Data Supplement). Iso did significantly induce interstitial fibrosis for the MCM+Tam control group but not the mAKAPfl/fl;MCM and mAKAP CKO cohorts (Figure IV in the Data Supplement).
Figure 2.

Muscle A-kinase anchoring protein β (mAKAPβ) is important for catecholamine and exercise-induced hypertrophy. A–F, mAKAP CKO (mAKAPfl/fl;MCM+tamoxifen [Tam]) and control MCM+Tam and mAKAPfl/fl;MCM mice were infused for 2 weeks with saline±isoproterenol (Iso). A, M-mode echocardiography for left ventricular posterior wall thickness in diastole (LVPW;d); cf Table III in the Data Supplement. B, Biventricular weight indexed to body weight; cf Table IV in the Data Supplement. C and D, Myocyte cross section area was determined by wheat germ agglutinin staining. Bar=50 μm (n=3–6). E and F, Isolated adult myocytes were measured for width and length for the mAKAP CKO and MCM+Tam cohorts. n=3 to 7 mice; >100 myocytes assayed per mouse. G, mAKAP CKO and MCM+Tam mice were forced to swim or rested for 5 weeks. Biventricular weight indexed to body weight is shown; cf Table VI in the Data Supplement. *Vs saline-infused or resting mice of same genotype; †vs MCM+Tam; ‡vs mAKAPfl/fl;MCM.
Pressure overload and catecholamine stimulation are models for heart disease. The heart also undergoes physiological hypertrophy (eg, during pregnancy and strenuous exercise).16 To test mAKAPβ’s relevance to physiological hypertrophy, mAKAP CKO and MCM+Tam control mice were subjected to a 5-week swimming regimen. Athletic conditioning of both cohorts was evident by a lower heart rate (Table V in the Data Supplement). Swimming induced a mild hypertrophy in control mice significant only by gravimetric analysis that was greater than for mAKAP CKO mice (13% versus 5%; Figure 2G; Figure V and Tables V and VI in the Data Supplement). Taken together with the aforementioned results, mAKAPβ is important for both physiological and pathological cardiac hypertrophy.
Role of mAKAPβ in the Development of Heart Failure
Long-term cardiac stress that causes LV hypertrophy can induce CHF. To test whether mAKAPβ targeting might prevent CHF, additional mAKAP CKO and MCM+Tam control mice were subjected to prolonged pressure overload (LT-TAC). All mice tolerated LT-TAC well for a period of time. Mice that began to exhibit decreased spontaneous movement decompensated rapidly (abnormal, labored, or tachypnic respirations and weight loss) and died within a few days of initial clinical distress if not euthanized beforehand. During the 16-week study, 29% of control LT-TAC mice died (Figure 3A; P=0.003 versus sham control mice by Mantel–Cox test). In contrast, only 6% of mAKAP CKO LT-TAC mice died, demonstrating a clear survival benefit to the loss of mAKAPβ scaffolding (P=0.02 versus control LT-TAC mice).
Figure 3.

Muscle A-kinase anchoring protein β (mAKAPβ) knockout protects against heart failure induced by long-term pressure overload (LT-transverse aortic constriction [TAC]). Control mice were MCM+tamoxifen (Tam). A, Kaplan–Meier survival curves. n: control sham, 26; control LT-TAC, 24; mAKAP CKO sham, 39; and mAKAP CKO LT-TAC, 31. P values determined by Mantel–Cox log-rank test; P=0.0003 for comparison across all cohorts. B–I, Data for mice alive 16 weeks after TAC. B, Representative mouse hearts. Bar, 3 mm. C–E, Biventricular, wet lung, and left atrial weights indexed to body weights; cf Table VII in the Data Supplement. F, mAKAPβ knockout inhibited left ventricular cell death as measured by TUNEL assay (n=4–7). G–I, mAKAPβ knockout attenuated interstitial fibrosis. G, Trichrome-stained sections. H, Gray scale images of picrosirius red–stained sections imaged by polarized light microscopy. Collagen fibers appear bright by this technique. Montages were assembled using Photoshop. Bar (G and H), 1 mm. I, Collagen content was quantified using picrosirius red–stained sections (n=4–6). *Vs sham-operated mice for same genotype; †vs control mice.
Consistent with the significant LT-TAC mortality, control mice alive after 16 weeks of pressure overload had pulmonary edema (30% greater indexed wet lung weight), a cardinal sign of left-sided CHF (Figure 3D; Table VII in the Data Supplement). In contrast, indexed wet lung weight was not greater for mAKAP CKO LT-TAC mice than for sham-operated. Ejection fraction was only 14% lower for the surviving control LT-TAC mice than control LT-sham (P<0.05; Table VIII in the Data Supplement), analogous to human heart failure with preserved ejection fraction because of aortic stenosis.1,17 That diastolic dysfunction contributed to the CHF in this control cohort was suggested by the 2-fold greater left atrial weight (Figure 3B and 3E; Table VII in the Data Supplement). Consistent with their improved status, left atrial weight was only 35% greater for mAKAP CKO LT-TAC mice compared with sham-operated mice.
Pressure overload–induced CHF is characterized by myocyte hypertrophy associated with myocyte apoptosis and interstitial fibrosis.17 By serial echocardiography (Figure VI and Table VIII in the Data Supplement), LV concentric hypertrophy was progressively induced by LT-TAC, with mAKAP CKO mice consistently exhibiting less hypertrophy than controls. Postmortem examination of mice surviving the duration of the study corroborated this result (Figure 3B and 3C). Compared with sham-operated mice, biventricular weight was 54% greater for LT-TAC control but only 29% for mAKAP CKO mice. Histologically, myocyte cross section area was 34% greater for control but only 16% for mAKAP CKO mice (Figure VII in the Data Supplement). Importantly, the myocardial apoptosis and collagen deposition induced by LT-TAC were 75% and 78% less, respectively, for mAKAP CKO mice (Figure 3F–3I; Figure VII in the Data Supplement). In sum, the improved survival and CHF of mAKAP CKO LT-TAC mice was consistent with the attenuated remodeling of their myocardium, including less myocyte hypertrophy and apoptosis and interstitial fibrosis.
mAKAPβ and Canonical Signaling Pathways
Hypertrophic signaling was studied in mAKAP CKO and MCM+Tam control mice subjected to pressure overload, both during the early phase of compensated hypertrophy and the later phase of CHF. Ventricular expression of extracellular signal–regulated kinase types 1/2 (ERK1/2) and 5 (ERK5) was higher both 2 and 16 weeks after TAC (Figure 4; Figures VIII and IX in the Data Supplement), with the upregulation significantly inhibited by mAKAPβ knockout only for 2-week TAC. ERK5 activation loop phosphorylation (indicative of activated enzyme) was significantly greater for LT-TAC control mice, whereas increased ERK1/2 phosphorylation was not detected for these time points. The ERK-effector p90 ribosomal S6 kinase (RSK) was more highly phosphorylated, but not changed in expression, in control mice for both short- and long-term TAC, with the attenuation in RSK phosphorylation by mAKAPβ knockout significant for 2-week TAC. Glycogen synthase kinase-3β phosphorylation and expression were similar for all cohorts.
Figure 4.

Muscle A-kinase anchoring protein β (mAKAPβ) knockout affects extracellular signal–regulated kinase (ERK) signaling. Control MCM+tamoxifen (Tam) and mAKAP CKO left ventricular (LV) extracts were analyzed by immunoblot after (A) 2-week (S, sham-operated; T, transverse aortic constriction [TAC]) and (B) 16-week TAC (LS, sham-operated and LT-TAC). Ponceau S total protein staining confirmed the equal loading of whole ventricular extracts. For quantification, see Figures VIII and IX in the Data Spplement. Note that the 2- and 16-week Westerns were performed separately, preventing comparison between time points.
A mAKAPβ Protein Kinase D1–Histone Deacetylase 4 Signaling Module
Protein kinase D (PKD) phosphorylation of type IIA histone deacetylases (HDAC-IIA, ie, HDACs 4/5/7/9) induces HDAC-IIA nuclear export, derepressing gene expression.18 We now show that like PKD and its upstream activators phospholipase Cε (PLCε) and protein kinase Cε,19 HDAC4 binds mAKAPβ in the heart (Figure 5A). When expressed in heterologous cells, myc-tagged mAKAPβ bound PKD1 and Flag-tagged HDAC4 (Figure 5B and 5C). In contrast, PKD1–HDAC4 binding was consistently detected only when both mAKAPβ was coexpressed, and the cells were stimulated with the phorbol ester phorbol-12-myristate-13-acetate (PMA) that activated the kinase (phosphorylation of Ser-744/748 on the kinase domain activation loop). Without PMA, PKD1–HDAC4 coimmuno-precipitation was also possible after alanine substitution of the HDAC4 PKD1 phosphorylation sites (Ser-246/467/632), presumably through stabilization of HDAC4 binding to the PKD1 catalytic cleft (Figure 5D). Together, these results show that PKD1 and HDAC4 can form a complex with mAKAPβ in cells.
Figure 5.

Muscle A-kinase anchoring protein β (mAKAPβ) is the scaffold for a histone deacetylase 4 (HDAC4)–protein kinase D (PKD) signaling complex. A, Protein complexes were immunoprecipitated from rat heart extract with VO54 α-mAKAP or preimmune (PI) serum. B–D, Extracts from COS-7 cells (a fibroblast-like, kidney cell line) expressing myc-mAKAPβ, Flag-HDAC4 (wildtype or Ser-246/467/632-Ala S3A mutant), and hemagglutinin-tagged–PKD1 were immunoprecipitated using the indicated antibodies. C, +PMA indicates cell stimulation with 1 μmol/L okadaic acid, 1 μg/mL cyclosporine A, and 200 nmol/L phorbol myristate acetate. n≥3 for each blot. E, As in Figure 4, MCM+tamoxifen (Tam) and mAKAP CKO left ventricular (LV) extracts were analyzed by immunoblot after 2- and 16-week transverse aortic constriction (TAC). F, HDAC4 and HDAC5 were assayed after protein immunoprecipitation with the corresponding antibody (Figure X in the Data Supplement). For quantification, see Figures XI and XII in the Data Supplement.
The relevance of HDAC4-PKD-mAKAPβ complexes was demonstrated using the aforementioned mice subjected to pressure overload. PKD Ser-744/748 phosphorylation required the scaffold’s expression after both 2 and 16 weeks of pressure overload (Figure 5E; Figure XI in the Data Supplement). PKD autophosphorylation (Ser-916) was also enhanced via an mAKAPβ-dependent mechanism after short-term TAC. Accordingly, HDAC4 phosphorylation was significantly greater only for the control cohort at Ser-467/632 after both 2 and 16 weeks and at Ser-246 after 16 weeks of pressure overload (Figure 5F; Figure XII in the Data Supplement). Although mAKAP CKO significantly attenuated HDAC4 Ser-467/632 phosphorylation after LT-TAC, mAKAPβ CKO did not inhibit the contemporaneous change in HDAC5 Ser-259 phosphorylation. Together, these results reveal that mAKAPβ facilitates PKD-catalyzed HDAC4 phosphorylation during pressure overload, defining a new mAKAPβ signaling module important for the regulation of a chromatin modifier.
Loss of mAKAPβ Prevents Genetic Remodeling
Hypertrophic gene expression depends on both HDAC-IIA nuclear export and the activation of relevant transcription factors. MEF2D binds its activator the Ca2+/calmodulin-dependent phosphatase calcineurin in a mAKAPβ-dependent manner in myocytes.5 After LT-TAC, MEF2D migrated faster in SDS-PAGE, consistent with MEF2D activation by dephosphorylation (Figure 6B).20,21 Although total MEF2D levels were 23% less (P<0.005) after LT-TAC, the ratio of faster to slower migrating MEF2D forms was 1.6-fold greater (P<0.005; Figure XIV in the Data Supplement). These changes were mAKAPβ dependent (P<0.05). NFATc transcription factors are also activated by mAKAPβ-bound calcineurin in cultured myocytes.6 Accordingly, the ratio of faster to slower migrating NFATc2 forms was significantly greater after both short- and long-term TAC only for control mice (Figure 6A and 6B; Figures XIII and XIV in the Data Supplement). Likewise, mAKAPβ knockout inhibited the in vivo activity of a NFAT-dependent luciferase reporter induced by 2-week TAC (Figure XV in the Data Supplement). Although mAKAPβ-associated ubiquitin E3-ligases regulated HIF-1α levels in vitro,7 we did not detect any changes in ventricular HIF-1α levels at these time points (Figure 6A and 6B).
Figure 6.

Muscle A-kinase anchoring protein β (mAKAPβ) knockout inhibits transverse aortic constriction (TAC)–induced gene expression. A and B, As in Figure 4, MCM+tamoxifen (Tam) and mAKAP CKO left ventricular (LV) extracts were analyzed by immunoblot after 2- and 16-week TAC. For quantification, see Figures XIII and XIV in the Data Supplement. Control MCM+Tam and mAKAP CKO total heart RNA were assayed by Nanostring after 2-week TAC; cf Table IX in the Data Supement. C, Genes associated with pathological hypertrophy. D, Genes associated with interstitial fibrosis. *Vs sham-operated control; †vs control TAC mice. n=3 for each cohort.
MEF2 and NFATc, in conjunction with HDAC-IIA nuclear export, affect gene expression in cardiac myocytes and, through paracrine mediators, in the adjacent cardiac fibroblasts.1 To show the functional relevance of mAKAPβ-dependent transcription factor activation, we assayed the expression of genes associated with myocyte signaling, hypertrophy, and interstitial fibrosis using LV tissue from 2-week TAC mice (Table IX in the Data Supplement). Overall, TAC-induced changes in gene expression were attenuated by mAKAPβ knockout (P<0.001 for control versus mAKAP CKO TAC). Among the TAC-induced genes, the MEF2D targets Nppa (atrial natriuretic factor),22 Acta1 (skeletal muscle α-actin),23 and Actc1 (cardiac muscle α-actin),24 and the NFATc-target Rcan1 (regulator of calcineurin 1)25 were induced in a mAKAPβ-dependent manner (Figure 6C). Consistent with the diminished myocardial fibrosis in LT-TAC mice lacking mAKAPβ, the induction of genes that either regulate or participate in the extracellular matrix was reversed by mAKAPβ myocyte-specific knockout (Figure 6D).
Discussion
Studies in neonatal myocytes have established that mAKAPβ is a scaffold that organizes multiple signaling modules through the dynamic binding of diverse enzymes and effectors.4 mAKAPβ is best characterized for its function in cAMP signaling. By binding type 5 adenylyl cyclase, the cAMP targets Epac1 and protein kinase A (for whose anchoring mAKAPβ is named), and the cAMP-specific phosphodiesterase 4D3 (PDE4D3), mAKAPβ orchestrates a complete cAMP module that regulates local cAMP levels through a series of integrated feedback loops.10,26 We have proposed that the main function of mAKAPβ signalosomes is to regulate myocyte growth and remodeling.4 For example, PKA phosphorylation of mAKAPβ-bound ryanodine receptors may enhance the local release of Ca2+, activating mAKAPβ-bound calcineurin.9 In addition, mAKAPβ-bound Epac1 can activate a PLCε–PKD hypertrophic pathway.19 Besides cAMP-related signaling, mAKAPβ signalosomes includes modules for other pathways, including those featuring mitogen-activated protein kinases, RSK, and HIF-1α.7,10,14 In this study, we show that mAKAPβ organizes a PKD1–HDAC4 complex and is required in vivo for the activation of those signalosome effectors necessary for cardiac remodeling (Figure 7). We provide the first evidence that targeting of a scaffold can attenuate the development of CHF and improve survival in chronic heart disease.
Figure 7.
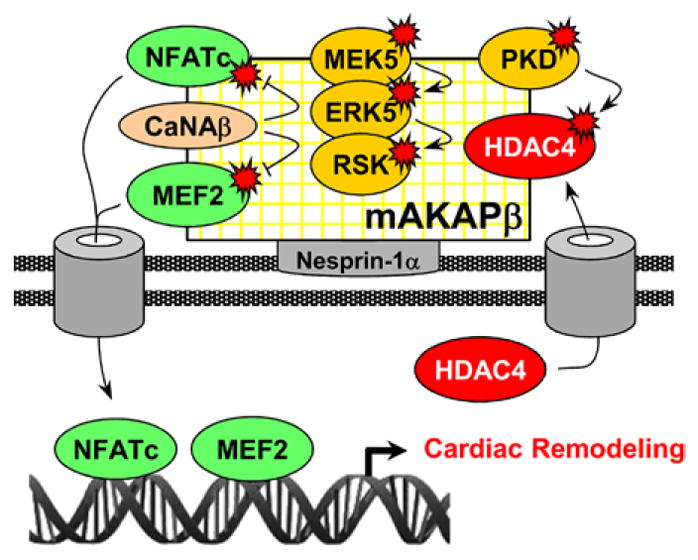
Model for muscle A-kinase anchoring protein β (mAKAPβ) signalosome-regulated remodeling. mAKAPβ anchored by nesprin-1α at the nuclear envelope coordinates the activity of transcription factors that transiently dock the scaffold. CaNAβ dephosphorylates and activates nuclear factor of activated T-cell (NFATc) and myocyte-enhancer factor (MEF) 2. Mitogen-activated protein kinase/ ERK kinase 5 (MEK5) and extracellular signal–regulated kinase (ERK) 5 activate ribosomal S6 kinase (RSK). Protein kinase D (PKD) phosphorylates and inactivates histone deacetylase (HDAC) 4, derepressing hypertrophic gene expression.
ERK5 is a mitogen-activated protein kinase indirectly bound to mAKAPβ through PDE4D3.10 Although PDE4D3 also binds ERK1, we have yet to detect ERK1/2–mAKAPβ complexes in the heart. Although implicated in eccentric myocyte growth, ERK5 is also required for pressure overload–induced concentric hypertrophy.27 In contrast, ERK1/2 preferentially induces concentric myocyte growth.28 Both ERK1/2 and ERK5 activate RSK,29 whereas ERK5 also activates MEF2 transcriptional activity.27 Recently, we showed that RSK3 binds mAKAPβ and is required for pathological hypertrophy.14 The relevant RSK3 substrates remain unknown. Although TAC induced ERK1/2 and ERK5 expression, increased activation was only significant for ERK5 after LT-TAC. However, there were important limitations to the Western blot data presented in Figures 4 to 6. Although the use of flash-frozen ventricular tissue presumably avoided any confounding effects associated with myocyte isolation protocols, heart tissue includes other cell types besides myocytes that express the proteins of interest. Second, our studies focused on time points representing compensated hypertrophy and heart failure. Like the transient, early induction of glycogen synthase kinase-3β phosphorylation and HIF-1α expression,30–32 ERK1/2 is most highly phosphorylated immediately after the initiation of pressure overload and not at the time points studied (Jinliang Li, unpublished observations, 2014). Nevertheless, RSK was activated by both short- and long-term TAC only in hearts expressing mAKAPβ.
Located at the nuclear envelope, mAKAPβ signalosomes are poised to serve as gatekeepers for cardiac myocyte–specific gene expression, orchestrating the post-translational modifications of gene regulatory proteins that transiently dock at the scaffold. The Smrcka group recently discovered that both Epac1 and upstream endothelin-1 signaling activate mAKAPβ-bound PLCε.19 Using cultured myocytes, they showed that PLCε hydrolyzes perinuclear phosphatidylinositol 4-phosphate, activating protein kinase Cε and its target PKD. PLCε and PKD1 knockout both inhibited pressure overload hypertrophy.19,33 We now demonstrate that the mAKAPβ scaffold is required for PKD activation in vivo. PKD also binds AKAP-lbc, another scaffold implicated in hypertrophy. However, AKAP-lbc–PKD anchoring was not required for Iso-induced hypertrophy.34 Phosphorylation and oxidation of key residues inhibit type IIA HDACs, promoting hypertrophic gene expression.18,35 mAKAPβ knockout inhibited HDAC4 phosphorylation, consistent with our new observation that mAKAPβ is the scaffold for a PKD1–HDAC4 signaling module. Together, these data reveal that mAKAPβ coordinates signaling regulating chromatin structure in hypertrophy.
We previously demonstrated that calcineurin Aβ (CaNAβ), MEF2, and NFATc bind mAKAPβ.5,6 CaNAβ–mAKAPβ binding was required for neonatal myocyte hypertrophy in vitro, as well as NFATc and MEF2 transcriptional activity.5,6 In vivo, CaNAβ, NFATc2, NFATc3, and MEF2D are required for pressure overload–associated remodeling.36–39 Accordingly, our data reveal that NFATc- and MEF2-dependent gene expression is induced by pressure overload via mAKAPβ-dependent mechanisms.
Besides pathological models, mAKAPβ was required for swimming-induced hypertrophy. Although calcineurin–NFAT signaling has an established role in pathological remodeling, there is evidence suggesting that this pathway may serve a role in physiological hypertrophy.16,40 In addition, a HDAC-IIA knockout (HDAC5/9 double knockout) resulted in both a spontaneous increase in indexed heart weight and expression of hypertrophic markers.41 Future work will be required to determine how mAKAPβ controls normal heart growth, including which mAKAPβ-binding partners are relevant to physiological hypertrophy.
A major challenge in the development of heart failure therapies is that many candidate drug targets regulating hypertrophy are pleiotropic, resulting in unacceptable side effects.2 In contrast, signalosomes containing cell-type specific combinations of ubiquitous enzymes may be dedicated to distinct cellular processes. In addition, because signalosomes integrate signals transduced by multiple pathways, signalosome targeting may allow the inhibition of pathological signaling in a concerted manner. mAKAPβ is the first example of a scaffold whose ablation confers a survival benefit, reducing mouse mortality 79% after long-term pressure overload. In fact, the potential benefits of mAKAPβ targeting may be greater because Tam-induced mAKAPβ knockout was not always complete (Figure IX in the Data Supplement). The improved cardiac function of patients with aortic stenosis after valve replacement surgery correlates with decreased LV mass and fibrosis.42 mAKAPβ knockout similarly attenuated the progression of ventricular hypertrophy and interstitial fibrosis during LT-TAC. Future work will address whether inhibition of mAKAPβ-orchestrated signaling can prevent or treat human heart failure (eg, the use of viral vectors for in vivo RNA interference).43 Potential hurdles include a possible mAKAPβ function in long-term cardiac eutrophy, given mAKAPβ’s role in physiological hypertrophy. Chronic mAKAPβ targeting may also have unanticipated effects because mAKAPβ binds the ryanodine receptor and has been detected by others in additional subcellular compartments.44,45 As an alternative to inhibiting mAKAPβ expression, transduction of peptides that uncouple select mAKAPβ signalosome modules may be effective. Key targets include mAKAPβ-bound CaNAβ and RSK3 for which mAKAPβ-derived anchoring disruptor peptides effectively inhibited the hypertrophy of cultured neonatal myocytes.6,14
Supplementary Material
CLINICAL PERSPECTIVE.
Heart failure is a syndrome of major public health significance accountable for ≈300 000 deaths each year in the United States. Pathological cardiac remodeling, including left ventricular hypertrophy, myocyte apoptosis, and interstitial fibrosis, contributes to the development of both systolic and diastolic dysfunction when the heart is chronically stressed, whether by pressure overload such as in aortic stenosis or hypertension or by volume overload such as after a myocardial infarction. Cardiac remodeling is regulated by an intricate intracellular signaling network. It is increasingly recognized that within myocytes, this signaling network includes multimolecular protein complexes organized by scaffold proteins. We have proposed that these scaffold proteins might be alternative therapeutic targets to conventional targets such as G-protein–coupled plasmalemmal receptors (eg, β-blockers). In this article, we show that signaling involving signaling complexes or signalosomes organized by the scaffold protein muscle A-kinase anchoring protein β contributes to pathological remodeling. In fact, conditional knockout of this scaffold protein conferred a survival benefit in mice stressed by long-term pressure overload. The data presented herein suggest that disruption of these intracellular protein complexes might be useful therapeutically, especially because muscle A-kinase anchoring protein β knockout in unstressed mice was benign. These results should inspire future translational research directed at disrupting muscle A-kinase anchoring protein β signalosomes in the prevention or treatment of heart failure.
Acknowledgments
Sources of Funding
This work was supported by National Institutes of Health grant HL075398 (Dr Kapiloff) and an American Heart Association award (Dr Li, Scientific Development Grant).
Footnotes
The Data Supplement is available at http://circheartfailure.ahajournals.org/lookup/suppl/doi:10.1161/CIRCHEARTFAILURE.114.001266/-/DC1.
Disclosures
Drs Kapiloff, Kritzer, Li, Passariello, and Dodge-Kafka are coinventors of intellectual property concerning the use of muscle A-kinase anchoring protein β–derived peptides for the treatment of heart failure, for which a patent is pending and which may yield future royalties. This patent is currently assigned to anchored ribosomal S6 kinase 3 inhibitors, LLC, in which Dr Kapiloff holds equity. He and the University of Miami may gain royalties from future commercialization.
The other authors report no conflicts.
References
- 1.Burchfield JS, Xie M, Hill JA. Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation. 2013;128:388–400. doi: 10.1161/CIRCULATIONAHA.113.001878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 3.Good MC, Zalatan JG, Lim WA. Scaffold proteins: hubs for controlling the flow of cellular information. Science. 2011;332:680–686. doi: 10.1126/science.1198701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kritzer MD, Li J, Dodge-Kafka K, Kapiloff MS. AKAPs: the architectural underpinnings of local cAMP signaling. J Mol Cell Cardiol. 2012;52:351–358. doi: 10.1016/j.yjmcc.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li J, Vargas MA, Kapiloff MS, Dodge-Kafka KL. Regulation of MEF2 transcriptional activity by calcineurin/mAKAP complexes. Exp Cell Res. 2013;319:447–454. doi: 10.1016/j.yexcr.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li J, Negro A, Lopez J, Bauman AL, Henson E, Dodge-Kafka K, Kapiloff MS. The mAKAPbeta scaffold regulates cardiac myocyte hypertrophy via recruitment of activated calcineurin. J Mol Cell Cardiol. 2010;48:387–394. doi: 10.1016/j.yjmcc.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong W, Goehring AS, Kapiloff MS, Langeberg LK, Scott JD. mAKAP compartmentalizes oxygen-dependent control of HIF-1alpha. Sci Signal. 2008;1:ra18. doi: 10.1126/scisignal.2000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vargas MA, Tirnauer JS, Glidden N, Kapiloff MS, Dodge-Kafka KL. Myocyte enhancer factor 2 (MEF2) tethering to muscle selective A-kinase anchoring protein (mAKAP) is necessary for myogenic differentiation. Cell Signal. 2012;24:1496–1503. doi: 10.1016/j.cellsig.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pare GC, Bauman AL, McHenry M, Michel JJ, Dodge-Kafka KL, Kapiloff MS. The mAKAP complex participates in the induction of cardiac myocyte hypertrophy by adrenergic receptor signaling. J Cell Sci. 2005;118(pt 23):5637–5646. doi: 10.1242/jcs.02675. [DOI] [PubMed] [Google Scholar]
- 10.Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature. 2005;437:574–578. doi: 10.1038/nature03966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L, Malik S, Kelley GG, Kapiloff MS, Smrcka AV. Phospholipase C epsilon scaffolds to muscle-specific A kinase anchoring protein (mAKAPbeta) and integrates multiple hypertrophic stimuli in cardiac myocytes. J Biol Chem. 2011;286:23012–23021. doi: 10.1074/jbc.M111.231993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res. 2001;89:20–25. doi: 10.1161/hh1301.092687. [DOI] [PubMed] [Google Scholar]
- 13.Moses KA, DeMayo F, Braun RM, Reecy JL, Schwartz RJ. Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genesis. 2001;31:176–180. doi: 10.1002/gene.10022. [DOI] [PubMed] [Google Scholar]
- 14.Li J, Kritzer MD, Michel JJ, Le A, Thakur H, Gayanilo M, Passariello CL, Negro A, Danial JB, Oskouei B, Sanders M, Hare JM, Hanauer A, Dodge-Kafka K, Kapiloff MS. Anchored p90 ribosomal S6 kinase 3 is required for cardiac myocyte hypertrophy. Circ Res. 2013;112:128–139. doi: 10.1161/CIRCRESAHA.112.276162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas JA, Marks BH. Plasma norepinephrine in congestive heart failure. Am J Cardiol. 1978;41:233–243. doi: 10.1016/0002-9149(78)90162-5. [DOI] [PubMed] [Google Scholar]
- 16.Chung E, Yeung F, Leinwand LA. Calcineurin activity is required for cardiac remodelling in pregnancy. Cardiovasc Res. 2013;100:402–410. doi: 10.1093/cvr/cvt208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ozkan A, Kapadia S, Tuzcu M, Marwick TH. Assessment of left ventricular function in aortic stenosis. Nat Rev Cardiol. 2011;8:494–501. doi: 10.1038/nrcardio.2011.80. [DOI] [PubMed] [Google Scholar]
- 18.Monovich L, Vega RB, Meredith E, Miranda K, Rao C, Capparelli M, Lemon DD, Phan D, Koch KA, Chapo JA, Hood DB, McKinsey TA. A novel kinase inhibitor establishes a predominant role for protein kinase D as a cardiac class IIa histone deacetylase kinase. FEBS Lett. 2010;584:631–637. doi: 10.1016/j.febslet.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 19.Zhang L, Malik S, Pang J, Wang H, Park KM, Yule DI, Blaxall BC, Smrcka AV. Phospholipase Cε hydrolyzes perinuclear phosphatidylinositol 4-phosphate to regulate cardiac hypertrophy. Cell. 2013;153:216–227. doi: 10.1016/j.cell.2013.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shalizi A, Gaudillière B, Yuan Z, Stegmüller J, Shirogane T, Ge Q, Tan Y, Schulman B, Harper JW, Bonni A. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science. 2006;311:1012–1017. doi: 10.1126/science.1122513. [DOI] [PubMed] [Google Scholar]
- 21.Grégoire S, Tremblay AM, Xiao L, Yang Q, Ma K, Nie J, Mao Z, Wu Z, Giguère V, Yang XJ. Control of MEF2 transcriptional activity by coordinated phosphorylation and sumoylation. J Biol Chem. 2006;281:4423–4433. doi: 10.1074/jbc.M509471200. [DOI] [PubMed] [Google Scholar]
- 22.Morin S, Charron F, Robitaille L, Nemer M. GATA-dependent recruitment of MEF2 proteins to target promoters. EMBO J. 2000;19:2046–2055. doi: 10.1093/emboj/19.9.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slepak TI, Webster KA, Zang J, Prentice H, O’Dowd A, Hicks MN, Bishopric NH. Control of cardiac-specific transcription by p300 through myocyte enhancer factor-2D. J Biol Chem. 2001;276:7575–7585. doi: 10.1074/jbc.M004625200. [DOI] [PubMed] [Google Scholar]
- 24.Molinari S, Relaix F, Lemonnier M, Kirschbaum B, Schäfer B, Buckingham M. A novel complex regulates cardiac actin gene expression through interaction of Emb, a class VI POU domain protein, MEF2D, and the histone transacetylase p300. Mol Cell Biol. 2004;24:2944–2957. doi: 10.1128/MCB.24.7.2944-2957.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Rooij E, Doevendans PA, de Theije CC, Babiker FA, Molkentin JD, de Windt LJ. Requirement of nuclear factor of activated T-cells in calcineurin-mediated cardiomyocyte hypertrophy. J Biol Chem. 2002;277:48617–48626. doi: 10.1074/jbc.M206532200. [DOI] [PubMed] [Google Scholar]
- 26.Kapiloff MS, Piggott LA, Sadana R, Li J, Heredia LA, Henson E, Efendiev R, Dessauer CW. An adenylyl cyclase-mAKAPbeta signaling complex regulates cAMP levels in cardiac myocytes. J Biol Chem. 2009;284:23540–23546. doi: 10.1074/jbc.M109.030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kimura TE, Jin J, Zi M, Prehar S, Liu W, Oceandy D, Abe J, Neyses L, Weston AH, Cartwright EJ, Wang X. Targeted deletion of the extracellular signal-regulated protein kinase 5 attenuates hypertrophic response and promotes pressure overload-induced apoptosis in the heart. Circ Res. 2010;106:961–970. doi: 10.1161/CIRCRESAHA.109.209320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kehat I, Davis J, Tiburcy M, Accornero F, Saba-El-Leil MK, Maillet M, York AJ, Lorenz JN, Zimmermann WH, Meloche S, Molkentin JD. Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ Res. 2011;108:176–183. doi: 10.1161/CIRCRESAHA.110.231514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol. 2008;9:747–758. doi: 10.1038/nrm2509. [DOI] [PubMed] [Google Scholar]
- 30.Lorenz K, Schmitt JP, Schmitteckert EM, Lohse MJ. A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat Med. 2009;15:75–83. doi: 10.1038/nm.1893. [DOI] [PubMed] [Google Scholar]
- 31.Sugden PH, Fuller SJ, Weiss SC, Clerk A. Glycogen synthase kinase 3 (GSK3) in the heart: a point of integration in hypertrophic signalling and a therapeutic target? A critical analysis. Br J Pharmacol. 2008;153(suppl 1):S137–S153. doi: 10.1038/sj.bjp.0707659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, Shimizu I, Asahara T, Hamada H, Tomita S, Molkentin JD, Zou Y, Komuro I. p53-induced inhibition of HIF-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–448. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- 33.Fielitz J, Kim MS, Shelton JM, Qi X, Hill JA, Richardson JA, Bassel-Duby R, Olson EN. Requirement of protein kinase D1 for pathological cardiac remodeling. Proc Natl Acad Sci U S A. 2008;105:3059–3063. doi: 10.1073/pnas.0712265105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spindler MJ, Burmeister BT, Huang Y, Hsiao EC, Salomonis N, Scott MJ, Srivastava D, Carnegie GK, Conklin BR. AKAP13 Rho-GEF and PKD-binding domain deficient mice develop normally but have an abnormal response to β-adrenergic-induced cardiac hypertrophy. PLoS One. 2013;8:e62705. doi: 10.1371/journal.pone.0062705. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Matsushima S, Kuroda J, Ago T, Zhai P, Park JY, Xie LH, Tian B, Sadoshima J. Increased oxidative stress in the nucleus caused by Nox4 mediates oxidation of HDAC4 and cardiac hypertrophy. Circ Res. 2013;112:651–663. doi: 10.1161/CIRCRESAHA.112.279760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim Y, Phan D, van Rooij E, Wang DZ, McAnally J, Qi X, Richardson JA, Hill JA, Bassel-Duby R, Olson EN. The MEF2D transcription factor mediates stress-dependent cardiac remodeling in mice. J Clin Invest. 2008;118:124–132. doi: 10.1172/JCI33255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilkins BJ, De Windt LJ, Bueno OF, Braz JC, Glascock BJ, Kimball TF, Molkentin JD. Targeted disruption of NFATc3, but not NFATc4, reveals an intrinsic defect in calcineurin-mediated cardiac hypertrophic growth. Mol Cell Biol. 2002;22:7603–7613. doi: 10.1128/MCB.22.21.7603-7613.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bourajjaj M, Armand AS, da Costa Martins PA, Weijts B, van der Nagel R, Heeneman S, Wehrens XH, De Windt LJ. NFATc2 is a necessary mediator of calcineurin-dependent cardiac hypertrophy and heart failure. J Biol Chem. 2008;283:22295–22303. doi: 10.1074/jbc.M801296200. [DOI] [PubMed] [Google Scholar]
- 39.Bueno OF, Wilkins BJ, Tymitz KM, Glascock BJ, Kimball TF, Lorenz JN, Molkentin JD. Impaired cardiac hypertrophic response in Calcineurin Abeta -deficient mice. Proc Natl Acad Sci U S A. 2002;99:4586–4591. doi: 10.1073/pnas.072647999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eto Y, Yonekura K, Sonoda M, Arai N, Sata M, Sugiura S, Takenaka K, Gualberto A, Hixon ML, Wagner MW, Aoyagi T. Calcineurin is activated in rat hearts with physiological left ventricular hypertrophy induced by voluntary exercise training. Circulation. 2000;101:2134–2137. doi: 10.1161/01.cir.101.18.2134. [DOI] [PubMed] [Google Scholar]
- 41.Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol. 2004;24:8467–8476. doi: 10.1128/MCB.24.19.8467-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Di Bello V, Giorgi D, Viacava P, Enrica T, Nardi C, Palagi C, Grazia Delle Donne M, Verunelli F, Mariani MA, Grandjean J, Dell’Anna R, Di Cori A, Zucchelli G, Romano MF, Mariani M. Severe aortic stenosis and myocardial function: diagnostic and prognostic usefulness of ultrasonic integrated backscatter analysis. Circulation. 2004;110:849–855. doi: 10.1161/01.CIR.0000138930.12773.41. [DOI] [PubMed] [Google Scholar]
- 43.Prasad KM, Xu Y, Yang Z, Acton ST, French BA. Robust cardiomyocyte-specific gene expression following systemic injection of AAV: in vivo gene delivery follows a Poisson distribution. Gene Ther. 2011;18:43–52. doi: 10.1038/gt.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 45.Yang J, Drazba JA, Ferguson DG, Bond M. A-kinase anchoring protein 100 (AKAP100) is localized in multiple subcellular compartments in the adult rat heart. J Cell Biol. 1998;142:511–522. doi: 10.1083/jcb.142.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.