

Abstract
Bone Morphogenetic Protein (BMP) receptors mediate a diverse range of signals to regulate both development and disease. BMP activity has been linked to both tumor promoting and suppressive functions in both tumor cells and their surrounding microenvironment. We sought to investigate the requirement for BMPR2 in stromal fibroblasts during mammary tumor formation and metastasis. We utilized FSP1 (Fibroblast Specific Protein‐1) promoter driven Cre to genetically delete BMPR2 in mice expressing the MMTV.PyVmT mammary carcinoma oncogene. We found that abrogation of stromal BMPR2 expression via FSP1 driven Cre resulted in increased tumor metastasis. Additionally, similar to epithelial BMPR2 abrogation, stromal loss of BMPR2 results in increased inflammatory cell infiltration. We proceeded to isolate and establish fibroblast cell lines without BMPR2 and found a cell autonomous increase in inflammatory cytokine secretion. Fibroblasts were co‐implanted with syngeneic tumor cells and resulted in accelerated tumor growth and increased metastasis when fibroblasts lacked BMPR2. We observed that the loss of BMPR2 results in increased chemokine expression, which facilitates inflammation by a sustained increase in myeloid cells. The chemokines increased in BMPR2 deleted cells correlated with poor outcome in human breast cancer patients. We conclude that BMPR2 has tumor suppressive functions in the stroma by regulating inflammation.
Keywords: Bone morphogenetic proteins, Fibroblasts, Metastasis, Chemokines, Inflammation, Breast cancer, Tumor microenvironment, Carcinoma‐associated‐fibroblast
Highlights
Stromal deletion of BMPR2 increases metastasis in a breast cancer mouse model.
Fibroblasts with deletion of BMPR2 have elevated cytokines and chemokines.
BMPR2 KO fibroblasts promote breast cancer metastasis and myeloid cell infiltration.
Chemokines from BMPR2 deleted fibroblasts correlate with invasive breast cancer.
1. Introduction
Bone Morphogenetic Proteins (BMPs) belong to the TGFβ family of cytokines and growth factors and are known to elicit diverse and complex functions in development and disease (Miyazono et al., 2010). BMP ligands are secreted, require processing, and are facilitated by co‐receptors to bind their cognate serine/threonine kinase receptors. Through this process, they canonically mediate phosphorylation of Smads 1, 5 and 9(human)‐8(mouse). Activation of BMP signaling induces the transcription of known target genes Id1, Smad6 and Smad7 (Miyazono et al., 2010, 2005). Induction of Smad6 and Smad7 transcription results in a strong negative feedback that self‐limits the pathway from over activation. BMPs can have both tumor suppressive and tumor promoting roles (Alarmo and Kallioniemi, 2010; Ehata et al., 2013). It is clear that they can suppress growth of the tumor epithelium, yet they can also enhance cell migration and invasion (Ketolainen et al., 2010). Less is known about the effect of BMP pathway in the tumor stroma, but the impact of BMP on the tumor microenvironment appears to promote tumor progression and metastasis. However, one of the many secreted inhibitors of BMP, DAND5 (COCO), has been found to promote metastasis (Gao et al., 2012). The paracrine nature of the BMP/TGFβ signaling pathways require careful dissection of context and function in many cell types that are capable of signal transduction via this diverse family (Pickup et al., 2013a).
The tumor microenvironment is a critical mediator of cancer development and outcome (Finak et al., 2008). A myriad of factors surrounding the tumor have the profound ability to alter the course of tumor progression (Mueller and Fusenig, 2004). In breast cancer, changes in fibroblasts can affect the formation, progression and metastatic dissemination of cancers (Morales et al., 2011). Through the creation and remodeling of the extracellular matrix, secretion of numerous growth factors and other cytokines, as well as directing epithelial cell migration and invasion, fibroblasts promote the growth and metastasis of breast cancer in addition to other neoplasms (Pickup et al., 2013, 2013). An important signaling pathway mediating the functional activities of fibroblasts includes the TGFβ signaling pathway. Experiments that seek to either increase or remove TGFβ and other paracrine signaling systems can worsen the outcome of a given cancer (Barlow et al., 2003; Bhowmick et al., 2004b). Given the conflicting data surrounding BMP's role in tumor progression and intriguing potential interactions between the TGFβ and BMP signaling, there is significant potential for stromal BMP signaling to be a mediator in determining tumor progression.
Genetic disruption of BMP receptors in epithelial cells has the effect of inducing neoplasms and accelerating tumor growth (Owens et al., 2012). Loss of BMP signaling most commonly results in benign neoplasia (hamartomas) in the colon (Friedl et al., 2002; Howe et al., 2001). When mice were targeted for deletion of BMPR2 in the colon, neoplastic growths were observed (similar to the polyps that develop in humans) (Beppu et al., 2008). This phenomenon has been seen in the case of TGFβ signaling loss in the stroma, whereby epithelial transformation is initiated and progressed by loss of TGFβ in the adjacent stromal cells (Bhowmick et al., 2004a; Cheng et al., 2008; Pickup et al., 2013b). We have shown that stimulation of fibroblasts by secreted BMP ligands can promote tumor cell invasion and increased inflammatory cytokine production (Owens et al., 2013). We have sought to understand how BMP signaling in stromal fibroblasts in the mammary tumor microenvironment dictate progression and metastasis. Unfortunately, it is unclear to what effect stromal loss of BMP signaling may have on metastasis, which is the primary underlying pathology driving morbidity in cancer patients. In this study, we found that the loss of BMPR2 in the stroma of mice expressing the oncogene PyVmT in the mammary epithelium, increases tumor metastasis and is accompanied by heightened cytokine secretion and myeloid inflammatory cell infiltration.
2. Materials and methods
2.1. Ethical issues, mice, surgeries, sample collection and staining of lung whole mounts
All animal experiments were performed at Vanderbilt University and approved by IACUC (Internal protocol #M/07/331). All animals were used within the standards as prescribed by “Guidelines for the welfare and use of animals in cancer research” (Workman et al., 2010). C57BL6 mice were purchased from Charles River Laboratories and were used to maintain transgenes used in this study. PCR genotyping was performed as previously described for mice harboring the MMTV.PyVmT and FSP1.Cre transgenes as well as the BMPR2 and mTom/mGFP cre reporter alleles (Beppu et al., 2005; Bhowmick et al., 2004a; Guy et al., 1992; Muzumdar et al., 2007; Owens et al., 2012). Mice were weaned at three weeks of age and female mice were genotyped for the PyVmT transgene and then palpated for tumors at least twice weekly. Implantation of PyVmT tumor cells were combined with isolated fibroblasts with and without BMPR2 expression. 1 × 105 carcinoma cells were resuspended in a collagen I plug with 2.5 × 105 fibroblasts of either genotype. These collagen plugs were implanted into the #4 mammary gland of a non‐tumorigenic syngeneic C57/B6 mouse (Harlan). Implanted tumor size was followed for progression and tumor weight until tumors reached 2 cm in size. Tumor tissue was collected by dissecting tumors and snap‐freezing tissue in LN2, OCT and formalin fixation for paraffin embedding. Lungs were inflated with 2–3 ml of heparin (50 μg/ml), fixed in 10% neutral buffered formalin and dehydrated, cleared in xylene, rehydrated, and stained with Mayer's hematoxylin, dehydrated and metastatic lung foci quantitated. Lungs were then embedded in paraffin and sectioned for histology, stained with H&E to confirm metastases.
2.2. Cell culture, establishment of primary fibroblasts and growth/viability assays
Mammary glands from 12‐week‐old virgin females were dissected, minced and placed into a digestion buffer. Digestion buffer consisted of DNaseI (125 μg/ml), Collagenase 3 (500 μg/ml), Neutral Protease (Dispase) (10 μg/ml) (Worthington Bio). Mammary glands were digested for 2 h at 37° while shaken at 300 rpm. Cells were strained with a 40 μM cell strainer (Fisher Scientific) and rinsed in fibroblast media [DMEM + 10%FBS + Triple antibiotic (Gibco)] and plated into a T‐75 culture flask. BMPR2 cKO cells containing the Cre reporter transgene and expressing membrane bound GFP were sorted for Cre recombined cells (Muzumdar et al., 2007). Control cells were passage matched to cKO regardless of cell growth rates, which were monitored by counting cells and viability using the Countess (Life Technologies) according to manufacturer's instructions.
2.3. Immunohistochemistry, immunofluorescence, ELISA and cytokine array
Paraffin tissues were embedded and sectioned at 5 μM and dewaxed in xylene and rehydrated in alcohol with citrate antigen retrieval as previously described (Owens et al., 2012). Standard Mayer's hematoxylin and eosin (H&E) was performed. Cleaved Caspase‐3 (Cell Signaling Cat#9661, 1:200), MECA‐32 (BD Cat#550563 1:200), F4/80 (Invitrogen #MF48000 1:50), Vimentin (Covance Cat#PCK‐594P 1:500), αSMA (Sigma Cat#A2547 1:500), FSP‐1 (EMD Millipore Cat#07‐2274), Phalloidin (Molecular Probes Cat#), Gr‐1 (BD Cat#557979 1:200), BrdU (BD Cat#563445 1:100), B220 (BD Cat#550539 1:200), CD4 (BioLegend Cat#100401 1:50), CD8a (BioLegend Cat#100801 1:100). Paraffin derived sections were counterstained with hematoxylin (Vector Labs QS) and mounted with Cytoseal. Immunofluorescence staining was performed with primary and secondary antibodies diluted in 12% Fraction‐V BSA (Pierce) and slides were mounted in SlowFade mounting medium containing DAPI (Invitrogen). All fluorescent secondary antibodies were highly cross‐adsorbed, produced in goat and used at a dilution of 1:200 for 20 min (Molecular Probes). Quantification of IHC and IF was performed using NIH ImageJ (http://rsbweb.nih.gov/ij/docs/examples/stained‐sections/index.html) as previously described (Owens et al., 2010). Conditioned medium was collected 48 h after equal cell numbers were plated into 6‐well culture plates. Supernatant was spun to remove cells and debris and 50 μl were used per well for ELISA (RnD Systems Cat#'s MCS00 and DY478‐05) and mouse cytokine array panel A was performed following manufactures instructions (RnD Systems Cat# ARY006).
2.4. Flow cytometry
Single‐cell suspensions were made from primary tumors as previously described (Novitskiy et al., 2011). Cells were stained with fluorescence‐conjugated antibodies (BioLegend, eBioscience, BD) and isotype matched IgG controls. The cells were analyzed on an LSRII flow cytometer (BD) EpCAM+CD45‐, CD45+EpCAM‐, CD45‐EpCAM‐, DAPI was used to exclude dead cells.
2.5. RNA isolation, cDNA synthesis and qPCR
RNA isolation of snap‐frozen tissue was performed by placing tissue directly into Trizol (Invitrogen) and purified by chloroform and alcohol precipitation. Trizol isolated RNA was then subjected to cleanup with RNeasy purification including DNAseI treatment. Equal amounts of RNA were synthesized into cDNA using the VILO cDNA synthesis kit (Invitrogen). LuminoCt (Sigma) 2X SYBR mastermix was combined with 1 μM of both a forward and reverse primer sequence (full table of sequences is listed in Supplemental Table 1) into 20 μl reactions and cycled for 95°‐10 s to 60° for 30 s for 40 cycles followed by a melting curve. BioRad CFX96 was used and instrument provided software was used to determine relative normalized expression to Gapdh expression. Inflammation genes were analyzed from cDNA of fibroblast cell lines with the Inflammatory Response & Autoimmunity PCR Array (sabiosciences/Qiagen Cat#PAMM‐3803Z) and performed according to manufacturer's instructions. Fold regulation of gene expression is listed in Supplemental Table 2.
2.6. Database utilization and statistical analysis
Analysis of the human breast cancer stroma microarray dataset presented in Finak et al. (Finak et al., 2008) was analyzed using Oncomine (Rhodes et al., 2004). The stroma from normal (n = 3) and invasive ductal carcinoma (n = 54) patients was collected using laser capture microdissection for RNA extraction and microarray analysis of gene expression changes specifically in the stroma. These data were queried for changes in gene expression in CSF3 and CCL5 in the stroma of these human patients. Statistical analysis was performed using Excel (Microsoft), Prism (Graphpad), and FlowJo (TreeStar) software. Statistical significance was deemed for any comparison where p < 0.05.
3. Results
3.1. Stromal deletion of BMPR2 increases lung metastasis
To address whether the stromal loss of BMPR2 could affect metastasis, we assembled genetically engineered mouse models together to target BMPR2 loss to the tumor microenvironment. All mouse strains were in the C57/BL6 background. First, we used mice harboring the transgene in which the MMTV promoter drives Polyoma middle T antigen expression (MMTV.PyVmT) and bred them with mice expressing FSP1 promoter driven Cre to target stromal cells. Next, these were combined with mice that had loxP sites in the BMPR2 gene. Males with a single allele for the oncogene (PyVmT), Cre and heterozygous floxed BMPR2 allele, were bred to female mice homozygous for floxed BMPR2 alleles (Figure 1A). Mice were palpated for tumor formation weekly and no difference was detected for tumor onset. In order to avoid sacrificing mice without any metastases we used 2 cm primary tumor size in conjunction with IACUC standards for our euthanasia/survival. Once mice presented with 2 cm tumors they were sacrificed. Knockout (cKO) and heterozygous mice took longer to form 2 cm tumors, but this difference was not statistically significant (Figure 1B). We analyzed 27 control mice, 12 heterozygous mice and 13 cKO mice. All mice formed typical adenocarcinomas as reported previously for this oncogene and background (Guy et al., 1992; Owens et al., 2012) as shown in Figure 1C–H. IHC for BrdU was performed to determine rates of proliferation in tumors, and no significant differences were observed (Figure 1I–J). IHC for cleaved caspase‐3, a marker of apoptosis, revealed that control tumors had typical amounts of cell death (Figure 1K), while cKO tumors displayed very little positive staining for cleaved caspase‐3 (Figure 1L). Statistical quantification of the area of positive cleaved caspase‐3 staining demonstrated a significant decrease in cell death in cKO tumors (Figure 1N), yet not in the number of BrdU+ cells (Figure 1M). Strikingly, cKO tumor had a much greater number of lung metastases (approximately 5 fold) than either control mice or heterozygous mice (Figure 1O).
Figure 1.
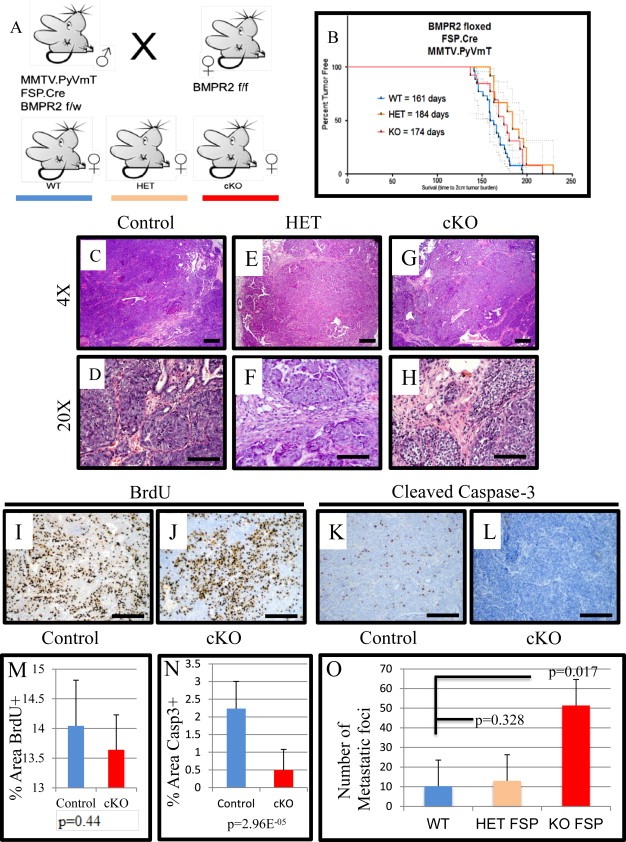
Stromal deletion of BMPR2 increases pulmonary metastasis of mammary carcinomas. A) Mouse breeding strategy used pure strain C57BL6 mice that contained the oncogene PyVmT driven by the mammary epithelium specific MMTV promoter (MMTV.PyVmT) combined with Cre recombinase under the control of the FSP1 promoter (FSP1.Cre) and mice with BMPR2 floxed alleles. Animals with no Cre act as controls (n = 27), mice with only one BMPR2 floxed allele are heterozygous‐HET (n = 12) and knockout‐cKO (n = 13) are homozygous floxed alleles for BMPR2 in FSP1 expressing cells. B) Kaplan–Meier analysis for the time to reach maximum tumor burden (2 cm). C–H) Representative images of Hematoxylin and Eosin (H&E) staining of spontaneous tumors for Control and cKO. I–J) Representative images of BrdU, injected prior to sacrifice, immunohistochemistry comparing control and cKO. K–L) Representative images of immunohistochemistry for cleaved caspase‐3 to detect apoptotic cells in control tumors compared with cKO tumors. M−N) Quantification of BrdU and cleaved caspase‐3 IHC comparing control and cKO tumors. O) At the time of sacrifice lungs were removed and metastatic foci were counted. Significance was determined by students t‐test. Scale bars C, E, G = 200 μM, D, F, H = 50 μM I–L = 100 μM. Error bars indicate SD.
3.2. Stromal deletion of BMPR2 results in inflammation
To examine the tumor microenvironment for inflammation, we performed IHC for the granulocyte (neutrophils, eosinophils and myeloid derided suppressor cells) marker Gr‐1, which in the tumor microenvironment marks myeloid derived suppressor cells (MDSCs) (Yang et al., 2008a). We observed a significant increase in Gr‐1 positive cells in cKO tumors compared with controls (Figure 2A–B&E). In addition, we stained for macrophages by F4/80 and found a significant increase in cKO tumors (Figure 2C–D&F). Interestingly, we did not observe significant changes in tumor B or T cells by IHC staining (Figure S1A–F). However, FSP1 mediated Cre recombination of the stroma has been implicated in lineages not exclusive to the cancer associated fibroblasts, and is not expressed in all fibroblasts (Boomershine et al., 2009; Osterreicher et al., 2011). Thus, the variations in immune cell infiltrates could be explained by a myriad of genetic mechanisms such as Cre recombination in the myeloid populations inducing differential cell infiltration or altered chemokine expression by another cell population. To test misexpression of FSP Cre we bred our mice to a transgenic reporter that constitutively expressed mTomato and upon Cre activity excises mTomato and activates GFP expression, thus allowing analysis of Cre activity by mGFP expression (Muzumdar et al., 2007). We found that in tumors there existed populations of cells positive for both GFP and Tomato that were single and double positive for antibody staining to CD45 (immune cells) as well as EpCAM (epithelial cells) (Figure S2A–B). Specifically looking at the cells that were GFP+, and thus recombined for BMPR2 via FSP1 driven Cre we found that 10% of mammary tumors contained GFP expressing cells, and approximately 80% of CD45+ cells, 5% of EpCAM+ cells and double negative representing ∼3%. This final double negative population is thought to include the fibroblasts (Figure S2C). Fibroblasts are derived from CD45+ cells and additionally tumors that undergo EMT may no longer express EpCAM (Gorges et al., 2012).
Figure 2.
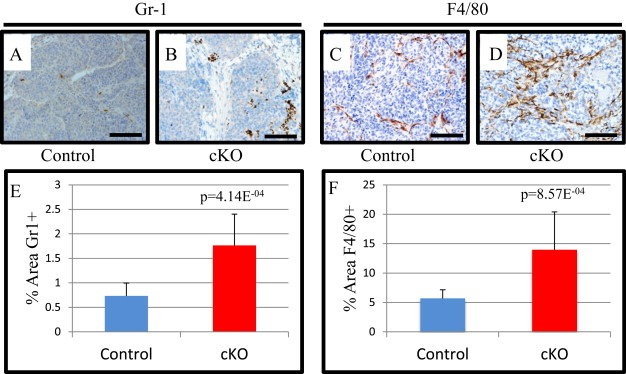
FSP1.cre mediated deletion of BMPR2 results in increased GR1+ and F4/80 positive cells. A–B) Representative images for control and cKO MMTV.PyVmT tumors stained for Ly6c/g(Gr‐1). C–D) Representative images of control and cKO tumors stained for the macrophage marker F4/80. E) Quantification of % area Gr‐1 immunohistochemistry in control and cKO tumors. F) Quantification of F4/80% area in control and cKO tumors. Scale bars = 100 μM. Significance was determined by students t‐test. Error bars indicate SD.
3.3. Mammary gland fibroblasts deleted for BMPR2 express more inflammatory genes
Given the propensity for FSP1 to heterogeneously direct Cre recombination in numerous cell populations, we made fibroblast cell lines from our mouse model to specifically evaluate fibroblast contribution to the observed tumorigenic phenotypes. Cell lines were validated by performing PCR similar to genotyping (Beppu et al., 2005; Bhowmick et al., 2004a; Muzumdar et al., 2007) but also included PCR to detect whether recombination had occurred (Figure S3A). Immunoflourescent staining was performed for Phalloidin (Actin stain), Vimentin (Stromal intermediate filament), αSMA (activated myofibroblast marker) and FSP1 and found that both control and cKO cells displayed equal staining for fibroblast markers as well as maintained a similar morphology (Figure 3A–H). qPCR analysis of fibroblast markers FSP1, Vimentin, αSMA, and FGFR2 showed no significant difference in expression levels (Figure 3I). We further examined the canonical signaling response to BMP signaling in our fibroblasts by treating them with rBMP2 and measuring expression of canonical BMP target genes Id1, Smad6, and Smad7. We found that only control cells had a significant induction of canonical target genes and that cKO cells had an insignificant response (Figure S3B). We further examined the growth and viability of our cells by counting and determining their viability. While cKO cells initially grew slower, they still had similar growth rates to control cells and no change in viability was observed (Figure S4A–B). While the cKO cells have an initial lag from the first day they have a doubling rate that is similar after the second day of being plated into culture. We next investigated changes in the cells BMP signaling components and targets of BMP signaling. We found that Bmp2 expression was elevated in knockouts (Figure S5A). We also detected modest increases in Bmp4, Tmeff2, Grem1 and Grem2 in BMPR2 cKO fibroblasts by qPCR (Figure S5B). With the increased ligand production in knockouts we saw that canonical BMP target genes Id1, Smad6 and Smad7 were also significantly increased in BMPR2 cKO fibroblasts (Figure S5C). These findings indicate that the BMP pathway has lost its negative feedback and results in increased BMP secretion and activity, both autocrine and paracrine. We have previously reported that BMP4 can stimulate mammary fibroblasts to express higher levels of Il6 and Mmp3 (Owens et al., 2013) and found that this was also the case in BMPR2 cKO which express higher levels of both Bmp2 and Bmp4 ligands (Figure S5D). Because we had observed significant changes in inflammatory cells in our tumors, we performed an inflammation focused qPCR array to determine any changes in gene expression. We found a large number of chemokines and inflammatory molecules overexpressed in cKO cells compared to control (Supplemental Table 1). Interestingly, we found that BMPR2 cKO fibroblasts had mostly elevated expression of cytokines. We next sought to examine the protein levels of secreted cytokines by performing a comparison of conditioned medium from our control and BMPR2 cKO fibroblasts on a panel of mouse cytokines (Figure 3J). Following normalization to reference spot controls we found several distinct secreted protein changes in BMPR2 cKO cells including much higher levels of G‐CSF (also known as CSF3), IP‐10 (also known as CXCL10) and RANTES (also known as CCL5). While the qPCR data show us relative difference, the protein changes are more indicative of the magnitude of paracrine factors secreted from BMPR2 cKO mammary fibroblasts (Figure 3K).
Figure 3.
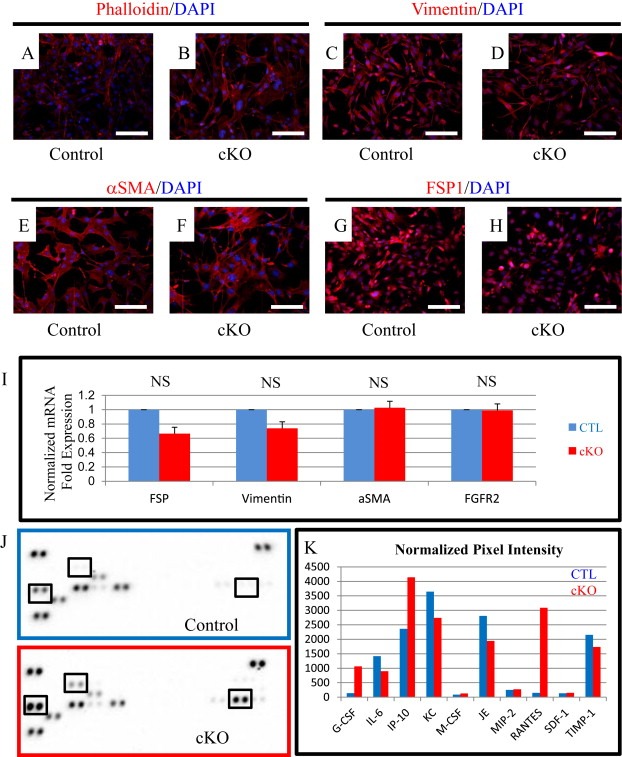
BMPR2 cKO fibroblasts have elevated inflammatory gene expression. A–F) Fibroblast cell lines were generated from control and cKO mammary glands and stained with Phalloidin (A–B), Vimentin (C–D), αSMA (E–F) and FSP1 (G–H) in red and counterstained with DAPI (blue). I) SYBR qPCR for fibroblast markers Fsp1, Vimentin, αSma, and Fgfr2 in control and cKO fibroblast cells. J–K) Mouse panel A cytokine array from RnD Systems with conditioned medium from control (blue) and BMPR2 cKO (red). K) Pixel intensity is quantified and normalized to average intensity of reference spots. mRNA is normalized to Gapdh levels and relative to control cells. Scale bars = 50 μM. Error bars indicate SEM. NS = Not Significant.
3.4. Implantation of BMPR2 cKO fibroblasts accelerates tumor growth and metastasis
We were concerned that our analysis in spontaneous tumors was not reflective of cancer associated fibroblasts due to the heterogeneous expression of FSP1.Cre in the resulting PyMT tumors (Boomershine et al., 2009; Osterreicher et al., 2011). Additionally we wanted to determine whether the changes in cytokines from the BMPR2 cKO fibroblasts could recapitulate the phenotype observed in spontaneous tumors. We utilized our validated cell lines to perform a co‐implantation that was both syngeneic and orthotopic to the #4 mammary gland (Figure 4A). We observed that implanted tumors with BMPR2 cKO fibroblasts grow at a faster rate and reach 2 cm in a shorter period of time compared to control fibroblast tumors (Figure 4B). We allowed control tumors to reach 2 cm in size for comparison with our previous results (Figure 1). Histologically the tumors appeared similar, both containing regions of typical adenocarcinoma and necrotic regions typical of 2 cm implanted tumors (Figure 4C–F). Interestingly, even though cKO implanted tumors had faster growth, we did not observe a quantitative significant change in BrdU+ or Cleaved Caspase‐3+ cells indicating no difference in proliferation and cell death respectively (Figure 4G–L). We observed a three‐fold increase in lung metastasis in the cKO tumors compared to 2 cm size matched tumors (Figure 4M). The metastasis increase is remarkable given that the control fibroblasts containing tumors had an additional 3 weeks to metastasize given the different growth rates leading to delayed time to 2 cm (Figure 4B).
Figure 4.
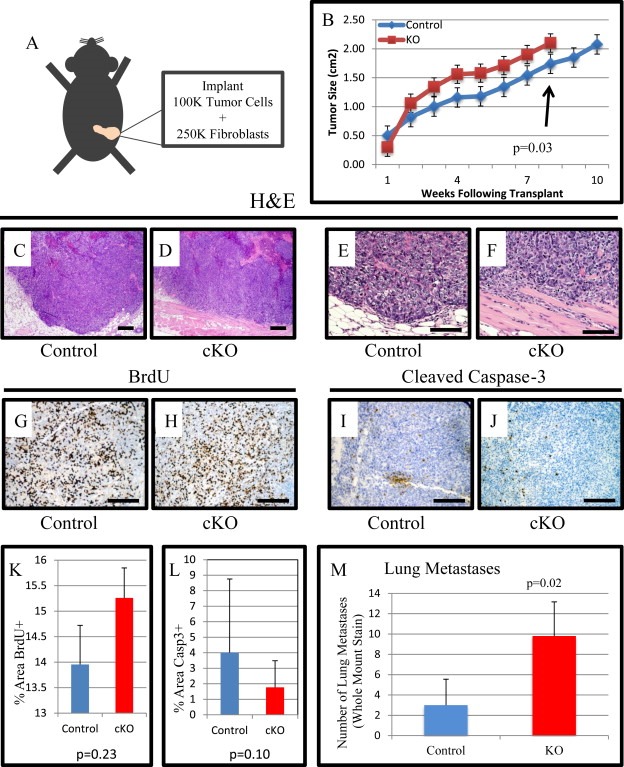
Tumor implantation with fibroblasts accelerates tumor growth and metastases. A) Schematic representing implantation approach. 2.5 × 105 fibroblasts were combined with 1 × 105 PyVmT tumor cells and orthotopically implanted into 12‐week‐old virgin female mice left #4 mammary fat pads. B) Mice were palpated weekly and when palpable tumors were discovered they were measured with digital calipers for size. Mice continued to be measured until they reached the IACUC limit of 2 cm in primary tumor size and euthanized. C–F) Representative images of H&E staining for control and cKO implant tumors. G–H) Representative images of BrdU IHC for control and cKO implant tumors. I–J) Representative images of cleaved caspase‐3 IHC for control and cKO implant tumors. K–L) Quantification of BrdU and cleaved caspase‐3 IHC comparing control and cKO tumors. M) Average number of lung metastases per experimental group. Scale bars C–D = 200 μM, E–F = 50 μM G–J = 100 μM. Significance was determined by a students t‐test. Error bars indicate SD.
3.5. Implantation of BMPR2 cKO fibroblasts results in inflammation
As we observed inflammation in our spontaneous model and elevated chemokines in our cell lines, we performed IHC for immune cells in our tumor implants. We found a significant increase in Gr‐1 positive cells in cKO tumors mimicking the spontaneous model (Figure 5A–B&E). Interestingly, we observed a trending, yet non‐significant increase in macrophage infiltration (p = 0.085). Similar to the spontaneous tumor, we did not observe any significant changes in the primary tumor's composition of B and CD4 or CD8 T cells, as evaluated by IHC analysis (Figure S6A–E). Orthotopic implants are known to be limited by angiogenesis (Hanahan and Weinberg, 2011) and it has been previously shown that BMP2 from prostate fibroblasts can promote angiogenesis (Yang et al., 2008b). BMP2 was elevated in our cKO cells (Figure S5A) and so we performed IHC for the pan‐endothelial marker MECA‐32 and found no significant changes in blood vessels in either control or cKO fibroblast tumor implants (Figure S7).
Figure 5.
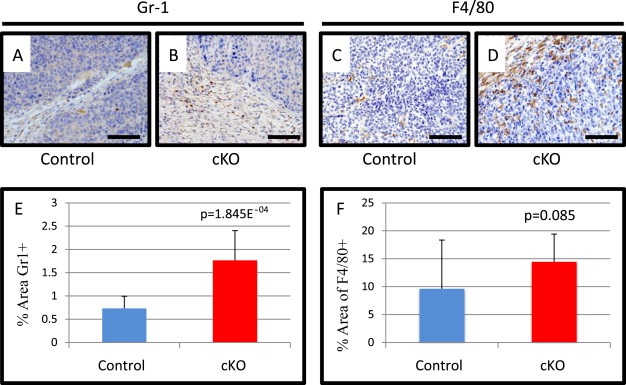
Tumors implanted with fibroblasts deleted for BMPR2 have increased inflammatory cells. A) Representative images of PyVmT tumors with either control or cKO fibroblasts stained by IHC for Ly6c/g(Gr‐1). C–D) Representative images of F4/80 IHC of implanted PyVmT tumors with either control or cKO fibroblasts. E) Quantification of Gr‐1+ % area of control and cKO tumors. F) Quantification of F4/80% area in control and cKO tumors. Scale bars = 50 μM. Significance was determined by students t‐test. Error bars indicate SD.
3.6. BMPR2 cKO fibroblasts have increased cytokine production and correlate with stromal changes seen for human patients with breast cancer
We explored two of the cytokines most elevated in BMPR2 cKO fibroblasts relative to control mammary gland fibroblasts: CSF3 (G‐CSF) and CCL5 (RANTES). We found that Ccl5 mRNA was largely unaffected by either recombinant BMP2 stimulation or blocking with a BMP2/4 neutralizing antibody and the 10 fold increase was maintained in BMPR2 cKO compared to control mammary gland fibroblasts (Figure 6A). We also investigated gene expression of Csf3 and found that in control cells adding recombinant BMP2 could reduce Csf3 levels as could blocking with a neutralizing antibody (Figure 6B). While in BMPR2 cKO cells treatment with recombinant BMP2 had no significant effect, the neutralizing antibody significantly reduced the expression of Csf3 (Figure 6B). We next performed ELISA protein analysis on CCL5 and found we could not detect CCL5 in normal fibroblast media or conditioned media from control cells regardless of stimulation or blocking antibody treatment. However, we could detect significant quantities of CCL5 secreted into conditioned medium in BMPR2 cKO fibroblasts. These levels were unchanged with the acute treatment with either BMP2 or neutralizing antibody (Figure 6C). We then performed ELISA analysis for CSF3 and found that we could detect small amounts in both the media alone as well as the conditioned media from control fibroblasts. Consistent with qPCR data we found a large increase in CSF3 secretion in BMPR2 cKO fibroblast conditioned medium, which similar to CCL5 was unchanged with acute treatment with recombinant BMP2 or neutralizing antibody (Figure 6D).
Figure 6.
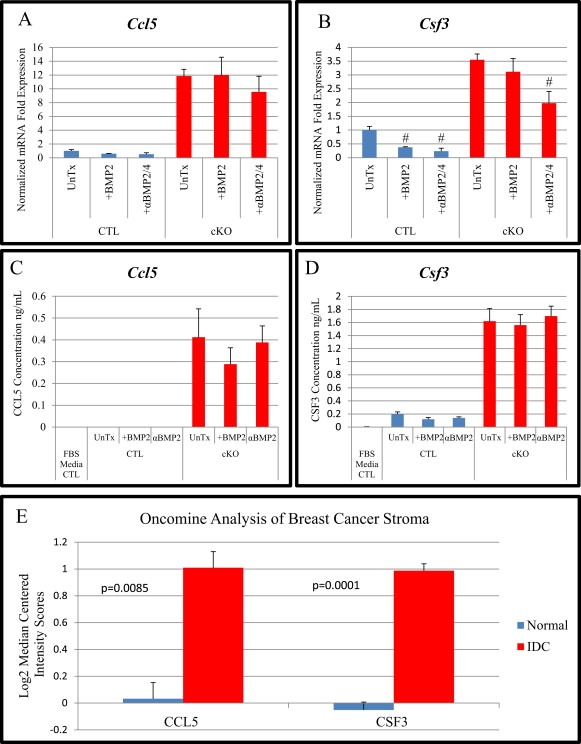
BMPR2 cKO fibroblasts have increased cytokine production and is correlated with stromal changes seen for human patients with breast cancer. A–B) SYBR qPCR for Ccl5 (RANTES) and Csf3 (G‐CSF) in control and BMPR2 cKO cells after no treatment, 100 ng/mL BMP2 and 100 ng/mL BMP2 combined with neutralizing antibody to BMP2/4 at 1 μg/mL for 24 h and mRNA gene expression was determined. C–D) ELISA from conditioned medium of samples analyzed in A&B was performed in triplicate. Normal media was analyzed for presence of cytokines as control. E) Data from GSE9054 analyzing laser captured stroma from Normal and IDC (Invasive Ductal Carcinoma) showing gene expression levels of human CCL5 and CSF3. Analysis performed using Oncomine. # indicates statistical significance from untreated sample. mRNA is normalized to Gapdh levels and relative to control cells. Error bars indicate SEM.
Having identified several gene targets of BMP action in the stroma, we sought to identify whether these genes held any influence over breast cancer presentation in patients. Using the online microarray analysis software Oncomine (Rhodes et al., 2004), we were able to identify significant increases in expression of CCL5 and CSF3 among patients with invasive ductal carcinoma (IDC) compared with normal patients (Figure 6E).
4. Discussion
The tumor microenvironment has been widely appreciated as a key determinant for tumor initiation, progression, metastasis, and therapeutic intervention (Mueller and Fusenig, 2004; Pickup et al., 2013a). This is emphasized by work in which stromal cell variability (in particular fibroblast activation and macrophage polarization) has been shown to influence the progression of the disease. Additionally, informatics data supports a causal role for the stroma in the progression of numerous cancers (Finak et al., 2008). Important breakthroughs in understanding the role of the stroma on tumor progression have been made through the use of genetically engineered mouse models that target the stromal cell populations (Bhowmick et al., 2004a). It is now possible to specifically target a number of immune and vascular components of the tumor stroma. Using a model targeting Cre expression to an FSP1 expressing cell population, we depleted BMPR2 from FSP1 expressing cells in mice challenged with PyVmT tumors. Targeting this signaling pathway in FSP1 expressing cells did not appreciably alter initiation or growth of the PyVmT tumors. Notably, there was a significant difference in the cleavage of caspase‐3, which marks apoptotic cells. This was not appreciated in the gross tumor measurements due to representation by such a small population of cells. Additionally, the lack of differences in tumor size with apoptosis differences could be made up by the increase in immune cell infiltrates. A significant phenotypic difference was observed in the metastatic potential of the cells (Figure 1L). Tumors in which BMPR2 was recombined in FSP1 expressing cells had a significant increase in lung metastasis compared to control animals. Potentially contributing to this phenotype, a significant difference in immune cell infiltration into the tumors was induced upon knockout of BMPR2 in FSP1 expressing cells.
Unfortunately, many of the mouse models including FSP1 directed Cre, do not target all fibroblasts, and can induce recombination in other cell types (Boomershine et al., 2009; Osterreicher et al., 2011). For this reason, we proceeded to generate cell lines that we could ensure BMPR2 knockout only in the fibroblast population (Figure 3 & Figure S3). However, these cKO cells are still generated via the FSP1 driven Cre, which may reflect an unknown functional subset of fibroblasts in normal tissues and cancer initiation and promotion (Kong et al., 2013; Sugimoto et al., 2006).
Previously it has been shown that nestin promoter driven deletion of BMPR2 in the intestinal stroma lacks the ability to form metastatic cancers (Beppu et al., 2008). While these polyps did not form overt metastases, the role of BMP/TGFβ/Smad4 loss is typically associated in cancer as an event later in tumor progression (Vogelstein et al., 2013). We found that BMPR2 loss in the fibroblast co‐implantation model recapitulated and drove pro‐tumorigenic myeloid cell infiltrates that enhanced metastasis in both spontaneous and implant models. Mechanisms whereby myeloid cells, including MDSCs enhance tumor progression include suppression of immune surveillance and alterations of extracellular matrix by factors secreted by the myeloid cells (Pickup et al., 2013b; Yang et al., 2008a). We previously found that expression of a dominant negative form of BMPR2 accelerated metastasis, when expressed in the tumor epithelia via the myeloid chemokine Ccl9 (Owens et al., 2012). We now find that BMP signaling through BMPR2 is required to suppress inflammation regardless of whether BMPR2 is deleted in FSP1 directed cell types in our spontaneous model or in a fibroblast specific culture model. Our data suggests that BMP acts to suppress the expression of CCL5 and CSF3 in fibroblasts found within the tumor stroma. Relieving this suppressive function, enhanced expression of these chemokines, which likely plays a significant role in the recruitment of myeloid cells to the tumor microenvironment. While expression of both CCL5 and CSF3 are enhanced in tumor stroma, CSF3 in particular has been shown to play an essential role in promoting granulocyte infiltration. CCL5 derived from the stroma has been demonstrated to drive breast cancer metastasis (Karnoub et al., 2007). Given that TGFβ and BMP are family members, it is interesting to speculate that both could be acting on fibroblasts to ultimately suppress infiltration of granulocytic MDSCs into the tumor to promote tumor progression (Figure 2, (Achyut et al., 2013)) This may indicate a global function of the BMP pathway to regulate inflammation and homeostasis in normal tissues that can be disrupted during cancer progression.
BMPR2 mutations and dysfunction are the genetic basis for Pulmonary Arterial Hypertension (PAH), which is characterized by elevation of inflammatory cells and cytokine/chemokine secretion (Austin et al., 2011; Song et al., 2008; Tuder et al., 2013). Interestingly, loss of BMPR2 signaling can lead to alternate BMP signaling through Activin receptors and still produce active BMP signaling (Yu et al., 2005). Deletion of BMPR2, hypothesized to be required for bone formation, did not result in skeletal defects when targeted in bone (Gamer et al., 2011). Adding more complexity to BMP signaling, it is apparent that elevated BMP stimulation of fibroblasts can increase inflammation and mammary tumor invasion (Owens et al., 2013). BMP stimulation can also promote angiogenesis in either fibroblasts or immune cells (Kwon et al., 2014; Yang et al., 2008b). Not only can BMP promote tumors via angiogenesis, but also it has been shown that stimulation of macrophage cells results in the polarization to ‘M2‐like’ alternate‐tumor promoting macrophages (Lee et al., 2013). Recently, expression of the dominant negative BMPR2 in macrophages also resulted in elevated inflammation derived from a model of Pulmonary Arterial Hypertension (PAH) (Talati et al., 2014).
The paradox where loss of function phenotypically resembles the gain of function experiment is not unique to BMP signaling and has been a common theme in the TGFβ family (Bierie and Moses, 2006). It should not come as a surprise then that BMP signaling, like TGFβ, has both tumor promoting and suppressive roles. Previously we have shown that stimulation of BMP signaling promotes tumor cell invasion via fibroblasts, while we are currently presenting data which supports BMP suppressing tumor progression through modulation of inflammatory infiltrates (Owens et al., 2013). The implications of the results reported in this study suggest that BMP signaling is centrally required for homeostasis of tissues as they interact with their stromal and immune cell counterparts. As has also been proposed for TGFβ signaling, this paradoxical effect is derived from different mechanisms leading to similar phenotypes. In this case, BMP stimulation could promote a cytokine expression profile with directly signaling through the epithelium to promote tumor cell migration and invasion while suppressing an inflammatory chemokine profile. However, loss of a BMP receptor abrogates the direct pro‐tumorigenic signaling between fibroblasts and epithelial cells and instead promotes tumor progression through the modulation of the inflammatory tumor microenvironment. Particularly, given that the expression of the BMP suppressed chemokines are wildly altered in IDC, abrogation of these pro‐tumorigenic functions could significantly promote patient care and survival. Such a hypothesis would support new and innovative approaches to targeting the BMP pathway outside of a direct signaling inhibition. Given the suppressive function BMP signaling has been shown to have in fibroblasts, our work provides compelling evidence that perhaps induction of the BMP pathway could be targeted to these fibroblasts to alleviate the pro‐tumorigenic inflammation established as a hallmark of cancer progression (Hanahan and Weinberg, 2011). Further work in these pathways will continue to reveal specific signaling nodes within the pathway that both integrate and specify distinct disease processes and potential therapies.
Conflict of interest
The authors have no conflicts of interest to declare.
Supporting information
The following are the supplementary data related to this article:
Supplemental Figure 1. IHC for B&T cells does not demonstrate significant changes. A–F) Spontaneous tumor paraffin sections were IHC stained for markers of B (CD45R/B220) and T cells (CD4 and CD8a) in control tumors and BMPR2 FSP1 mediated cKO tumors. Scale bars = 100 μM.
Supplemental Figure 2. Lineage tracing of FSP1 expression from Cre recombination GFP reporter. A–B) Representative flow plot from digested tumors of BMPR2 cKO mice containing mTom/mGFP cre reporter. FITC indicates GFP expressing cells which specify Cre mediated recombination of BMPR2 and PE axis indicate mTomato expressing cells not expressing Cre. B) Staining for both CD45 (immune cells) and EpCam (epithelial cells) indicate Cre expression from box inset of panel A. C) The total percentage of GFP+ cells in the BMPR2 cKO (three tumors) and the relative percentage of immune (CD45), epithelial (EpCam) and double negative. Error bars indicate SD.
Supplemental Figure 3. Confirmation of BMPR2 deletion in cell lines. A) PCR genotyping for the Cre reporter transgene mTomato/mGFP was only present in cKO cells. B) SYBR based qPCR for BMP target genes Id1, Smad6, and Smad7 in control (white bars) and cKO (black bars) fibroblasts analyzed after 24 h of BMP2 stimulation at 100 ng/ml. Fold expression was normalized to Gapdh and untreated cells. # indicates statistical significance p = <0.01 by students t‐test. NS indicates Not Significant. Error bars indicate SEM.
Supplemental Figure 4. BMPR2 cKO fibroblast cells had decreased growth rate and unaffected viability. A) Average cell counts for control and cKO fibroblasts every 24 h. B) Viability of cells counted at each time point determined by trypan blue dye exclusion. Cell counts and viability were performed using the Life Technologies Countess automated cell counter.
Supplemental Figure 5. BMP pathway changes in BMPR2 cKO fibroblasts. A) SYBR qPCR for Bmp2 ligand revealed over 700 fold change increase relative to untreated control. B) Additional components of the BMP pathway significantly altered in BMPR2 cKO fibroblast by SYBR qPCR. C) SYBR qPCR for BMP activation gene targets reveals that BMPR2 cKO cells have elevated canonical BMP transcriptional response of Id1, Smad6 and Smad7. Previously known BMP signaling targets in mouse mammary fibroblasts Il‐6 and Mmp3 were also found to be significantly upregulated from control fibroblasts. Fold expression was normalized to Gapdh and untreated cells were relative to fold change from untreated cells. # indicates statistical significance p = <0.01 by students t‐test. NS indicates Not Significant. Error bars indicate SEM.
Supplemental Figure 6. IHC for B&T cells does not demonstrate significant changes. A–B) Representative images of PyVmT tumors implanted with control and cKO fibroblasts IHC stained for B cells. C–D) Representative images of PyVmT tumors implanted with control cKO fibroblasts IHC stained for CD4+ T cells. E‐F) Representative images of PyVmT tumors with control and cKO fibroblasts IHC stained for CD8+ T cells. Scale bars = 100 μM. Error bars indicate SD.
Supplemental Figure 7. Angiogenesis was not significantly altered in tumors. A–D) IHC for the pan‐endothelial marker MECA‐32 was performed on both spontaneous tumors and implanted tumors. E–F) Quantification of MECA‐32 IHC in PyVmT tumors with either control or cKO fibroblasts as a percent area of each image analyzed. Scale bars = 100 μM. Error bars indicate SD.
Supplementary Table 1. Control and cKO cell line cDNA was analyzed by SABio PCR array for inflammation (Catalog #3803). Red indicates the fold change increase and blue represents fold change decrease in cKO cells relative to control cells.
Supplementary Table 2. SYBR qPCR primer sequences. Mouse primer sequences were selected from Primer Bank (http://pga.mgh.harvard.edu/primerbank/) that contained validation data. All primer sequences are directed against mouse sequences. All qPCR was performed with melting curves that verified specificity to single target amplicon.
Acknowledgments
We would like to thank the members of the Moses laboratory for critically reading the manuscript. We would like to thank Dr. James West for discussion of BMPR2 in pulmonary hypertension. Flow Cytometry experiments were performed in the VMC Flow Cytometry Shared Resource. The VMC Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404). The Vanderbilt Antibody and Protein Resource. We would like acknowledge assistance from Vanderbilt Translational Pathology Shared Resource (TPSR), which is funded by the NIH/NCI Vanderbilt Cancer Center Support Grant (2P30 CA068485‐14). P.O. has been supported by DoD BCRP postdoctoral fellowship grant number W81XWH‐09‐1‐0421. This work is supported by NIH grants CA085492, CA102162, the Robert J. and Helen C. Kleberg Foundation and the T.J. Martell Foundation to HLM. Grant number CA068485 provided core laboratory support.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.08.004.
Pickup Michael W., Hover Laura D., Polikowsky Eleanor R., Chytil Anna, Gorska Agnieszka E., Novitskiy Sergey V., Moses Harold L., Owens Philip, (2015), BMPR2 loss in fibroblasts promotes mammary carcinoma metastasis via increased inflammation, Molecular Oncology, 9, doi: 10.1016/j.molonc.2014.08.004.
References
- Achyut, B.R. , Bader, D.A. , Robles, A.I. , Wangsa, D. , Harris, C.C. , Ried, T. , Yang, L. , 2013. Inflammation-mediated genetic and epigenetic alterations drive cancer development in the neighboring epithelium upon stromal abrogation of TGF-beta signaling. PLoS Genet. 9, e1003251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarmo, E.L. , Kallioniemi, A. , 2010. Bone morphogenetic proteins in breast cancer: dual role in tumourigenesis?. Endocr. Relat. Cancer. 17, R123–R139. [DOI] [PubMed] [Google Scholar]
- Austin, E.D. , Menon, S. , Hemnes, A.R. , Robinson, L.R. , Talati, M. , Fox, K.L. , Cogan, J.D. , Hamid, R. , Hedges, L.K. , Robbins, I. , Lane, K. , Newman, J.H. , Loyd, J.E. , West, J. , 2011. Idiopathic and heritable PAH perturb common molecular pathways, correlated with increased MSX1 expression. Pulm. Circ. 1, 389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow, J. , Yandell, D. , Weaver, D. , Casey, T. , Plaut, K. , 2003. Higher stromal expression of transforming growth factor-beta type II receptors is associated with poorer prognosis breast tumors. Breast Cancer Res. Treat. 79, 149–159. [DOI] [PubMed] [Google Scholar]
- Beppu, H. , Lei, H. , Bloch, K.D. , Li, E. , 2005. Generation of a floxed allele of the mouse BMP type II receptor gene. Genesis. 41, 133–137. [DOI] [PubMed] [Google Scholar]
- Beppu, H. , Mwizerwa, O.N. , Beppu, Y. , Dattwyler, M.P. , Lauwers, G.Y. , Bloch, K.D. , Goldstein, A.M. , 2008. Stromal inactivation of BMPRII leads to colorectal epithelial overgrowth and polyp formation. Oncogene. 27, 1063–1070. [DOI] [PubMed] [Google Scholar]
- Bhowmick, N.A. , Chytil, A. , Plieth, D. , Gorska, A.E. , Dumont, N. , Shappell, S. , Washington, M.K. , Neilson, E.G. , Moses, H.L. , 2004. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 303, 848–851. [DOI] [PubMed] [Google Scholar]
- Bhowmick, N.A. , Neilson, E.G. , Moses, H.L. , 2004. Stromal fibroblasts in cancer initiation and progression. Nature. 432, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierie, B. , Moses, H.L. , 2006. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer. 6, 506–520. [DOI] [PubMed] [Google Scholar]
- Boomershine, C.S. , Chamberlain, A. , Kendall, P. , Afshar-Sharif, A.R. , Huang, H. , Washington, M.K. , Lawson, W.E. , Thomas, J.W. , Blackwell, T.S. , Bhowmick, N.A. , 2009. Autoimmune pancreatitis results from loss of TGFbeta signalling in S100A4-positive dendritic cells. Gut. 58, 1267–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, N. , Chytil, A. , Shyr, Y. , Joly, A. , Moses, H.L. , 2008. Transforming growth factor-beta signaling-deficient fibroblasts enhance hepatocyte growth factor signaling in mammary carcinoma cells to promote scattering and invasion. Mol. Cancer Res. 6, 1521–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehata, S. , Yokoyama, Y. , Takahashi, K. , Miyazono, K. , 2013. Bi-directional roles of bone morphogenetic proteins in cancer: another molecular Jekyll and Hyde?. Pathol. Int. 63, 287–296. [DOI] [PubMed] [Google Scholar]
- Finak, G. , Bertos, N. , Pepin, F. , Sadekova, S. , Souleimanova, M. , Zhao, H. , Chen, H. , Omeroglu, G. , Meterissian, S. , Omeroglu, A. , Hallett, M. , Park, M. , 2008. Stromal gene expression predicts clinical outcome in breast cancer. Nat. Med. 14, 518–527. [DOI] [PubMed] [Google Scholar]
- Friedl, W. , Uhlhaas, S. , Schulmann, K. , Stolte, M. , Loff, S. , Back, W. , Mangold, E. , Stern, M. , Knaebel, H.P. , Sutter, C. , Weber, R.G. , Pistorius, S. , Burger, B. , Propping, P. , 2002. Juvenile polyposis: massive gastric polyposis is more common in MADH4 mutation carriers than in BMPR1A mutation carriers. Hum. Genet. 111, 108–111. [DOI] [PubMed] [Google Scholar]
- Gamer, L.W. , Tsuji, K. , Cox, K. , Capelo, L.P. , Lowery, J. , Beppu, H. , Rosen, V. , 2011. BMPR-II is dispensable for formation of the limb skeleton. Genesis. 49, 719–724. [DOI] [PubMed] [Google Scholar]
- Gao, H. , Chakraborty, G. , Lee-Lim, A.P. , Mo, Q. , Decker, M. , Vonica, A. , Shen, R. , Brogi, E. , Brivanlou, A.H. , Giancotti, F.G. , 2012. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell. 150, 764–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorges, T.M. , Tinhofer, I. , Drosch, M. , Rose, L. , Zollner, T.M. , Krahn, T. , von Ahsen, O. , 2012. Circulating tumour cells escape from EpCAM-based detection due to epithelial-to-mesenchymal transition. BMC Cancer. 12, 178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy, C.T. , Cardiff, R.D. , Muller, W.J. , 1992. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol. Cell. Biol. 12, 954–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan, D. , Weinberg, R.A. , 2011. Hallmarks of cancer: the next generation. Cell. 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Howe, J.R. , Bair, J.L. , Sayed, M.G. , Anderson, M.E. , Mitros, F.A. , Petersen, G.M. , Velculescu, V.E. , Traverso, G. , Vogelstein, B. , 2001. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat. Genet. 28, 184–187. [DOI] [PubMed] [Google Scholar]
- Karnoub, A.E. , Dash, A.B. , Vo, A.P. , Sullivan, A. , Brooks, M.W. , Bell, G.W. , Richardson, A.L. , Polyak, K. , Tubo, R. , Weinberg, R.A. , 2007. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 449, 557–563. [DOI] [PubMed] [Google Scholar]
- Ketolainen, J.M. , Alarmo, E.L. , Tuominen, V.J. , Kallioniemi, A. , 2010. Parallel inhibition of cell growth and induction of cell migration and invasion in breast cancer cells by bone morphogenetic protein 4. Breast Cancer Res. Treat. 124, 377–386. [DOI] [PubMed] [Google Scholar]
- Kong, P. , Christia, P. , Saxena, A. , Su, Y. , Frangogiannis, N.G. , 2013. Lack of specificity of fibroblast-specific protein 1 in cardiac remodeling and fibrosis. Am. J. Physiol. Heart Circulatory Physiol. 305, H1363–H1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon, S.J. , Lee, G.T. , Lee, J.H. , Iwakura, Y. , Kim, W.J. , Kim, I.Y. , 2014. Mechanism of pro-tumorigenic effect of BMP-6: neovascularization involving tumor-associated macrophages and IL-1a. The Prostate. 74, 121–133. [DOI] [PubMed] [Google Scholar]
- Lee, J.H. , Lee, G.T. , Woo, S.H. , Ha, Y.S. , Kwon, S.J. , Kim, W.J. , Kim, I.Y. , 2013. BMP-6 in renal cell carcinoma promotes tumor proliferation through IL-10-dependent M2 polarization of tumor-associated macrophages. Cancer Res. 73, 3604–3614. [DOI] [PubMed] [Google Scholar]
- Miyazono, K. , Kamiya, Y. , Morikawa, M. , 2010. Bone morphogenetic protein receptors and signal transduction. J. Biochem. 147, 35–51. [DOI] [PubMed] [Google Scholar]
- Miyazono, K. , Maeda, S. , Imamura, T. , 2005. BMP receptor signaling: transcriptional targets, regulation of signals, and signaling cross-talk. Cytokine Growth Factor Rev. 16, 251–263. [DOI] [PubMed] [Google Scholar]
- Morales, M. , Planet, E. , Arnal-Estape, A. , Pavlovic, M. , Tarragona, M. , Gomis, R.R. , 2011. Tumor-stroma interactions a trademark for metastasis. Breast. 20, (Suppl. 3) S50–S55. [DOI] [PubMed] [Google Scholar]
- Mueller, M.M. , Fusenig, N.E. , 2004. Friends or foes – bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer. 4, 839–849. [DOI] [PubMed] [Google Scholar]
- Muzumdar, M.D. , Tasic, B. , Miyamichi, K. , Li, L. , Luo, L. , 2007. A global double-fluorescent Cre reporter mouse. Genesis. 45, 593–605. [DOI] [PubMed] [Google Scholar]
- Novitskiy, S.V. , Pickup, M.W. , Gorska, A.E. , Owens, P. , Chytil, A. , Aakre, M. , Wu, H. , Shyr, Y. , Moses, H.L. , 2011. TGF-beta receptor II loss promotes mammary carcinoma progression by Th17 dependent mechanisms. Cancer Discov. 1, 430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterreicher, C.H. , Penz-Osterreicher, M. , Grivennikov, S.I. , Guma, M. , Koltsova, E.K. , Datz, C. , Sasik, R. , Hardiman, G. , Karin, M. , Brenner, D.A. , 2011. Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc. Natl. Acad. Sci. U S A. 108, 308–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens, P. , Engelking, E. , Han, G. , Haeger, S.M. , Wang, X.J. , 2010. Epidermal Smad4 deletion results in aberrant wound healing. Am. J. Pathol. 176, 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens, P. , Pickup, M.W. , Novitskiy, S.V. , Chytil, A. , Gorska, A.E. , Aakre, M.E. , West, J. , Moses, H.L. , 2012. Disruption of bone morphogenetic protein receptor 2 (BMPR2) in mammary tumors promotes metastases through cell autonomous and paracrine mediators. Proc. Natl. Acad. Sci. U S A. 109, 2814–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens, P. , Polikowsky, H. , Pickup, M.W. , Gorska, A.E. , Jovanovic, B. , Shaw, A.K. , Novitskiy, S.V. , Hong, C.C. , Moses, H.L. , 2013. Bone morphogenetic proteins stimulate mammary fibroblasts to promote mammary carcinoma cell invasion. PloS One. 8, e67533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickup, M. , Novitskiy, S. , Moses, H.L. , 2013. The roles of TGFbeta in the tumour microenvironment. Nat. Rev. Cancer. 13, 788–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickup, M.W. , Laklai, H. , Acerbi, I. , Owens, P. , Gorska, A.E. , Chytil, A. , Aakre, M. , Weaver, V.M. , Moses, H.L. , 2013. Stromally derived lysyl oxidase promotes metastasis of transforming growth factor-beta-deficient mouse mammary carcinomas. Cancer Res. 73, 5336–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes, D.R. , Yu, J. , Shanker, K. , Deshpande, N. , Varambally, R. , Ghosh, D. , Barrette, T. , Pandey, A. , Chinnaiyan, A.M. , 2004. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 6, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, Y. , Coleman, L. , Shi, J. , Beppu, H. , Sato, K. , Walsh, K. , Loscalzo, J. , Zhang, Y.Y. , 2008. Inflammation, endothelial injury, and persistent pulmonary hypertension in heterozygous BMPR2-mutant mice. Am. J. Physiol. Heart circulatory Physiol. 295, H677–H690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto, H. , Mundel, T.M. , Kieran, M.W. , Kalluri, R. , 2006. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 5, 1640–1646. [DOI] [PubMed] [Google Scholar]
- Talati, M. , West, J. , Zaynagetdinov, R. , Hong, C.C. , Han, W. , Blackwell, T. , Robinson, L. , Blackwell, T.S. , Lane, K. , 2014. BMP pathway regulation of and by macrophages. PloS One. 9, e94119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuder, R.M. , Archer, S.L. , Dorfmuller, P. , Erzurum, S.C. , Guignabert, C. , Michelakis, E. , Rabinovitch, M. , Schermuly, R. , Stenmark, K.R. , Morrell, N.W. , 2013. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J. Am. Coll. Cardiol. 62, D4–D12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein, B. , Papadopoulos, N. , Velculescu, V.E. , Zhou, S. , Diaz, L.A. , Kinzler, K.W. , 2013. Cancer genome landscapes. Science. 339, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman, P. , Aboagye, E.O. , Balkwill, F. , Balmain, A. , Bruder, G. , Chaplin, D.J. , Double, J.A. , Everitt, J. , Farningham, D.A. , Glennie, M.J. , Kelland, L.R. , Robinson, V. , Stratford, I.J. , Tozer, G.M. , Watson, S. , Wedge, S.R. , Eccles, S.A. , Committee of the National Cancer Research Institute2010. Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer. 102, 1555–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, L. , Huang, J. , Ren, X. , Gorska, A.E. , Chytil, A. , Aakre, M. , Carbone, D.P. , Matrisian, L.M. , Richmond, A. , Lin, P.C. , Moses, H.L. , 2008. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 13, 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, S. , Pham, L.K. , Liao, C.P. , Frenkel, B. , Reddi, A.H. , Roy-Burman, P. , 2008. A novel bone morphogenetic protein signaling in heterotypic cell interactions in prostate cancer. Cancer Res. 68, 198–205. [DOI] [PubMed] [Google Scholar]
- Yu, P.B. , Beppu, H. , Kawai, N. , Li, E. , Bloch, K.D. , 2005. Bone morphogenetic protein (BMP) type II receptor deletion reveals BMP ligand-specific gain of signaling in pulmonary artery smooth muscle cells. J. Biol. Chem. 280, 24443–24450. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplemental Figure 1. IHC for B&T cells does not demonstrate significant changes. A–F) Spontaneous tumor paraffin sections were IHC stained for markers of B (CD45R/B220) and T cells (CD4 and CD8a) in control tumors and BMPR2 FSP1 mediated cKO tumors. Scale bars = 100 μM.
Supplemental Figure 2. Lineage tracing of FSP1 expression from Cre recombination GFP reporter. A–B) Representative flow plot from digested tumors of BMPR2 cKO mice containing mTom/mGFP cre reporter. FITC indicates GFP expressing cells which specify Cre mediated recombination of BMPR2 and PE axis indicate mTomato expressing cells not expressing Cre. B) Staining for both CD45 (immune cells) and EpCam (epithelial cells) indicate Cre expression from box inset of panel A. C) The total percentage of GFP+ cells in the BMPR2 cKO (three tumors) and the relative percentage of immune (CD45), epithelial (EpCam) and double negative. Error bars indicate SD.
Supplemental Figure 3. Confirmation of BMPR2 deletion in cell lines. A) PCR genotyping for the Cre reporter transgene mTomato/mGFP was only present in cKO cells. B) SYBR based qPCR for BMP target genes Id1, Smad6, and Smad7 in control (white bars) and cKO (black bars) fibroblasts analyzed after 24 h of BMP2 stimulation at 100 ng/ml. Fold expression was normalized to Gapdh and untreated cells. # indicates statistical significance p = <0.01 by students t‐test. NS indicates Not Significant. Error bars indicate SEM.
Supplemental Figure 4. BMPR2 cKO fibroblast cells had decreased growth rate and unaffected viability. A) Average cell counts for control and cKO fibroblasts every 24 h. B) Viability of cells counted at each time point determined by trypan blue dye exclusion. Cell counts and viability were performed using the Life Technologies Countess automated cell counter.
Supplemental Figure 5. BMP pathway changes in BMPR2 cKO fibroblasts. A) SYBR qPCR for Bmp2 ligand revealed over 700 fold change increase relative to untreated control. B) Additional components of the BMP pathway significantly altered in BMPR2 cKO fibroblast by SYBR qPCR. C) SYBR qPCR for BMP activation gene targets reveals that BMPR2 cKO cells have elevated canonical BMP transcriptional response of Id1, Smad6 and Smad7. Previously known BMP signaling targets in mouse mammary fibroblasts Il‐6 and Mmp3 were also found to be significantly upregulated from control fibroblasts. Fold expression was normalized to Gapdh and untreated cells were relative to fold change from untreated cells. # indicates statistical significance p = <0.01 by students t‐test. NS indicates Not Significant. Error bars indicate SEM.
Supplemental Figure 6. IHC for B&T cells does not demonstrate significant changes. A–B) Representative images of PyVmT tumors implanted with control and cKO fibroblasts IHC stained for B cells. C–D) Representative images of PyVmT tumors implanted with control cKO fibroblasts IHC stained for CD4+ T cells. E‐F) Representative images of PyVmT tumors with control and cKO fibroblasts IHC stained for CD8+ T cells. Scale bars = 100 μM. Error bars indicate SD.
Supplemental Figure 7. Angiogenesis was not significantly altered in tumors. A–D) IHC for the pan‐endothelial marker MECA‐32 was performed on both spontaneous tumors and implanted tumors. E–F) Quantification of MECA‐32 IHC in PyVmT tumors with either control or cKO fibroblasts as a percent area of each image analyzed. Scale bars = 100 μM. Error bars indicate SD.
Supplementary Table 1. Control and cKO cell line cDNA was analyzed by SABio PCR array for inflammation (Catalog #3803). Red indicates the fold change increase and blue represents fold change decrease in cKO cells relative to control cells.
Supplementary Table 2. SYBR qPCR primer sequences. Mouse primer sequences were selected from Primer Bank (http://pga.mgh.harvard.edu/primerbank/) that contained validation data. All primer sequences are directed against mouse sequences. All qPCR was performed with melting curves that verified specificity to single target amplicon.