

Abstract
Directed evolution of toluene ortho-monooxygenase (TOM) of Burkholderia cepacia G4 previously created the hydroxylase α-subunit (TomA3) V106A variant (TOM-Green) with increased activity for both trichloroethylene degradation (twofold enhancement) and naphthalene oxidation (six-times-higher activity). In the present study, saturation mutagenesis was performed at position A106 with Escherichia coli TG1/pBS(Kan)TOMV106A to improve TOM activity for both chloroform degradation and naphthalene oxidation. Whole cells expressing the A106E variant had two times better naphthalene-to-1-naphthol activity than the wild-type cells (Vmax of 9.3 versus 4.5 nmol · min−1 · mg of protein−1 and unchanged Km), and the regiospecificity of the A106E variant was unchanged, with 98% 1-naphthol formed, as was confirmed with high-pressure liquid chromatography. The A106E variant degrades its natural substrate toluene 63% faster than wild-type TOM does (2.12 ± 0.07 versus 1.30 ± 0.06 nmol · min−1 · mg of protein−1 [mean ± standard deviation]) at 91 μM and has a substantial decrease in regiospecificity, since o-cresol (50%), m-cresol (25%), and p-cresol (25%) are formed, in contrast to the 98% o-cresol formed by wild-type TOM. The A106E variant also has an elevated expression level compared to that of wild-type TOM, as evidenced by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Another variant, the A106F variant, has 2.8-times-better chloroform degradation activity based on gas chromatography (Vmax of 2.61 versus 0.95 nmol · min−1 · mg of protein−1 and unchanged Km) and chloride release (0.034 ± 0.002 versus 0.012 ± 0.001 nmol · min−1 · mg of protein−1). The A106F variant also was expressed at levels similar to those of wild-type TOM and 62%-better toluene oxidation activity than wild-type TOM (2.11 ± 0.3 versus 1.30 ± 0.06 nmol · min−1 · mg of protein−1). A shift in regiospecificity of toluene hydroxylation was also observed for the A106F variant, with o-cresol (28%), m-cresol (18%), and p-cresol (54%) being formed. Statistical analysis was used to estimate that 292 colonies must be screened for a 99% probability that all 64 codons were sampled during saturation mutagenesis.
Toluene ortho-monooxygenase (TOM) is encoded by the Burkholderia cepacia G4 gene cluster tomA012345 (33) and belongs to an evolutionarily related family of nonheme diiron hydroxylases found in a wide range of bacteria (16). Examples of the family include soluble methane monooxygenase (sMMO) from methanotrophic bacteria (6, 37) as well as other toluene monooxygenases, such as toluene 4-monooxygenase from Pseudomonas mendocina KR1 (40), toluene 3-monooxygenase from Ralstonia pickettii PKO1 (23), and toluene/o-xylene monooxygenase from Pseudomonas stutzeri OX1 (1). TOM is a three-component complex consisting of a 211-kDa hydroxylase (tomA1A3A4) with two binuclear iron centers in the (αβγ)2 quaternary structure, a 40-kDa NADH-oxidoreductase (tomA5), and a 10.4-kDa regulatory protein (tomA2) involved in the electron transfer between the hydroxylase and reductase (21). The (αβγ)2 component contains the active site for substrate binding and hydroxylation reaction (21).
TOM catalyzes the NADH- and O2-dependent hydroxylation of toluene to form 3-methylcatechol through the intermediate o-cresol (34). Since most naturally occurring toluene monooxygenases have a relaxed substrate range, it is not surprising to find that TOM is also able to oxidize naphthalene to 1-naphthol (4), which makes it a potentially attractive biological catalyst. Biocatalytic processes proceed under mild reaction conditions, decrease by-product formation because of their high selectivity (chemo-, regio-, and stereospecificity) (12, 15), reduce energy requirements, and increase process safety. Aqueous biocatalytic processes also eliminate the need for strong, potentially hazardous acids, require low metal concentrations, and do not require solvents; thus, they are environmentally friendly. Over 15,000 tons of 1-naphthol are produced annually in the United States, and most commercial processes use chemical methods requiring acids, bases, and metal catalysts (38). Hence, the application of biocatalytic processes for 1-naphthol production may have a significant beneficial environmental impact.
TOM also has the potential for bioremediation, since B. cepacia G4 expressing this enzyme was the first strain isolated that oxidizes trichloroethylene (TCE), converting it primarily to Cl− and CO2 in vivo (17, 20). TOM also aerobically degrades other chlorinated aliphatic compounds, including chloroform (33, 35, 36). Like TCE, chloroform is a widely used chlorinated solvent and a common groundwater contaminant due to improper disposal and its accumulation from anaerobic dehalogenation of carbon tetrachloride at contaminated sites (10). Chloroform is also a suspected carcinogen (18). Whole cells expressing TOM are also able to degrade mixtures of chlorinated aliphatic compounds, including TCE and chloroform (36).
The cometabolic nature of chlorinated ethene metabolism and the toxic chlorinated epoxyethanes generated as the primary initial metabolites (39) make the evolution of TOM by natural selection for higher activity toward chlorinated ethenes unlikely. However, genetic engineering techniques such as directed evolution may lead to variants that can break down these compounds more quickly. Previously, directed evolution was used to improve TOM for both green chemistry and bioremediation, creating a variant, the V106A variant, that synthesized 1-naphthol six times faster and degraded TCE two times faster than the wild-type enzyme (4). Only one amino acid substitution in the α-subunit (tomA3) of the hydroxylase (V106A) was required for enhanced enzymatic activity. This amino acid has been proposed to act as a smaller gate (Fig. 1) to control substrate and product transport to the diiron active center (4).
FIG. 1.

Active site of the TomA3 α-subunit showing mutations (in red) at position 106 for wild-type TOM (V106) (A), the V106A variant (TOM-Green) (B), the A106F variant (C), and the A106E variant (D). Residues in yellow (E110, E140, H143, E201, E235, and H238) are coordinate residues anchoring the diiron binding sites (pink spheres). Portions of the four-helix bundle of TomA3 (helix B, A93 to H123; helix C, E127 to K156; helix E, P189 to N219; and helix F, S223 to I246) anchoring the diiron active site are shown in white and terminate at K100 to G116 (helix B), C133 to M148 (helix C), A195 to S214 (helix E), and G228 to I243 (helix F).
Since a single beneficial mutation was identified by screening 20,000 randomly mutated colonies, we sought to fine-tune the enzyme by evaluating all possible amino acids at position A106 of tomA3. Site-specific saturation mutagenesis can be used to introduce all possible mutations at key sites or adjacent sites to explore a larger fraction of the protein sequence space (29). It can provide much more comprehensive information than can be gained by single amino acid substitutions as well as overcome the drawbacks of random mutagenesis, in that a single mutation randomly placed in codons generates on average only 5.6 out of 19 possible substitutions (2). In this study, saturation mutagenesis was used to seek further improvements in TOM activity for both the regiospecific hydroxylation of naphthalene and the degradation of chlorinated aliphatic compounds.
MATERIALS AND METHODS
Chemicals.
Naphthalene, chloroform, TCE, and kanamycin sulfate were purchased from Fisher Scientific Company (Pittsburgh, Pa.); cis-1,2-dichloroethylene (cis-DCE) and trans-1,2-dichloroethylene(trans-DCE) were purchased from TCI America, Inc. (Portland, Oreg.); and tetrazotized o-dianisidine, 1-naphthol, and 2-naphthol were purchased from Sigma Chemical Co. (St. Louis, Mo.). All materials used were of the highest purity available and were used without further purification.
Bacterial strains and growth conditions.
Escherichia coli strain TG1 [supE hsdΔ5 thi Δ(lac-proAB) F′(traD36 proAB+ lacIq lacZΔM15)] was utilized as the host for gene cloning and expression (30). TG1 was routinely cultivated at 37°C in Luria-Bertani (LB) medium (30), with kanamycin (100 μg/ml) added to maintain the vector pBS(Kan)TOMV106A (4), which expresses tomA012345 from a constitutive lac promoter. Exponential-phase cultures were used in all experiments by diluting cells grown overnight to an optical density at 600 nm (OD) of 0.05 to 0.10 and growing them to an OD of 1.0 (except for the chloroform gas chromatography [GC]-based experiments for determining Vmax, in which the cells were cultured to an OD of 2.5). The exponentially growing cells were washed either once for the naphthol experiments with 1 volume of 50 mM Tris-HCl, pH 7.4 (to remove metabolic by-products), or three times for the degradation experiments with chloroform and other chlorinated ethenes with 1 volume of 50 mM Tris-HNO3, pH 7.0 (to remove all traces of chloride and metabolic by-products), unless otherwise stated. Standard conditions were employed throughout for the cell preparation. For each experiment, the conditions used for the cell preparations (e.g., harvest OD and contact OD) were the same for each of the strains tested.
Protein analysis and molecular techniques.
The Total Protein kit (Sigma Chemical Co.) was used to determine the total cellular protein for the calculation of whole-cell-specific activities. Cellular protein samples were analyzed on standard Laemmli discontinuous sodium dodecyl sulfate (SDS)-12% polyacrylamide gels (30). Plasmid DNA was isolated by using a Midi or Mini kit (QIAGEN, Inc., Chatsworth, Calif.), and DNA fragments were isolated from agarose gels by using the GeneClean III kit (Bio 101, Vista, Calif.) or the QIAquick gel extraction kit (QIAGEN, Inc.).
Saturation mutagenesis of tomA3 in the V106A variant.
A gene library encoding all possible amino acids at position A106 of tomA3 in the V106A variant in pBS(Kan)TOMV106A was constructed by replacing the target codon with NNN (where N is A, G, C, or T) via overlap extension PCR (29). Two degenerate primers, 106F (5′-CCTCGGTGTGCCATATACTCCAACGGTGTNNNACCCTGG-3′) and 106R (5′-GTCAACGCACTCAAGGTGTTCATCCAGGGTNNNACACCG-3′), were designed to randomize position A106 in the nucleotide sequence. The two additional primers for cloning were BsiWI Front (5′-CCGATGGAGAAAGTGTTTCCGTACGAC-3′) and SphI Rear (5′-GTTGTAGTGCGAGAGAGCATGCATTTC-3′), where the BsiWI and SphI restriction enzyme sites, respectively, are underlined; these two sites occur naturally in tomA3 upstream and downstream from position A106. Vent DNA polymerase (New England Biolabs, Beverly, Mass.) was used in the PCR to minimize random point mutations, and pBS(Kan)TOMV106A was used as the template. The 256-bp DNA fragment was amplified using the primers BsiWI Front and 106R, and the 179-bp DNA fragment was amplified using SphI Rear and 106F. After being purified from agarose gels, the two fragments were combined at a 1:1 ratio as templates to obtain the full-length PCR product with the BsiWI Front and SphI Rear primers. A PCR program of 30 cycles of 94°C for 45 s, 55°C for 45 s, and 72°C for 1 to 2 min, with a final extension of 72°C for 7 min, was used. The resulting randomized PCR product (374 bp) was cloned into pBS(Kan)TOMV106A after double digestion with BsiWI and SphI, replacing the corresponding fragment in the original plasmid. The resulting plasmid library was electroporated into E. coli TG1 competent cells using a GenePulser pulse controller (Bio-Rad Laboratories, Hercules, Calif.) at 15 kV/cm, 25 μF, and 200 Ω.
Statistical analysis for screening.
To determine the number of independent clones from saturation mutagenesis that need to be screened to ensure that each possible codon has been tested, a multinomial distribution equation was used. For saturation mutagenesis at one position, it is assumed that each of the 64 possible outcomes has the same probability based on the random synthesis of the primers and the fact that electroporation and plating should have no bias. A program in C language was developed to solve the following equation (28), which was used to determine the number of colonies that should be tested to make sure that the probability that each of the 64 outcomes has been sampled at least once is around 1.0:
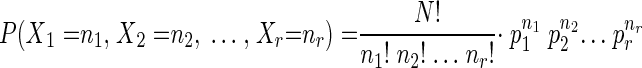 |
where p1, p2, and pr are the respective probabilities of one, two, or any one of r possible outcomes (e.g., r is 1 of 64 possible codons) resulting from independent and identical experiments A1, A2,…, Ar (e.g., A1 might be the codon CAC); ∑pi is equal to 1; P(X1 = n1, X2 = n2,…, Xr = nr) is the probability that A1 happens n1 times, A2 happens n2 times, and so on, and Ar happens nr times in N experiments (e.g., N equals 300 colonies); Σni is equal to N; and Xi is the number of n experiments that result in outcome number i (e.g., an X5 of 10 means codon 5 was seen 10 times in the population). In the saturation mutagenesis of one site, there are 64 possible outcomes, and the program calculates the number of colonies so that each Xi is ≥1, which means that every possible codon has been sampled at least once.
With two sites subjected to simultaneous saturation mutagenesis, two independent multinomial distributions apply. The probability that all possible mutants are sampled is found by multiplying the two independent probabilities, and the determination of the number of colonies needed to be sampled is based on P1(X1 = n1, X2 = n2,…, Xr = nr} · P2(X1 = n1, X2 = n2,…, Xr = nr) being equal to 1.0.
Screening for naphthol formation and chloroform mineralization.
Whole-cell synthesis of naphthol from naphthalene was determined with a spectrophotometric assay and a 96-well plate format as described previously (4). The inorganic chloride generated from the mineralization of chloroform by whole cells was also measured spectrophotometrically in 96-well plates (4). Cells in 96-well plates were contacted with shaking at 37°C in an airtight chamber, 23 by 20 by 23 cm, with either chloroform vapor (0.5 ml) for 16 h or naphthalene vapor for 1.5 h.
Naphthalene oxidation rates and high-pressure liquid chromatography (HPLC) product analysis.
The initial naphthalene oxidation rates were determined spectrophotometrically (4) using 2.5 ml of washed cells (OD of 2.0) in 10 μM to 5 mM naphthalene (dissolved in dimethyl formamide [DMF]) in 15-ml serum vials sealed with a Teflon-coated septum and an aluminum crimp seal. The vials were shaken at 37°C and 250 rpm on a Vibrax-VXR shaker (IKA-Works, Inc., Cincinnati, Ohio), and samples were taken every 5 min for 20 min. The naphthol concentrations were determined by reacting the samples with tetrazotized o-dianisidine and measuring the absorbance at 540 nm. Strain TG1/pBS(Kan) containing no monooxygenase was used as the negative control. This procedure was done at least three times for each strain tested, and results are shown in Table 1. The parameters apparent Km and Vmax were measured twice (at ODs of 1 and 2).
TABLE 1.
Toluene hydroxylation rate and regiospecificity, naphthol formation rate and regiospecificity, and chloroform degradation rate of wild-type TOM and variants of tomA3
| Enzymea | Relative approx expression levelb | Toluenec
|
Naphthalened
|
Chloroform degradation rate,e nmol of Cl− · min−1 mg of protein−1 (mean ± SD) | ||||
|---|---|---|---|---|---|---|---|---|
| Degradation rate, nmol · min−1 · mg of protein−1 (mean ± SD) | % Regiospecificity:
|
Naphthol formation rate, nmol · min−1 · mg of protein−1 (mean ± SD) | % Regiospecificity for 1-naphthol | |||||
| o-Cresol | m-Cresol | p-Cresol | ||||||
| Wild-type TOM | 1.0 | 1.30 ± 0.06 | >98 | 0 | 0 | 0.80 ± 0.06 | 98 | 0.012 ± 0.002 |
| V106A variant | 1.0 | 2.8 ± 0.5 | 50 | 33 | 17 | 3.6 ± 0.4 | 97 | 0.012 ± 0.002 |
| A106E variant | 5.0 | 2.12 ± 0.07 | 50 | 25 | 25 | 7.4 ± 0.4 | 98 | 0.005 ± 0.004 |
| A106F variant | 1.0 | 2.11 ± 0.3 | 28 | 18 | 54 | 2.4 ± 0.1 | 99 | 0.034 ± 0.001 |
Expressed in E. coli TG1 whole cells.
Determined via SDS-polyacrylamide gel electrophoresis.
The degradation rate was determined via GC over 20 min with 91 μM toluene calculated based on Henry's law (8), and regiospecificity was determined by GC.
The naphthol formation rate was determined in 15 min via spectrophotometric assay at 5 mM naphthalene (naphthalene solubility is 0.23 mM in water [25]), while regiospecificity for 1-naphthol was determined by HPLC.
Determined via chloride release over 16 h with 50 μM chloroform based on Henry's law (7).
The regiospecificity of naphthalene hydroxylation was determined by using HPLC as reported previously (4). The HPLC system (515 HPLC pump; Waters Corp., Milford, Mass.) was equipped with a 996 photodiode array detector (Waters Corp.), a Novapak C18 column (Waters Corp.), and Millenium32 Chromatography Manager software (Waters Corp.). A gradient system from 1:3 methanol-water to 100% methanol over 1 h was used to separate the naphthol products. The products generated were compared to the standards 1-naphthol and 2-naphthol.
Toluene degradation rates.
Washed cell suspensions adjusted to an OD of 10 were added to toluene (250 μM if all the toluene is in liquid phase); the actual initial liquid concentration was 91 μM based on Henry's law (8), and toluene degradation was measured by GC every 5 min by using a Hewlett-Packard model 6890 gas chromatograph equipped with an EC-WAX column (30 m by 0.25 mm, 0.25-μm thickness; Alltech Associates, Inc., Deerfield, Ill.) and a flame ionization detector. The injector and detector were maintained at 250 and 275°C, respectively, and a split ratio of 3:1 was used. The He carrier flow rate was maintained at 0.8 ml/min. The temperature program was 80°C for 5 min, 80 to 205°C for 25 min (increasing at a rate of 5°C/min), 205 to 280°C for 5 min (increasing at a rate of 15°C/min), and 280°C for 5 min. Under these conditions, the retention times for toluene, o-cresol, p-cresol, and m-cresol were 4.2, 27.6, 29.3, and 29.5 min, respectively. Retention times were determined by comparisons to neat standards.
Rates and extent of mineralization of chlorinated aliphatic compounds.
After being washed with Tris-HNO3 buffer, 2.5 ml of cell suspension adjusted to an OD of 5.0 was added into 15-ml serum vials, which were sealed. The supernatant chloride ion concentrations generated from mineralizing the chlorinated aliphatic compounds (chloroform, TCE, cis-DCE, trans-DCE, and 1,1-DCE) were measured after specific times of incubation (16 h for chloroform, 4 h for cis-DCE, 3 h for trans-DCE, and 40 min for TCE and 1,1-DCE). TCE and the three dichloroethylenes were added at an initial concentration of 200 μM, and chloroform was added at 80 μM as if all the volatile organics were in the liquid phase (DMF was the diluent); actual initial liquid concentrations based on Henry's law (7, 8) were 67 μM for TCE, 108 μM for cis-DCE, 69 μM for trans-DCE, 32 μM for 1,1-DCE, and 50 μM for chloroform. After contact, the enzyme activity was quenched by heating the vials in boiling water for 90 s. The chloride ion concentrations in 500 μl of supernatant were measured spectrophotometrically as described elsewhere (4) using the negative control of DMF addition (no chlorinated aliphatic compounds) for each strain.
To determine the kinetics of chloroform degradation, GC was used to monitor chloroform depletion as described previously (36). Cells harvested at an OD of 2.5 after 2 h of growth with 1.0 mM IPTG (isopropyl-β-d-thiogalactopyranoside) were washed and resuspended in chloride-free modified minimal salts (M9) medium (30) supplemented with succinate [with 0.24 g of Mg(SO4)2/liter and 0.17 g of Ca(SO4)2/liter replacing MgCl2 and CaCl2], sealed in 15-ml glass serum vials (2.5 ml, OD of 2.0), and allowed to react with chloroform at initial liquid concentrations ranging from 6.5 to 258 μM at 37°C and 250 rpm. Concentrations were monitored every hour for 3 h, and the activities of the cells remained relatively constant over this period. Apparent Km and Vmax were determined from the initial rate at each concentration.
DNA sequencing.
A dye terminator cycle sequencing protocol based on the dideoxy method of sequencing DNA developed by Sanger et al. (31) was used to sequence both strands of the subcloned region (374 bp) in the A106E and A106F variants by using the BsiWI Front and SphI Rear primers and the ABI Prism BigDye terminator cycle sequencing ready reaction kit (Perkin-Elmer, Wellesley, Mass.). A PE Biosystems (Perkin-Elmer) ABI 373 DNA sequencer was used for analyzing the fluorescently labeled DNA fragments by gel electrophoresis. Sequence data were analyzed using the VectorNTI suite sequencing alignment editor (InforMax, Inc., Frederick, Md.).
Homology structure modeling of tomA3.
Part of the amino acid sequence of the wild-type TOM α-subunit (residues 95 to 250 of tomA3) was modeled into the known three-dimensional structure of the homologue sMMO hydroxylase from Methylococcus capsulatus (Bath) (Protein Data Bank accession code 1MTY) using the SWISS-MODEL server (14, 24, 32). The molecular visualization program Swiss-PdbViewer (14, 24, 32) was utilized to visualize and manipulate the molecular model by means of steric hindrance and energy minimization and by performing amino acid substitutions isosterically at V106 based on residue interactions, among other things.
RESULTS
Saturation mutagenesis.
By cloning DNA fragments from saturation mutagenesis back into the corresponding sequence of pBS(Kan)TOMV106A (4), a library of 1,600 clones containing a variety of differently colored colonies on LB medium-kanamycin (100 μg/ml) agar plates was obtained; the colors arise due to the various hydroxylation products of indole (formed as a breakdown product of tryptophan in E. coli) that spontaneously oxidize into indigoid compounds (9, 11). A total of 800 colonies of the mutant library were screened by using whole cells in a 96-well plate format for both naphthol synthesis from naphthalene and chloride ion production from the mineralization of chloroform.
Determination of the number of clones for screening.
The number of saturation mutagenesis colonies which must be screened (N) was determined using a C language software program that calculates via a multinomial distribution equation the probability that each codon will be sampled in a population of colonies; by trial and error, N was changed until the probability reached 0.99 or 1.0. Our program indicates that 922 independent clones are needed to ensure that the probability that all 64 possible outcomes of single-site random mutagenesis have been sampled is 1.0. If the probability is decreased to 0.99, only 292 colonies need to be screened. If two residues are subject to simultaneous saturation mutagenesis, 342 independent clones need to be sampled to ensure with 0.99 probability that all the possible outcomes have been checked. Thus, by screening 800 colonies, all of the 64 possible codons should have been sampled. Compared to the large library of colonies that must be screened for the random mutagenesis of a whole protein using DNA shuffling or error-prone PCR, such a small number is attractive; however, saturation mutagenesis requires that the important residues first be known.
Although amino acid substitutions are of more interest than codon changes, a solution to the multinomial equation based on 20 amino acid substitutions would be seriously flawed. The basis for the calculation was the assumption that the respective probabilities of any one of r possible outcomes were equal (p1 = p2 =… = pr), and only the 64 codons, not the 20 amino acids, have the same outcome probabilities, since the number of codons coding for each amino acid is not the same. In addition, it is difficult to incorporate all the factors necessary to solve such a multinomial distribution equation (such as the number of codons coding for each amino acid and codon usage).
Characterization of the best mutants.
The best mutants initially identified by screening for naphthol synthesis in 96-well plates were further examined in 15-ml vials for naphthol production with 5 mM naphthalene (naphthalene solubility is 0.23 mM in water [25]). Because TOM is a complex multicomponent enzyme system that requires NADH, whole-cell recombinant E. coli strains expressing TOM variants were used in all assays.
As shown in Table 1, the initial naphthol synthesis rate by TG1 expressing the A106E variant was twofold faster than that of the original V106A variant-containing strain after 15 min, and this rate of enhancement was corroborated by data at 30 and 60 min (data not shown). Both strands of the subcloned tomA3 DNA region (374 bp) in the A106E variant were sequenced and compared with the same region of the V106A variant. As expected, there was no base change except at the A106 position, the saturation mutation point for the A106E mutation (GCG at position 106 to GAG). The apparent constant Vmax for whole cells expressing the A106E and V106A variants with naphthalene as the substrate were 9.33 and 4.5 nmol · min−1 · mg of protein−1, respectively, and these two strains had similar Km values, which were 12.1 and 13.5 μM, respectively. These twofold-better Vmax results for the saturation A106E mutant corroborate the 15-min initial naphthol formation rates. The naphthol products of the A106E and V106A variants were determined by HPLC analysis to be greater than 98% 1-naphthol, with 2-naphthol as the minor product. Therefore, the A106E mutation leads to an increase in the naphthol synthesis rate without changing the regiospecific site of oxidation.
The A106F variant, the best mutant screened from the 96-well microtiter plates for chloroform mineralization, was also examined further in 15-ml vials, and the sequencing of both strands of the subcloned tomA3 DNA region (374 bp) indicated that the only base changes were GCG to TTT at position 106. The chloroform mineralization rate of whole cells expressing the A106F mutation, as indicated by the production of chloride, was 2.8-fold faster than that of the V106A mutation-containing strain after 16 h (Table 1). A very similar rate of enhancement (2.8-fold) was also observed with the Vmax values for whole cells expressing the A106F and V106A mutations, determined via GC (2.61 versus 0.95 nmol · min−1 · mg of protein−1), while Km values for both strains were almost identical (42 μM initial liquid concentration). However, the A106F variant did not show higher activity toward TCE, cis-DCE, trans-DCE, or 1,1-DCE than the V106A variant. For example, whole cells expressing the V106A mutation mineralized TCE at a rate of 2.6 nmol · min−1 · mg of protein−1, compared to a rate of 1.34 nmol · min−1 · mg of protein−1 by cells expressing the A106F variant. Naphthalene oxidation catalyzed by the A106F protein in whole cells was slower than the same reaction mediated by the V106A protein, but it was three times faster than that mediated by wild-type TOM (Table 1).
Toluene activity and regiospecificity.
By using GC, whole cells expressing the A106E and A106F proteins were tested for their activities and regiospecificities on the natural substrate toluene compared to those of wild-type TOM and the V106A variant. Both proteins oxidized toluene significantly more quickly than wild-type TOM (Table 1), and the three enzyme variants had distinct regiospecific hydroxylation patterns for toluene oxidation. Cells expressing wild-type TOM catalyzed toluene hydroxylation with high regiospecificity, yielding o-cresol as the primary product (>98% of the total product), whereas cells expressing the A106E variant were much less specific and formed o-cresol (50%), m-cresol (25%), and p-cresol (25%). The A106F variant formed even less o-cresol (28%), as well as m-cresol (18%) and p-cresol (54%), while the V106A variant also formed o-cresol (50%), m-cresol (33%), and p-cresol (17%). Hence, by changing position V106 of tomA3, the regiospecificity of toluene oxidation may be changed.
Expression level and specific growth rate.
As seen with naphthalene dioxygenase and para-nitrobenzyl esterase (19, 29), the A106E variant is an “expression up mutant” as evidenced by SDS-polyacrylamide gel electrophoresis; a single nucleotide change in one codon led to a much elevated protein expression level of the TomA3 and TomA1 subunits. The variation in protein expression level may be due to the modification of the primary amino acid sequence which leads to a decrease in protein lability, or the single nucleotide change may lead to decreased lability of the transcript. This increase in expression may partially explain the twofold increase in activity toward naphthalene oxidation. The expression level of the A106F variant remained approximately the same as that of the V106A variant. The specific growth rates of the three strains are approximately the same, 1.62 ± 0.06 h−1 (mean ± standard deviation) in LB medium-kanamycin. No sequencing changes in either the promoter or ribosomal binding site were found for TomA3 of the A106E variant.
TomA3 structural modeling.
The approximate three-dimensional coordinates for the TomA3 four-helix bundle anchoring the active site are shown in Fig. 1. The accuracy of the model was judged by the conservation of positions of the diiron coordinating residues in TOM (E110, E140, H143, E201, E235, and H238) compared to sMMO (the average distance of the Cα carbons of the model to sMMO for the metal binding residues is 0.07 Å). The structural alignment of the template and model also showed conserved spatial configurations.
Although there are limitations to homology modeling (13) and no absolute statement can be made due to the low homology between TOM and sMMO (30% amino acid identity in the modeled part), the model did help visualize the positions of the side chains for the A106F and A106E variants (Fig. 1). It can be seen that although both the A106E and A106F variants contain side chains with increased bulkiness compared to those of the wild type and the V106A variant, both side chains of the A106E and A106F variants are oriented such that they are located closer to the diiron center. This may explain the enhanced activities of these two mutants.
DISCUSSION
It is clearly shown in this paper that the saturation mutagenesis of TOM at the key site V106 in TomA3 results in further improvements in TOM activity for the regiospecific hydroxylation of naphthalene (by the V106E variant) and for the enhanced degradation of chloroform (by the V106F variant). In addition, it is shown that only a small library is required to adequately sample a population for saturation mutagenesis (292 colonies for ensuring a 99% probability that all outcomes have been checked).
It seems unusual that whole cells expressing wild-type TOM have lower activity toward toluene, the physiological substrate, than the V106A, A106E, and A106F mutants. However, the much higher regiospecificity of toluene oxidation mediated by TOM may reflect an evolutionary balance between activity and regiospecificity. Since wild-type TOM converts toluene to 3-methylcatechol via o-cresol, which is channeled into a productive metabolic pathway by catechol 2,3-dioxygenase (34), the production of p-cresol may not be productive for this meta cleavage pathway since p-cresol is converted to 4-methylcatechol by TOM (34) (note that m-cresol is also converted by TOM into 3-methylcatechol). Even though the V106A variant produces o-cresol at a higher rate than does wild-type TOM (Table 1), its production of p-cresol may waste cell energy.
Primary sequence alignment of TOM with other nonheme monooxygenases, including sMMO, other toluene monooxygenases, and phenol hydroxylase from Pseudomonas putida strain CF600 (22) reveals that the following comparable residues in all the monooxygenases are hydrophobic: Ile in toluene 3-monooxygenase tbuA1 (3), toluene 4-monooxygenase tmoA (40), and toluene/o-xylene monooxygenase touA (1); Leu in sMMO mmoX (5, 37); and Val in both phenol hydroxylase dmpN (22) and TOM tomA3 (4). There is, however, considerable variation in these residues, which may reflect the subtle differences between the various activities of the diiron monooxygenases. Based on the X-ray crystal structure analysis of sMMO from Methylococcus capsulatus (Bath) (26, 27), Leu 110, the residue comparable to Val 106 in TOM, is a constituent residue of the hydrophobic shell that surrounds the coordinating hydrophilic residues and has been proposed to act as a hydrophobic gate (Fig. 1), controlling the movement of substrate into and products out of the active sites (4, 26). The A106E variant appears to be the first example in this family of enzymes to accommodate a hydrophilic residue, Glu, at this position with full functionality. It seems that the active site within the alpha-helix bundle (Fig. 1) is a robust scaffold that can tolerate even nonconservative modifications.
As the V106 residue of tomA3 changed from Val (in wild-type TOM) to Ala (in the V106A variant) and then to Glu (in the A106E variant), the properties of this residue varied greatly in terms of size and hydrophobicity. Although there are limitations to homology modeling (32), if one considers the vicinity of the residue to the active site (Fig. 1) and the important functionality inferred from sMMO and evidenced by our mutagenesis experiments, such a change may lead to the variation in the active-site shape as the volume occupied by the side chain at the position of V106 is altered. The substantial decrease in the regiospecificities of toluene hydroxylation by the V106A, A106E, and A106F variants (Table 1) also indicates a change in the active site. However, the oxidation of naphthalene remained unchanged: 98% 1-naphthol is formed by all the TOM variants (Table 1). Although the change in the regiospecificity of toluene hydroxylation and unaltered naphthalene oxidation by the A106E variant seem contradictory, they may be explained by the different dimensions of the two substrates. Toluene is a smaller molecule than naphthalene, and variation in the shape of the active site may facilitate toluene binding to the active site in different orientations, resulting in the relaxed regiospecific oxidation. Naphthalene, however, is more rigid and bulky and may be more limited in its binding orientation relative to the diiron active site. This high regiospecific oxidation of naphthalene is attractive for a biocatalyst, since high purity is desirable.
The fact that TOM can tolerate various mutations at such an important position indicates that a strong framework, which can be varied to create new activity without impairing the catalytic robustness of the whole system, has been created. This may reflect the evolutionary potential used to evolve novel functions. Directed evolution is able to accelerate this evolution, and such a potential can be tested using different substrates as probes.
Acknowledgments
This research was supported by the National Science Foundation (grant BES-9911469).
We thank Ming-Hui Chen and Hai Xu of the University of Connecticut for help with the statistical analysis.
REFERENCES
- 1.Bertoni, G., M. Martino, E. Galli, and P. Barbieri. 1998. Analysis of the gene cluster encoding toluene/o-xylene monooxygenase from Pseudomonas stutzeri OX1. Appl. Environ. Microbiol. 64:3626-3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bylina, E. J., W. J. Coleman, M. R. Dilworth, S. J. Robles, M. A. Tanner, M. M. Yang, and D. C. Youvan. 2000. Solid-phase enzyme screening. ASM News 66:211-217. [Google Scholar]
- 3.Byrne, A. M., J. J. Kukor, and R. H. Olsen. 1995. Sequence analysis of the gene cluster encoding toluene-3-monooxygenase from Pseudomonas pickettii PKO1. Gene 154:65-70. [DOI] [PubMed] [Google Scholar]
- 4.Canada, K. A., S. Iwashita, H. Shim, and T. K. Wood. 2002. Directed evolution of toluene ortho-monooxygenase for enhanced 1-naphthol synthesis and chlorinated ethene degradation. J. Bacteriol. 184:344-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cardy, D. L. N., V. Laidler, G. P. C. Salmond, and J. C. Murrell. 1991. The methane monooxygenase gene cluster of Methylosinus trichosporium: cloning and sequencing of the mmoC gene. Arch. Microbiol. 156:477-483. [DOI] [PubMed] [Google Scholar]
- 6.Cardy, D. L. N., V. Laidler, G. P. C. Salmond, and J. C. Murrell. 1991. Molecular analysis of the methane monooxygenase (MMO) gene cluster of Methylosinus trichosporium OB3b. Mol. Microbiol. 5:335-342. [DOI] [PubMed] [Google Scholar]
- 7.Chauhan, S., P. Barbieri, and T. K. Wood. 1998. Oxidation of trichloroethylene, 1,1-dichloroethylene, and chloroform by toluene/o-xylene monooxygenase from Pseudomonas stutzeri OX1. Appl. Environ. Microbiol. 64:3023-3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dolfing, J., A. J. van den Wijngaard, and D. B. Janssen. 1993. Microbiological aspects of the removal of chlorinated hydrocarbons from air. Biodegradation 4:261-282. [DOI] [PubMed] [Google Scholar]
- 9.Eaton, R. W., and P. J. Chapman. 1995. Formation of indigo and related compounds from indolecarboxylic acids by aromatic acid-degrading bacteria: chromogenic reactions for cloning genes encoding dioxygenases that act on aromatic acids. J. Bacteriol. 177:6983-6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Egli, C., R. Scholtz, A. M. Cook, and T. Leisinger. 1987. Anaerobic dechlorination of tetrachloromethane and 1,2-dichloroethane to degradable products by pure cultures of Desulfobacterium sp. and Methanobacterium sp. FEMS Microbiol. Lett. 43:257-261. [Google Scholar]
- 11.Ensley, B. D., B. J. Ratzkin, T. D. Osslund, and M. J. Simon. 1983. Expression of naphthalene oxidation genes in Escherichia coli results in the biosynthesis of indigo. Science 222:167-169. [DOI] [PubMed] [Google Scholar]
- 12.Frost, J. W., and K. M. Draths. 1995. Biocatalytic syntheses of aromatics from d-glucose: renewable microbial sources of aromatic compounds. Annu. Rev. Microbiol. 49:557-579. [DOI] [PubMed] [Google Scholar]
- 13.Guex, N., A. Diemand, and M. C. Peitsch. 1999. Protein modelling for all. Trends Biochem. Sci. 24:364-367. [DOI] [PubMed] [Google Scholar]
- 14.Guex, N., and M. C. Peitsch. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714-2723. [DOI] [PubMed] [Google Scholar]
- 15.Hudlicky, T., D. Gonzalez, and D. T. Gibson. 1999. Enzymatic dihydroxylation of aromatics in enantioselective synthesis: expanding asymmetric methodology. Aldrichimica Acta 32:35-62. [Google Scholar]
- 16.Leahy, J. G., P. J. Batchelor, and S. M. Morcomb. 2003. Evolution of the soluble diiron monooxygenases. FEMS Microbiol. Rev. 27:449-479. [DOI] [PubMed] [Google Scholar]
- 17.Luu, P. P., C. W. Yung, A. K. Sun, and T. K. Wood. 1995. Monitoring trichloroethylene mineralization by Pseudomonas cepacia G4 PR1. Appl. Microbiol. Biotechnol. 44:259-264. [Google Scholar]
- 18.McClay, K., B. G. Fox, and R. J. Steffan. 1996. Chloroform mineralization by toluene-oxidizing bacteria. Appl. Environ. Microbiol. 62:2716-2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore, J. C., and F. H. Arnold. 1996. Directed evolution of a para-nitrobenzyl esterase for aqueous-organic solvents. Nat. Biotechnol. 14:458-467. [DOI] [PubMed] [Google Scholar]
- 20.Nelson, M. J. K., S. O. Montgomery, E. J. O'Neill, and P. H. Pritchard. 1986. Aerobic metabolism of trichloroethylene by a bacterial isolate. Appl. Environ. Microbiol. 52:383-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newman, L. M., and L. P. Wackett. 1995. Purification and characterization of toluene 2-monooxygenase from Burkholderia cepacia G4. Biochemistry 34:14066-14076. [DOI] [PubMed] [Google Scholar]
- 22.Nordlund, I., J. Powlowski, and V. Shingler. 1990. Complete nucleotide sequence and polypeptide analysis of multicomponent phenol hydroxylase from Pseudomonas sp. strain CF600. J. Bacteriol. 172:6826-6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olsen, R. H., J. J. Kukor, and B. Kaphammer. 1994. A novel toluene-3-monooxygenase pathway cloned from Pseudomonas pickettii PKO1. J. Bacteriol. 176:3749-3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peitsch, M. C. 1993. Protein modeling by e-mail. Bio/Technology 13:658-660. [Google Scholar]
- 25.Perry, R. H., and C. H. Chilton. 1973. Chemical engineers' handbook, 5th ed., p. 3-39. McGraw-Hill Book Company, New York, N.Y.
- 26.Rosenzweig, A. C., H. Brandstetter, D. A. Whittington, P. Nordlund, S. J. Lippard, and C. A. Frederick. 1997. Crystal structures of the methane monooxygenase hydroxylase from Methylococcus capsulatus (Bath): implications for substrate gating and component interactions. Proteins Struct. Funct. Genet. 29:141-152. [PubMed] [Google Scholar]
- 27.Rosenzwieg, A. C., P. Nordlund, P. M. Takahara, C. A. Frederick, and S. J. Lippard. 1995. Geometry of the soluble methane monooxygenase catalytic diiron center in two oxidation states. Chem. Biol. 2:409-418. [PubMed] [Google Scholar]
- 28.Ross, S. 1997. A first course in probability, 5th ed., p. 252. Prentice-Hall, Inc., Englewood Cliffs, N.J.
- 29.Sakamoto, T., J. M. Joern, A. Arisawa, and F. H. Arnold. 2001. Laboratory evolution of toluene dioxygenase to accept 4-picoline as a substrate. Appl. Environ. Microbiol. 67:3882-3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 31.Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74:5463-5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwede, T., J. Kopp, N. Guex, and M. C. Peitsch. 2003. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31:3381-3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shields, M. S., and S. C. Francesconi. August 1996. Microbial degradation of trichloroethylene, dichloroethylenes, and aromatic pollutants. U.S. patent 5,543,317.
- 34.Shields, M. S., S. O. Montgomery, P. J. Chapman, S. M. Cuskey, and P. H. Pritchard. 1989. Novel pathway of toluene catabolism in the trichloroethylene-degrading bacterium G4. Appl. Environ. Microbiol. 55:1624-1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shields, M. S., M. J. Reagin, R. R. Gerger, C. Somerville, R. Schaubhut, R. Campbell, and J. Hu-Primmer. 1994. Constitutive degradation of trichloroethylene by an altered bacterium in a gas-phase bioreactor, p. 50-65. In R. E. Hinchee, A. Leeson, L. Semprini, and S. K. Ong (ed.), Bioremediation of chlorinated and polycyclic aromatic hydrocarbon compounds. Lewis Publishers, Boca Raton, Fla.
- 36.Shim, H., and T. K. Wood. 2000. Aerobic degradation of mixtures of chlorinated aliphatics by cloned toluene-o-xylene monooxygenase and toluene o-monooxygenase in resting cells. Biotechnol. Bioeng. 70:693-698. [DOI] [PubMed] [Google Scholar]
- 37.Stainthorpe, A. C., V. Lees, G. P. C. Salmond, H. Dalton, and J. C. Murrell. 1990. The methane monooxygenase gene cluster of Methylococcus capsulatus (Bath). Gene 91:27-34. [DOI] [PubMed] [Google Scholar]
- 38.Talukder, M., and C. R. Kates. 1995. Naphthalene derivatives, p. 979-1017. In M. Howe-Grant (ed.), Kirk-Othmer encyclopedia of chemical technology, 4th ed., vol. 16. John Wiley & Sons, Inc., New York, N.Y. [Google Scholar]
- 39.van Hylckama Vlieg, J. E. T., and D. B. Janssen. 2001. Formation and detoxification of reactive intermediates in the metabolism of chlorinated ethenes. J. Biotechnol. 85:81-102. [DOI] [PubMed] [Google Scholar]
- 40.Yen, K.-M., M. R. Karl, L. M. Blatt, M. J. Simon, R. B. Winter, P. R. Fausset, H. S. Lu, A. A. Harcourt, and K. K. Chen. 1991. Cloning and characterization of a Pseudomonas mendocina KR1 gene cluster encoding toluene-4-monooxygenase. J. Bacteriol. 173:5315-5327. [DOI] [PMC free article] [PubMed] [Google Scholar]