

Abstract
Myasthenia gravis (MG) is a disease that affects the neuro-muscular junction resulting in classical symptoms of variable muscle weakness and fatigability. It is called the great masquerader owing to its varied clinical presentations. Very often, a patient of MG may present to the ophthalmologist given that a large proportion of patients with systemic myasthenia have ocular involvement either at presentation or during the later course of the disease. The treatment of ocular MG involves both the neurologist and ophthalmologist. Thus, the aim of this review was to highlight the current diagnosis, investigations, and treatment of ocular MG.
Keywords: Acetylcholine, autoimmune, neuro-muscular junction, ocular myasthenia gravis
Myasthenia gravis (MG) is a potentially serious, but treatable autoimmune disease affecting the neuro-muscular junction (NMJ) of the skeletal muscle. Ocular myasthenia gravis (OMG) can mimic isolated cranial nerve palsies, gaze palsies, internuclear ophthalmoplegia, blepharospasm, and even a stroke.
History of Myasthenia Gravis
Thomas Willis (1672) and Samuel Wilks (1877) along with their European colleagues, Erb and Goldflam, were the earliest to write about MG.[1,2] In 1895, the term “Myasthenia Gravis (MG) pseudo-paralytica” was used by German physician, Jolly. Treatment of MG became possible in 1934, when in an episode described as “The miracle at St. Alfege's,” Mary Walker treated a case of MG with physostigmine (a cholinesterase inhibitor) on the basis that MG symptoms were similar to those of curare poisoning.[1,2] Simpson and Nastuck later elaborated the role of the immune system in the pathophysiology of MG independently, and Patrick and Lindstrom (1973) showed that rabbits immunized with purified muscle-like acetylcholine (Ach) receptors developed MG-like symptoms.[3]
Epidemiology and Demographics
Myasthenia may affect any age group and shows no geographic predilection.[3,4] Onset of symptoms in the first decade or after the age of 70 years is less common.[2] The incidence ranges from 0.04 to 5/100 000/year and prevalence estimates of 0.5-12.5/100 000/year.[2] Generalized and OMG differ with respect to the demographics of the affected population; whilst the ratio of affected females: males is 3:2 or higher in generalized myasthenia gravis (GMG), more males are affected by purely OMG, more so over the age of 40.[5,6] Onset occurs at an earlier age in women (mean age 28 years) than in men (mean age 42 years).[3]
In India, MG is reportedly more common in males than in females; the age of onset in males is in the sixth to seventh decade, and that in females is seen to be in the third decade.[4]
Pathophysiology
The NMJ is the site of chemical communication between a nerve fiber and a muscle where motor nerve impulses are transmitted to the muscle cell. An action potential initiates neuro-muscular transmission and results in the release of ACh molecules at the NMJ, which then diffuse across the synapse, bind to receptors on the striated muscle and depolarize the postsynaptic membrane, resulting in muscle contraction.
Antiacetylcholine receptor antibodies (AChR-Abs) have been demonstrated in up to 99% of patients with generalized myasthenia and 40-77% of patients with OMG. AChR-Abs decrease the number of available AChRs by receptor blockade, complement-mediated membrane damage, and accelerated degradation of the receptors.[5] This results in defective transmission at the NMJ and subsequent muscle weakness.
Extraocular muscles (EOMs) are more commonly affected as twitch fibers in EOMs develop tension faster and have a higher frequency of synaptic firing than limb muscles. This makes them more susceptible to fatigue. Furthermore, tonic muscle fibers are necessary to sustain the gaze in any direction. This type of fiber has fewer ACh receptors, which makes them more susceptible to receptor loss or damage.[6] Differences in ACh receptor types expressed in extraocular versus typical skeletal muscle[5] may contribute to increased susceptibility. Furthermore, EOMs represent a distinct muscle allotype with differential expression of numerous genes, including those associated with the immune response.[7]
Clinical Features
Myasthenia gravis is characterized by a variable weakness of skeletal muscles, which improves on resting. Weakness is exacerbated by repetitive contraction.[5] Generalized myasthenia involves the bulbar, limb, and respiratory muscles; OMG is a subtype of MG where the weakness is clinically isolated to the EOMs, levator, and orbicularis oculi.[5] Expectedly, due to variable involvement of different EOMs, motility patterns are not characteristic of lesions of one or more nerves.[6] Ptosis and diplopia are the initial signs of the disease in over 50% of MG patients;[8] 50-80% of these patients go on to develop generalized disease.[7] In the majority of cases (90%), progression of OMG to its generalized form will occur within the first 2 years after ocular symptoms begin.[9]
Eyelid Manifestations
Variable ptosis is one of the most common manifestations of MG. Ptosis occurs primarily due to the involvement of the levator palpebrae superioris (LPS) complex. It may be unilateral or bilateral– in bilateral cases, it is often asymmetrical. Ptosis may increase after prolonged upgaze-referred to as the “lid fatigability test.” Another clinical sign described is the Cogan's lid twitch, a quick overshooting upward movement followed by a down-drift of the upper lid after the patient performs a saccade back to primary position from looking down for at least 15 seconds. Cogan's lid twitch is however, not specific to ocular MG.[10] When the ptotic eyelid is lifted manually, enhancement of ptosis of the contralateral eye may be noted, explained by Hering's law of equal innervation to yoke muscles.
Other eyelid manifestations of OMG include unilateral eyelid retraction and orbicularis weakness. Lid retraction may be seen with contralateral ptosis as a manifestation of Hering's law (due to increased innervation to the ptotic eyelid), or maybe a sign of co-existent thyroid eye disease, or maybe due to post-tetanic facilitation of the LPS after prolonged upgaze. Orbicularis tone can be evaluated by attempting to open the eyes against forced eyelid closure, which is easily achieved in patients with OMG. Even without forced opening, the eyelids tend to drift apart, and the underlying sclera can be seen – this is called the “peek sign.”
Extraocular Muscle Involvement
Diplopia is very common in cases with OMG since even slight weakness of the EOMs causes symptomatic diplopia and since the EOMs do not adapt to variable weakness like limb muscles.[6,11] Diplopia is, usually, seen with ptosis but maybe present as an isolated finding also. The most commonly affected EOM is the medial rectus followed by the superior rectus.[12] OMG, can mimic any comitant or incomitant strabismus ranging from nerve palsies, gaze palsies, unilateral or bilateral internuclear ophthalmoplegias to even complete ophthalmoplegia.[6] Clinically, OMG should be suspected in any variable incomitant strabismus, with or without ptosis.
Patients with OMG may display hypometric large saccades and hypermetric small saccades, which may be a result of CNS adaptation to EOM weakness.[13] These patients may also show intrasaccadic fatigue, a decline in the saccadic velocity during a long saccade.[14] In rare cases isolated nystagmus may be observed, which probably represents a gaze paretic nystagmus.
In most patients with OMG pupillary examination is, usually, normal, and this serves as a useful tool to distinguish OMG from conditions such as pupil involving third nerve palsy, Horner's syndrome and botulism. Pupillary abnormalities have been described in OMG[15] and pupillographic studies have confirmed reduced velocities of pupillary constriction.[16] There are also reports of fatigue of accommodation with improvement after edrophonium.[17]
Diagnosing Ocular Myasthenia Gravis
Tests in the clinic
Sleep test
This test measures the improvement in manifestations of OMG after a period of rest. The patient is asked to sleep or rest with his or her eyes closed for a period of about 30 min. Prior to the test, the patient is examined, and the ocular motility deficits and/or ptosis present are measured. The diagnosis of myasthenia can be confirmed by observing resolution of ptosis or ophthalmoparesis immediately after a 30-min period of sleep. Reappearance of the myasthenic signs over the next 30 s to 5 min adds further confirmation.
Ice test
The Ice test is a simple, but effective clinical test that can be used to confirm the diagnosis of MG. An icepack is placed over the patient's closed eyelids for a period of 2 min (for ptosis) to 5 min (for ophthalmoparesis) [Fig. 1].[5] The ocular motility deficits and ptosis must be measured before and after the test. Although there are no strict guidelines regarding the interpretation of this test,[1] it is, usually, considered positive when the upper eyelid elevates by at least 2 mm following ice application.[5] Cooling may reduce anticholinesterase (AChE) activity, which increases the amount of available ACh at the neuro-muscular junction.[14] There is thus an increase in the efficiency of ACh in eliciting depolarization at the motor end plate.[18]
Figure 1.
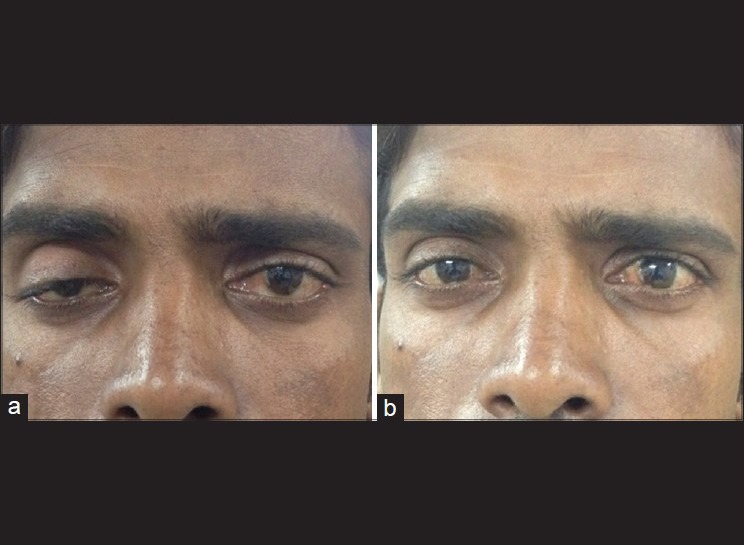
Ice test. A 28-year-old male who presented with ptosis of the right eye with a history of variability in the amount of ptosis. Before the ice test (a) and immediately after the ice test (b). Note the improvement in ptosis, thus helping in diagnosing ocular myasthenia gravis
If the ice pack is used for over 2 min, the test becomes uncomfortable for the patient and reduction of muscle fiber temperature below 22°C will reduce the contractile force of the muscle itself and create potential false-negatives.[19] Resolution of ptosis has been reported in over 90% of OMG patients after the ice test.[20] According to one study, the sensitivity and specificity of this test was 76.9% and 98.3%, respectively.[21] A combination of sleep test and the ice test produces a larger change in lid position than rest alone and is quite sensitive for MG.[20,22]
Pharmacological Testing and Laboratory Investigations
Edrophonium test
Edrophonium is a rapidly acting and quickly hydrolyzed AChE drug. It prevents the breakdown of ACh by competitively inhibiting the acetylcholinesterase at the NMJ. This results in an increase in synaptic ACh, providing maximum saturation of the limited available receptor population in myasthenia.[5] Intravenous (IV) edrophonium ameliorates the signs and symptoms of OMG. Edrophonium is available as a 10 mg/mL parenteral formulation and is given intravenously. Onset of action begins within 30-60 s after injection, and the effects resolve within 5-10 min. The dose of edrophonium chloride is 0.10 mg/kg in children and 10 mg or less in adults and 0.05-1.0 mg (subcutaneously) in infants. Ptosis and ocular motility deficits should be measured and documented photographically before administration. Initially, 1-2 mg IV test dose is given and if there is no idiosyncratic reaction, 3-4 mg is injected after 2 min. If eyelid position, ocular alignment, or motility do not improve within 1-min, the remaining 8-9 mg is injected, preferably in 2-4 mg increments, waiting 45-60 s between increments.[5] However, doses larger than 5 mg often do not, usually, produce a positive result if lower doses are ineffective. Paradoxical worsening of ocular motility has been reported in patients with MG (up to 25%) due to a depolarizing block caused by excess of ACh.[2,5] The edrophonium test is best-evaluated by observing for increased strength of a single muscle, such as the levator, rather than changes in relative strength of multiple muscles, as with ocular alignment.[23]
Edrophonium can cause various side effects due to the increased muscarinic activity–these include lacrimation, salivation, sweating, and abdominal cramping. Serious side effects include bradycardia, bronchospasm hypotension, and syncope; atropine should be readily available during testing and the testing should only be carried out with continuous blood pressure, pulse, and electrocardiographic monitoring. The test is relatively contraindicated in patients with bronchial asthma and cardiac diseases. The sensitivity of the test is 95% in generalized MG and about 86% for OMG.[11,24]
Neostigmine test
Neostigmine is a longer acting AChE being increasingly used as an alternative to edrophonium for diagnostic testing. The peak effect is achieved at about 30 min after an intramuscular injection, although the response may be seen within 15 min. The duration of effect may last for several hours. The usual dose in adults is 1.5 mg, and the drug is administered intramuscularly, in the deltoid muscle. The advantages of Neostigmine over edrophonium include it is a longer duration of action, which makes it more suited to detailed and prolonged examination of ocular motility, diplopia testing, etc.[5]
Immunologic testing
Elevated AChR-Ab titers confirm the diagnosis of MG. However, a normal titer does not exclude the disease. The presence of an elevated AChR-Ab titer helps to distinguish acquired MG from a congenital myasthenic syndrome since the latter is persistently seronegative. AChRAb testing also provides a baseline for future comparisons and response to immunomodulatory treatment.[5] The absolute antibody titer correlates poorly with the severity of the disease from the patient to patient, but in individual patients, changes in disease severity do tend to be associated with changes in antibody titer.[5]
Antiacetylcholine receptor antibodies have been demonstrated in as many as 80-99% of patients with generalized myasthenia and 30-77% of patients with OMG.[5,23,25] About 20% of generalized MG patients are seronegative for ACh receptor antibodies. Of these patients, 30% will have autoantibodies against muscle-specific kinase (anti MuSKAb) expressed on skeletal muscle.[6] Patients who are negative for both AChR and MuSKAbs are classified as “seronegative” MG.[26] Almost all myasthenic patients with thymoma have antibodies against skeletal muscle, these have also been found in up to 30% of patients without a thymoma.[27,28]
Electrophysiologic testing
In the diagnosis of MG, both repetitive nerve stimulation studies (RNS) and single-fiber electromyography (SFEMG) are recommended.[29]
Repetitive nerve stimulation studies
The nerve to be studied is electrically stimulated 6-10 times at 2 or 3 Hz (slow rate) with a supramaximal stimulus and the compound muscle action potential (CMAP) is recorded with surface electrodes. In MG, as the number of individual muscle fiber action potentials reduce, the CMAP also reduces in both amplitude and area with a resulting decremental response.[30] In MG, a characteristic decrement (>10%) in muscle action potential amplitude is typically seen by the fourth or fifth response in a series of low-frequency RNS, whereas the amplitude remains the same in normal individuals.[31] This decremental response is seen in only 33% of patients with purely OMG.[32] A decremental response to RNS is not specific and may also be seen in Lambert-Eaton myasthenic syndrome, motor neuron diseases, and myopathies.
Single-fiber electromyography
Single-fiber electromyography is the most sensitive diagnostic test for detecting abnormal neuro-muscular transmission. In SFEMG, a specialized concentric needle records individual muscle fiber action potentials generated by the same motor neuron with a 25 μm diameter recording surface and a 500 Hz high-frequency filter.
When potentials elicited by nerve stimulation are recorded with an SFEMG electrode, the latency from stimulus to response varies. This variation is the neuro-muscular jitter, most of which is produced by fluctuations in the time for end plate potentials at the NMJ to reach the AP threshold.[33] SFEMG has a sensitivity of 85-100% for OMG when used on the frontalis or orbicularis oculi muscle[34,35] and a sensitivity of 91–100% in generalized MG.
Imaging studies
As many as 70% of patients with myasthenia may have thymic hyperplasia, and 10-15% may harbor a thymoma.[36] Computerized tomography (CT) of the chest is advised in patients diagnosed with myasthenia to detect this association.
Others
Additional testing for thyroid dysfunction may also be considered in patients with myasthenia, since about 4-5% of patients with MG may have concurrent autoimmune thyroid disease.[37]
Treatment of myasthenia gravis
Treatment is chiefly medical and aims at improving muscle weakness (thereby alleviating symptoms of diplopia and ptosis), achieving disease remission, minimizing drug-induced side effects, and slowing or preventing progression to generalized MG.[38]
Acetycholinesterase inhibitors
Acetylcholinesterase inhibitors can serve to increase the duration of action of the neurotransmitter. These provide symptomatic improvement,[12] without modifying the long-term immunologic disease activity. Pyridostigmine bromide is the prototype – its onset of action ranges from 30 to 45 min after ingestion and lasts for up to 6 h. In tablet form (30 mg), it is, usually, administered two to four times a day, up to a maximum of 1500 mg/day. An increased incidence of hypotension and bradycardia has been reported when co-administered with beta-blockers or opiates.[6] Other contraindications include asthma and cardiac arrhythmias. While it is the first-line drug to improve symptoms and is generally well-tolerated, only about half of patients with ocular disease show an adequate response to pyridostigmine, with ptosis responding better than diplopia.[39,40] Neostigmine is an alternative drug of the same class but has a less favorable side effect profile. ACh receptor inhibitors, in general, produce side effects such as diarrhea, nausea, vomiting, and abdominal cramping.
Acetylcholinesterase inhibitors do not influence the natural course of the disease (36% of patients with OMG, who were treated with pyridostigmine and not steroids developed GMG within 2 years).[41] Immunosuppressive therapy may be considered in all patients with myasthenia irrespective of whether or not serum AChR-Abs are detectable[5] and are indicated when AChE inhibitors are intolerable or ineffective.[12]
Corticosteroids
Corticosteroids are the most widely used immune modulating agents in patients with MG. They chiefly act through their anti-inflammatory properties, additionally causing a reduction in cytokine expression, lymphocyte differentiation and proliferation, and also increase muscle AChR synthesis.[12]
Treatment, usually, starts at a dose of 20 mg per day of orally administered prednisolone. Dose optimization requires up-titration over several weeks, generally to a level of 1 mg/kg/day. This may be maintained for 6-12 weeks and then tapered slowly over months.[6,12] Initiating oral prednisolone therapy with high doses can result in a worsening of symptoms and even lead to myasthenic crisis in up to 15% of patients,[42] hence. patients on corticosteroids should be evaluated monthly.
Corticosteroids produce favorable response in OMG in 66-85% of patients.[41,43] However, patients rarely go into complete remission with oral corticosteroids alone. Steroids in the course of early OMG may reduce the likelihood of progression to GMG (according to some studies, from a rate of 36-83% in patients without to 7-17% of patients treated with prednisolone).[43,44,45] Prednisone may also delay generalization. Without prednisone, GMG develops in 50% of OMG patients, typically within 1-year.[46]
Common side effects of corticosteroid therapy include acne, obesity, hypertension, diabetes, osteoporosis, and steroid-induced myopathy. The risk of opportunistic infections is omnipresent, and tuberculosis needs to be ruled out before initiating therapy.
Immunosuppressive therapy
Azathioprine is a purine antagonist which inhibits DNA and RNA synthesis in fast dividing T- and B-cells. Azathioprine can be used both as monotherapy (for example, in steroid-resistant patients) as well as in conjunction with oral corticosteroids. The latter modality has been shown in randomized control trials to have a steroid-sparing effect and is associated with fewer failures and longer remissions.[47] The clinical response to azathioprine alone is, usually, delayed (>6 months), and is accompanied by a progressive fall in AChR-Ab titers. The full effect is seen after 2–3 years of continuous administration.[5,48]
In view of potential hepatotoxicity and bone marrow toxicity, blood counts and liver function tests should be done bi-weekly for the first 2 months after initiating treatment and monthly thereafter. Treatment should be discontinued if the white blood cells count falls below 3000/mm3. Dose reduction may be considered if it is below 3500/mm3.[12] Azathioprine is potentially teratogenic.[34]
Cyclosporine A inhibits calcineurin, which decreases the antigen-stimulated interleukin-2 production in T-cells.
It is a third-line drug and may be of particular use in patients who are dependent or intolerant of steroids and/or azathioprine. Treatment is initiated at a dose of 5 mg/kg/day in two to three divided doses and is subsequently modified is on the basis of serum creatinine levels and clinical response. Improvement in muscle strength and a reduction in AChR-Abs titers have been reported with cyclosporine.[49]
Less serious side effects include hirsutism, gingival hyperplasia, gastrointestinal disturbance, flu-like syndrome, myalgia, and hypertension. Renal failure can be fatal.[12] It should be avoided in patients using angiotensin-converting enzyme inhibitors or potassium-sparing diuretics due to the risk of hyperkalemia.[50] Some authors believe side effects make the risks of its use in OMG outweigh the benefits.[6]
Mycophenolate mofetil (MMF) selectively inhibits T- and B-lymphocyte proliferation by blocking purine synthesis exclusively in lymphocytes. It is a relatively new drug in the treatment of MG and has been used both as a steroid-sparing agent as well as monotherapy.[3]
Mycophenolate mofetil is administered orally in a dose between 1000 and 1500 mg twice a day. Clinical response is, usually, observed only 2 months after initiation of treatment. Treatment with MMF may reduce the rate of generalization of ocular disease. A dose of 1.0 g/day was safe and tolerable as a long-term immunosuppressant for OMG.[51] MMF with prednisone has not been found to be superior to prednisone alone in mild to moderate seropositive GMG patients.[52,53]
Rarely, opportunistic infections, myelosuppression, or hepatotoxicity may occur.[12]
Plasmapheresis has a role in the short-term management of acute and severe muscular weakness. The patient's plasma is separated from whole blood and replaced with saline, albumin, or plasma protein fraction, thereby reducing serum AChR-Ab levels.[3,34] Repeated exchanges (five over 5-10 days) are required to reduce AChR-Ab titers and the total IgG level.[5]
The role of plasmapheresis is limited to management of myasthenic exacerbations or crises. It can be used preoperatively to prepare patients for thymectomy or other surgical procedures. Some patients develop exacerbations of weakness when steroid therapy is initiated; plasmapheresis may be of value in these patients.
Intravenous immunoglobulin (IVIg) accelerates the catabolism of IgG in addition to suppressing antibody production and inhibiting complement activation and Fc receptor function. Its role is limited to the perioperative management of patients and treatment of myasthenic crisis.[2]
Thymectomy has for long been known to have a benefit in patients with MG.[54,55] However it may not be effective in pure OMG; on the contrary, it may have the same outcome as nonoperative management.[56,57] Thymectomy is, usually, recommended in patients with documented thymic enlargement on CT scans, in patients early in the course of their disease and those younger than 60 years of age.[3] Response to thymectomy may be delayed for several years.
Available data show that the thymectomy leads to clinical improvement in 70-80% of the patients, with approximately 35% reaching complete remission.[58,59,60] It has also been suggested that patients who underwent thymectomy, despite having no signs of thymoma on computed tomography, were less likely to progress to GMG and more likely to experience full remissions.[61,62]
Supportive measures include the use of prisms or occlusion therapy for those with persistent diplopia and crutch glasses for severe ptosis. Strabismus or lid surgery may be offered to selected patients who have stable findings for a period of at least 6 months.
Pediatric Myasthenia
MG in children can be classified based on the age at onset and disease pathogenesis – transient neonatal myasthenia, congenital myasthenia, and juvenile autoimmune myasthenia.[63,64] Neonatal transient myasthenia occurs due to transplacental transfer of maternal AChR-Abs. Infants may show ocular and systemic features; most recover spontaneously and usually need only supportive care.
Congenital myasthenia syndromes refer to a subset of children with myasthenia where the disease is caused by structural or functional, presynaptic or postsynaptic abnormalities. These may result in inadequate release of ACh or AChR dysfunction, such as slow channel or prolonged open channel syndrome. Since the primary pathology is not immune mediated, immunosuppressive drugs have little or no effect in these cases. AChE agents may be useful in some forms. The treatment in these cases is mainly supportive.[65,66] Juvenile MG is caused by AChR-Abs and like the adult form, may be ocular or systemic. The rates of generalization of ocular to systemic myasthenia are lower than in adults.[66] Treatment with AChE agents, steroids, and thymectomy has been described.[67] Thymectomy may be useful in juvenile OMG and may play a role in the prevention of generalization.[68]
Other concerns in the pediatric age group include variations in serological testing, technical difficulty in performing tests such as SFEMG and amblyopia evaluation – which are, unfortunately, often overlooked in these patients.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Conti-Fine BM, Milani M, Kaminski HJ. Myasthenia gravis: Past, present, and future. J Clin Invest. 2006;116:2843–54. doi: 10.1172/JCI29894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Brien MD. The miracle at St Alfege's: Seventy years on. J R Soc Med. 2007;100:257. doi: 10.1258/jrsm.100.6.257-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.López-Cano M, Ponseti-Bosch JM, Espin-Basany E, Sánchez-García JL, Armengol-Carrasco M. Clinical and pathologic predictors of outcome in thymoma-associated myasthenia gravis. Ann Thorac Surg. 2003;76:1643–9. doi: 10.1016/s0003-4975(03)01139-1. [DOI] [PubMed] [Google Scholar]
- 4.Singhal BS, Bhatia NS, Umesh T, Menon S. Myasthenia gravis: A study from India. Neurol India. 2008;56:352–5. doi: 10.4103/0028-3886.43455. [DOI] [PubMed] [Google Scholar]
- 5.Grigg J. Extraocular muscles: Relationship of structure and function to disease. Aust N Z J Ophthalmol. 1999;27:369–70. doi: 10.1046/j.1440-1606.1999.00253.x. [DOI] [PubMed] [Google Scholar]
- 6.Sommer N, Melms A, Weller M, Dichgans J. Ocular myasthenia gravis. A critical review of clinical and pathophysiological aspects. Doc Ophthalmol. 1993;84:309–33. doi: 10.1007/BF01215447. [DOI] [PubMed] [Google Scholar]
- 7.Antonio-Santos AA, Eggenberger ER. Medical treatment options for ocular myasthenia gravis. Curr Opin Ophthalmol. 2008;19:468–78. doi: 10.1097/ICU.0b013e328310da18. [DOI] [PubMed] [Google Scholar]
- 8.Grob D, Arsura EL, Brunner NG, Namba T. The course of myasthenia gravis and therapies affecting outcome. Ann N Y Acad Sci. 1987;505:472–99. doi: 10.1111/j.1749-6632.1987.tb51317.x. [DOI] [PubMed] [Google Scholar]
- 9.Oosterhuis HJ. Observations of the natural history of myasthenia gravis and the effect of thymectomy. Ann N Y Acad Sci. 1981;377:678–90. doi: 10.1111/j.1749-6632.1981.tb33766.x. [DOI] [PubMed] [Google Scholar]
- 10.Keane JR. Vertical diplopia. Semin Neurol. 1986;6:147–54. doi: 10.1055/s-2008-1041458. [DOI] [PubMed] [Google Scholar]
- 11.Kaminski HJ, Maas E, Spiegel P, Ruff RL. Why are eye muscles frequently involved in myasthenia gravis? Neurology. 1990;40:1663–9. doi: 10.1212/wnl.40.11.1663. [DOI] [PubMed] [Google Scholar]
- 12.Finelli PF, Hoyt WF. Myasthenic abduction mystagmus in a patient with hyperthyroidism. Neurology. 1976;26(6 PT 1):589–90. doi: 10.1212/wnl.26.6.589. [DOI] [PubMed] [Google Scholar]
- 13.Yee RD, Cogan DG, Zee DS, Baloh RW, Honrubia V. Rapid eye movements in myasthenia gravis. II. Electro-oculographic analysis. Arch Ophthalmol. 1976;94:1465–72. doi: 10.1001/archopht.1976.03910040299001. [DOI] [PubMed] [Google Scholar]
- 14.Sethi KD, Rivner MH, Swift TR. Ice pack test for myasthenia gravis. Neurology. 1987;37:1383–5. doi: 10.1212/wnl.37.8.1383. [DOI] [PubMed] [Google Scholar]
- 15.Lepore FE, Sanborn GE, Slevin JT. Pupillary dysfunction in myasthenia gravis. Ann Neurol. 1979;6:29–33. doi: 10.1002/ana.410060107. [DOI] [PubMed] [Google Scholar]
- 16.Yamazaki A, Ishikawa S. Abnormal pupillary responses in myasthenia gravis. A pupillographic study. Br J Ophthalmol. 1976;60:575–80. doi: 10.1136/bjo.60.8.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manson N, Stern G. Defects of near vision in myasthenia gravis. Lancet. 1965;1:935–7. doi: 10.1016/s0140-6736(65)91255-9. [DOI] [PubMed] [Google Scholar]
- 18.Hubbard JI, Jones SF, Landau EM. The effect of temperature change upon transmitter release, facilitation and post-tetanic potentiation. J Physiol. 1971;216:591–609. doi: 10.1113/jphysiol.1971.sp009542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kearsey C, Fernando P, D’Costa D, Ferdinand P. The use of the ice pack test in myasthenia gravis. JRSM Short Rep. 2010;1:14. doi: 10.1258/shorts.2009.090037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kubis KC, Danesh-Meyer HV, Savino PJ, Sergott RC. The ice test versus the rest test in myasthenia gravis. Ophthalmology. 2000;107:1995–8. doi: 10.1016/s0161-6420(00)00458-9. [DOI] [PubMed] [Google Scholar]
- 21.Chatzistefanou KI, Kouris T, Iliakis E, Piaditis G, Tagaris G, Katsikeris N, et al. The ice pack test in the differential diagnosis of myasthenic diplopia. Ophthalmology. 2009;116:2236–43. doi: 10.1016/j.ophtha.2009.04.039. [DOI] [PubMed] [Google Scholar]
- 22.Ellis FD, Hoyt CS, Ellis FJ, Jeffery AR, Sondhi N. Extraocular muscle responses to orbital cooling (ice test) for ocular myasthenia gravis diagnosis. J AAPOS. 2000;4:271–81. doi: 10.1067/mpa.2000.106204. [DOI] [PubMed] [Google Scholar]
- 23.Barton JJ, Fouladvand M. Ocular aspects of myasthenia gravis. Semin Neurol. 2000;20:7–20. doi: 10.1055/s-2000-6829. [DOI] [PubMed] [Google Scholar]
- 24.Phillips LH, 2nd, Melnick PA. Diagnosis of myasthenia gravis in the 1990s. Semin Neurol. 1990;10:62–9. doi: 10.1055/s-2008-1041255. [DOI] [PubMed] [Google Scholar]
- 25.Kelly JJ, Jr, Daube JR, Lennon VA, Howard FM, Jr, Younge BR. The laboratory diagnosis of mild myasthenia gravis. Ann Neurol. 1982;12:238–42. doi: 10.1002/ana.410120303. [DOI] [PubMed] [Google Scholar]
- 26.Kim JY, Park KD, Richman DP. Treatment of myasthenia gravis based on its immunopathogenesis. J Clin Neurol. 2011;7:173–83. doi: 10.3988/jcn.2011.7.4.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nastuk WL, Kessler HJ, Grynbaum A, Smith M, Herrmann C., Jr Immunological changes following thymectomy in myasthenia gravis. Arch Neurol. 1966;15:1–12. doi: 10.1001/archneur.1966.00470130005001. [DOI] [PubMed] [Google Scholar]
- 28.Strauss AJ, Kemp PG, Douglas SD. Myasthenia gravis. Lancet. 1966;1:772–3. doi: 10.1016/s0140-6736(66)90931-7. [DOI] [PubMed] [Google Scholar]
- 29.Katzberg HD, Bril V. A comparison of electrodiagnostic tests in ocular myasthenia gravis. J Clin Neuromuscul Dis. 2005;6:109–13. doi: 10.1097/01.cnd.0000155026.66153.f0. [DOI] [PubMed] [Google Scholar]
- 30.Juel VC, Massey JM. Myasthenia gravis. Orphanet J Rare Dis. 2007;2:44. doi: 10.1186/1750-1172-2-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ozdemir C, Young RR. The results to be expected from electrical testing in the diagnosis of myasthenia gravis. Ann N Y Acad Sci. 1976;274:203–22. doi: 10.1111/j.1749-6632.1976.tb47686.x. [DOI] [PubMed] [Google Scholar]
- 32.Costa J, Evangelista T, Conceição I, de Carvalho M. Repetitive nerve stimulation in myasthenia gravis – relative sensitivity of different muscles. Clin Neurophysiol. 2004;115:2776–82. doi: 10.1016/j.clinph.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 33.Rodríguez J, Dimitrova NA, Dimitrov GV, Gila L. Shape variability of potentials recorded by a single-fiber electrode and its effect on jitter estimation. Ann Biomed Eng. 2011;39:812–23. doi: 10.1007/s10439-010-0207-z. [DOI] [PubMed] [Google Scholar]
- 34.Padua L, Stalberg E, LoMonaco M, Evoli A, Batocchi A, Tonali P. SFEMG in ocular myasthenia gravis diagnosis. Clin Neurophysiol. 2000;111:1203–7. doi: 10.1016/s1388-2457(00)00307-2. [DOI] [PubMed] [Google Scholar]
- 35.Sanders DB, Howard JF. Disorders of neuromuscular transmission. In: Bradley WG, Daroff RB, Fenichel GM, Marsden CD, editors. Neurology in Clinical Practice. 1st ed. Boston: Butterworth-Heine-Mann; 1991. pp. 1819–42. [Google Scholar]
- 36.Osserman KE, Tsairis P, Weiner LB. Myasthenia gravis and thyroid disease: Clinical and immunologic correlation. J Mt Sinai Hosp N Y. 1967;34:469–83. [PubMed] [Google Scholar]
- 37.Chen CS, Lee AW, Miller NR, Lee AG. Double vision in a patient with thyroid disease: What's the big deal? Surv Ophthalmol. 2007;52:434–9. doi: 10.1016/j.survophthal.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 38.Gandhi RA, Nair AG. Ocular myasthenia gravis. In: Chaudhari Z, Vanathi M, editors. Postgraduate Ophthalmology. New Delhi: Jaypee Brothers Medical Publishers; 2011. pp. 1510–21. [Google Scholar]
- 39.Schlezinger NS, Fairfax WA. Evaluation of ocular signs and symptoms in myasthenia gravis. Arch Ophthalmol. 1959;62:985–90. doi: 10.1001/archopht.1959.04220060057010. [DOI] [PubMed] [Google Scholar]
- 40.Kupersmith MJ, Latkany R, Homel P. Development of generalized disease at 2 years in patients with ocular myasthenia gravis. Arch Neurol. 2003;60:243–8. doi: 10.1001/archneur.60.2.243. [DOI] [PubMed] [Google Scholar]
- 41.Kupersmith MJ, Moster M, Bhuiyan S, Warren F, Weinberg H. Beneficial effects of corticosteroids on ocular myasthenia gravis. Arch Neurol. 1996;53:802–4. doi: 10.1001/archneur.1996.00550080128020. [DOI] [PubMed] [Google Scholar]
- 42.Pascuzzi RM, Coslett HB, Johns TR. Long-term corticosteroid treatment of myasthenia gravis: Report of 116 patients. Ann Neurol. 1984;15:291–8. doi: 10.1002/ana.410150316. [DOI] [PubMed] [Google Scholar]
- 43.Sommer N, Sigg B, Melms A, Weller M, Schepelmann K, Herzau V, et al. Ocular myasthenia gravis: Response to long-term immunosuppressive treatment. J Neurol Neurosurg Psychiatry. 1997;62:156–62. doi: 10.1136/jnnp.62.2.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Monsul NT, Patwa HS, Knorr AM, Lesser RL, Goldstein JM. The effect of prednisone on the progression from ocular to generalized myasthenia gravis. J Neurol Sci. 2004;217:131–3. doi: 10.1016/j.jns.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 45.Mee J, Paine M, Byrne E, King J, Reardon K, O’Day J. Immunotherapy of ocular myasthenia gravis reduces conversion to generalized myasthenia gravis. J Neuroophthalmol. 2003;23:251–5. doi: 10.1097/00041327-200312000-00002. [DOI] [PubMed] [Google Scholar]
- 46.Kupersmith MJ. Ocular myasthenia gravis: Treatment successes and failures in patients with long-term follow-up. J Neurol. 2009;256:1314–20. doi: 10.1007/s00415-009-5120-8. [DOI] [PubMed] [Google Scholar]
- 47.Palace J, Newsom-Davis J, Lecky B. A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Myasthenia Gravis Study Group. Neurology. 1998;50:1778–83. doi: 10.1212/wnl.50.6.1778. [DOI] [PubMed] [Google Scholar]
- 48.Beekman R, Kuks JB, Oosterhuis HJ. Myasthenia gravis: Diagnosis and follow-up of 100 consecutive patients. J Neurol. 1997;244:112–8. doi: 10.1007/s004150050059. [DOI] [PubMed] [Google Scholar]
- 49.Tindall RS, Phillips JT, Rollins JA, Wells L, Hall K. A clinical therapeutic trial of cyclosporine in myasthenia gravis. Ann N Y Acad Sci. 1993;681:539–51. doi: 10.1111/j.1749-6632.1993.tb22937.x. [DOI] [PubMed] [Google Scholar]
- 50.Tindall RS, Rollins JA, Phillips JT, Greenlee RG, Wells L, Belendiuk G. Preliminary results of a double-blind, randomized, placebo-controlled trial of cyclosporine in myasthenia gravis. N Engl J Med. 1987;316:719–24. doi: 10.1056/NEJM198703193161205. [DOI] [PubMed] [Google Scholar]
- 51.Chan JW. Mycophenolate mofetil for ocular myasthenia. J Neurol. 2008;255:510–3. doi: 10.1007/s00415-008-0718-9. [DOI] [PubMed] [Google Scholar]
- 52.Muscle Study Group. A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis. Neurology. 2008;71:394–9. doi: 10.1212/01.wnl.0000312373.67493.7f. [DOI] [PubMed] [Google Scholar]
- 53.Sanders DB, Hart IK, Mantegazza R, Shukla SS, Siddiqi ZA, De Baets MH, et al. An international, phase III, randomized trial of mycophenolate mofetil in myasthenia gravis. Neurology. 2008;71:400–6. doi: 10.1212/01.wnl.0000312374.95186.cc. [DOI] [PubMed] [Google Scholar]
- 54.Schumacher CH, Roth P. ThymektomiebeieinemFall von Morbus Basedowimit Myasthenie. Mitt Grenzgeb Med Chirurgia. 1912;25:746–65. [Google Scholar]
- 55.Blalock A, Mason MF, Morgan HJ, Riven SS. Myasthenia gravis and tumors of the thymic region: Report of a case in which the tumor was removed. Ann Surg. 1939;110:544–61. doi: 10.1097/00000658-193910000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Evoli A, Batocchi AP, Provenzano C, Ricci E, Tonali P. Thymectomy in the treatment of myasthenia gravis: Report of 247 patients. J Neurol. 1988;235:272–6. doi: 10.1007/BF00314173. [DOI] [PubMed] [Google Scholar]
- 57.Hatton PD, Diehl JT, Daly BD, Rheinlander HF, Johnson H, Schrader JB, et al. Transsternal radical thymectomy for myasthenia gravis: A 15-year review. Ann Thorac Surg. 1989;47:838–40. doi: 10.1016/0003-4975(89)90015-5. [DOI] [PubMed] [Google Scholar]
- 58.Buckingham JM, Howard FM, Jr, Bernatz PE, Payne WS, Harrison EG, Jr, O’Brien PC, et al. The value of thymectomy in myasthenia gravis: A computer-assisted matched study. Ann Surg. 1976;184:453–8. doi: 10.1097/00000658-197610000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rodriguez M, Gomez MR, Howard FM, Jr, Taylor WF. Myasthenia gravis in children: Long-term follow-up. Ann Neurol. 1983;13:504–10. doi: 10.1002/ana.410130506. [DOI] [PubMed] [Google Scholar]
- 60.Olanow CW, Wechsler AS, Roses AD. A prospective study of thymectomy and serum acetylcholine receptor antibodies in myasthenia gravis. Ann Surg. 1982;196:113–21. doi: 10.1097/00000658-198208000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roberts PF, Venuta F, Rendina E, De Giacomo T, Coloni GF, Follette DM, et al. Thymectomy in the treatment of ocular myasthenia gravis. J Thorac Cardiovasc Surg. 2001;122:562–8. doi: 10.1067/mtc.2001.116191. [DOI] [PubMed] [Google Scholar]
- 62.Schumm F, Wiethölter H, Fateh-Moghadam A, Dichgans J. Thymectomy in myasthenia with pure ocular symptoms. J Neurol Neurosurg Psychiatry. 1985;48:332–7. doi: 10.1136/jnnp.48.4.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brodsky MC, Baker RS, Hamed LM. New York: Springer; 1996. Pediatric Neuro-ophthalmology; pp. 251–301. [Google Scholar]
- 64.Mullaney P, Vajsar J, Smith R, Buncic JR. The natural history and ophthalmic involvement in childhood myasthenia gravis at the hospital for sick children. Ophthalmology. 2000;107:504–10. doi: 10.1016/s0161-6420(99)00138-4. [DOI] [PubMed] [Google Scholar]
- 65.Middleton LT. Congenital myasthenic syndromes 34th ENMC International Workshop, 10-11 June 1995. Neuromuscul Disord. 1996;6:133–6. doi: 10.1016/0960-8966(95)00037-2. [DOI] [PubMed] [Google Scholar]
- 66.Ortiz S, Borchert M. Long-term outcomes of pediatric ocular myasthenia gravis. Ophthalmology. 2008;115:1245–48.e1. doi: 10.1016/j.ophtha.2007.10.022. [DOI] [PubMed] [Google Scholar]
- 67.Pineles SL, Avery RA, Moss HE, Finkel R, Blinman T, Kaiser L, et al. Visual and systemic outcomes in pediatric ocular myasthenia gravis. Am J Ophthalmol. 2010;150:453–9.e3. doi: 10.1016/j.ajo.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 68.Gadient P, Bolton J, Puri V. Juvenile myasthenia gravis: Three case reports and a literature review. J Child Neurol. 2009;24:584–90. doi: 10.1177/0883073808325651. [DOI] [PubMed] [Google Scholar]