

Abstract
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a genetic disease characterized by myocyte loss and fibro-fatty tissue replacement. Diagnosis of ARVC remains a clinical challenge mainly at its early stages and in patients with minimal echocardiographic right ventricular (RV) abnormalities. ARVC shares some common features with other cardiac diseases, such as RV outflow ventricular tachycardia, Brugada syndrome, and myocarditis, due to arrhythmic expressivity and biventricular involvement. The identification of ARVC can be often challenging, because of the heterogeneous clinical presentation, highly variable intra- and inter-family expressivity and incomplete penetrance. This genotype-phenotype “plasticity” is largely unexplained. A familial history of ARVC is present in 30% to 50% of cases, and the disease is considered a genetic cardiomyopathy, usually inherited in an autosomal dominant pattern with variable penetrance and expressivity; in addition, autosomal recessive forms have been reported (Naxos disease and Carvajal syndrome). Diagnosis of ARVC relays on a scoring system, with major or minor criteria on the Revised Task Force Criteria. Implantable cardioverter defibrillators (ICDs) are increasingly utilized in patients with ARVC who have survived sudden death (SD) (secondary prevention). However, there are few data available to help identifying ARVC patients in whom the prophylactic implantation of an ICD is truly warranted. Prevention of SD is the primary goal of management. Pharmacologic treatment of arrhythmias, catheter ablation of ventricular tachycardia, and ICD are the mainstay of treatment of ARVC.
Keywords: Arrhythmogenic right ventricular cardiomyopathy, Sudden cardiac death, Risk stratification, Genetic, Implantable cardioverter-defibrillator
Core tip: This manuscript constitutes an update on arrhythmogenic right ventricular cardiomyopathy (ARVC). Recently, molecular genetic studies have provided significant advances in the understanding the pathogenesis of ARVC. However, criteria on treatment with Implantable cardioverter defibrillators are still lacking. We believe that this topic can provide a useful instrument to physicians and guide them in their clinical practice.
INTRODUCTION: DEFINITION OF DISEASE, EPIDEMIOLOGY, AND CLINICAL FEATURES
Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC) is an inherited cardiomyopathy (CMP) characterized by fibro-fatty replacement of the right ventricular (RV) myocardium (Figures 1 and 2) that predisposes patients to life-threatening ventricular arrhythmias and slowly progressive ventricular dysfunction[1-4]. Biventricular and left-dominant forms of the disease are increasingly recognized[5-7].
Figure 1.
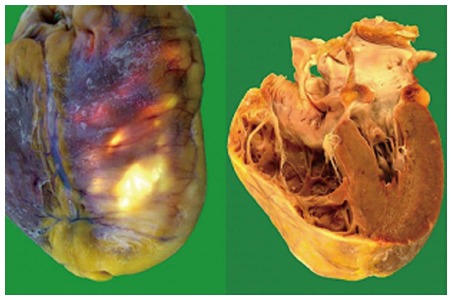
Gross anatomic specimens in a patient affected by arrhythmogenic right ventricular cardiomyopathy who died suddenly. Severe right ventricular enlargement and wall atrophy and fatty replacement are evident.
Figure 2.
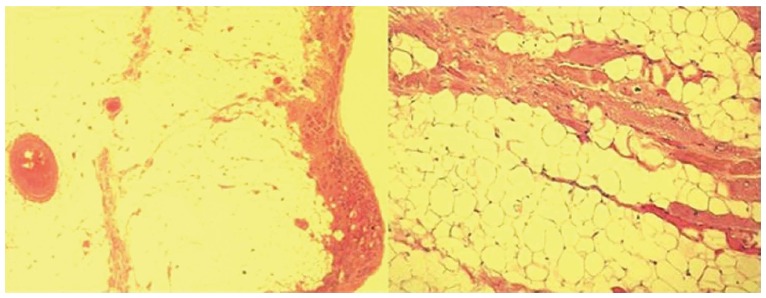
Histologic specimens of a case with arrhythmogenic right ventricular cardiomyopathy that show severe right ventricular fibro-fatty replacement and loss and degeneration of myocytes (Hematoxylin Eosin × 2.5, × 10).
The estimated prevalence of ARVC in the general population ranges from 1 in 2000 to 1 in 5000 individuals; men are more frequently affected than women, with an approximate ratio of 3:1[8]. ARVC is a leading cause of sudden cardiac death (SCD) in young people and in athletes, accounting for up to 10% of deaths from undiagnosed cardiac disease in patients less than 65 years old[2,9-11]. In particular, in young adults and athletes, ARVC has been reported as the second most frequent cause of SCD[11]. The disease expression is variable and the penetrance (the proportion of carriers manifesting the disease) appears age-related. According to Dalal et al[12], the median age at onset of the disease is 29 years, whereas it rarely manifests before the age of 12 or after the age of 60 years. The most common presenting symptoms are palpitations and syncope, found in 27% and 26% of patients, respectively. Importantly, life-threatening ventricular arrhythmias and SCD can be the first presentation of the disease[2,9-11].
ARVC has frequently a progressive course. In the early stage of the disease, structural changes may be absent or subtle and confined to a localized region of the RV. The 3 most common locations of the disease are: the anterior infundibulum, RV apex and subtricuspid infero-basal aspect of the RV, comprising the so-called “triangle of dysplasia”, considered a hallmark of ARVC[1]. ARVC leads to RV dilatation or aneurysms. With disease progression, further involvement of the RV free wall, and left ventricular (LV) involvement can occur[13-15].
The natural history of ARVC, in its classic “right dominant” form, has been classified into 4 distinct phases with progressive development of symptoms and structural abnormalities[3]: (1) concealed phase: a subclinical asymptomatic phase with mild or absence of identifiable structural RV abnormalities. SCD may still occur in this stage of disease[2,3,10,11]; (2) overt electrical disorder: with palpitations, syncope and typically with symptomatic ventricular arrhythmias of RV origin usually triggered by effort. Arrhythmias may vary from premature ventricular beats, to non-sustained ventricular tachycardia with left bundle branch block (LBBB) morphology up to ventricular fibrillation leading to cardiac arrest; (3) RV failure: progressive loss of RV myocardium due to fibro-fatty replacement impairs RV function and may result in pump failure; and (4) biventricular failure: an advanced stage with involvement of the interventricular septum and LV causing congestive heart failure (HF). Endocavitary mural thrombosis may occur, especially within RV aneurysm or in the atria if atrial fibrillation is present. The phenotype may eventually resemble an advanced dilated CMP, making the differential diagnosis difficult at this stage.
The identification of ARVC can be often challenging, because of the heterogeneous clinical presentation, highly variable intra- and inter-family expressivity and incomplete penetrance. This genotype-phenotype “plasticity” is largely unexplained. The frequent involvement of the LV[7,15], sometimes predominant, suggests that ARVC is not a unique entity, but a complex heterogeneous disease with a spectrum of phenotypes and three possible patterns of expression: the classic (39% of cases), the left dominant (5%) and the biventricular (56%) forms[5]. Consequently, according to some Authors, in this disease it may be more appropriate to use the term of “arrhythmogenic cardiomyopathy” instead of the more “restrictive” ARVC terminology[6], (see below, section “spectrum of disease”).
ETIOPATHOGENESIS AND GENETICS
A familial history of ARVC is present in 30% to 50% of cases, and the disease is considered a genetic CMP, usually inherited in an autosomal dominant pattern with variable penetrance and expressivity; in addition, autosomal recessive forms have been reported (Naxos disease and Carvajal syndrome)[16]. Its presumed pathomechanism is presently thought an inherited abnormality of myocytes adhesion caused by defects at the intercellular junctions, at the level of desmosomes, adherens junctions or gap junctions, together comprising the intercalated discs[17-21]. The role of other non-desmosomal genes is less well established[18].
The desmosomes have a complex structure that includes several families of adhesion molecules, as the cadherins (desmoglein-DSG and desmocollin-DSC), plakins [desmoplakin-desmoplakin (DSP)], and catenins (plakophillin-PKP, and plakoglobin-JUP). Their main functional role is to link intermediate filaments of the intramyocellular cytoskeleton to the extracellular desmosomal cadherins[21-23]. Mutations in several genes encoding proteins of the desmosome have been identified in ARVC, the majority of which are located in 5 genes: plakophilin-2 (PKP2), DSP, desmoglein-2 (DSG2), desmocollin-2 (DSC2) and plakoglobin (JUP), the last one causing the autosomal recessive ARVC (Naxos disease)[16,24].
More uncommonly, ARVC has been related to mutations in other non-desmosomal genes, as transforming growth factor β-3, cardiac ryanodine receptor, trans-membrane protein 43 (TMEM43), tumor protein p63 (TP63), desmin, lamin A/C (LMNA), alpha T-catenin (CTNNA3) and phospholamban[17,18]. Thus far, more than 800 genetic variants have been identified in 12 genes, although only around 300 of them have been classified as clearly pathogenic[17,18]. One review that analyzed pooled data from major ARVC studies noted an overall mutation detection rate of 39.2%[24,25]. The most frequently affected gene is PKP2 with a reported detection rate of 10%-45%[26].
Recently, Taylor et al[27] identified in 7 out of 38 ARVC families an ARVC overlap syndrome due to rare variants in the gene encoding the sarcomeric protein titin (TTN), the largest gene in mammals. The phenotype of TTN variant carriers was characterized by a frequent history of SCD (5 of 7 families), progressive myocardial dysfunction causing death or heart transplantation (8 of 14 cases), frequent conduction disease (11 of 14), and incomplete penetrance (86%)[27]. TTN filaments bridge the sarcomere along its longitudinal axis, overlapping end-to-end at the Z disc and M band at the amino and carboxyl ends, respectively, thus forming a contiguous filament along the myofibril. Interestingly, recent research showed that TTN is involved in cellular mechanics, specifically, in the “spring-like” properties of the sarcomere that underlie passive and restorative forces occurring after sarcomere lengthening or shortening[28-30]. In this ARVC “overlap” syndrome structural impairment of TTN probably leads to proteolysis and apoptosis, which could be hypothesized as a novel mechanism underlying myocardial remodeling and SCD.
SPECTRUM OF DISEASE (CLASSIC ARVC, BIVENTRICULAR, LEFT DOMINANT)
The well known “classic pattern” of ARVC is characterized by an increased RV to LV volume ratio and a more severe involvement of the RV, with LV involvement as a possible late complication of the disease[1-6]. Clinical hallmarks are negative anterior T waves, and ventricular arrhythmias with LBBB morphology.
Left-dominant arrhythmogenic cardiomyopathy (LDAC) is a novel entity recently described. LDAC is characterized by fibro-adipose replacement, which predominantly involves the LV and often occurs as a circumferential band in the outer one-third of the myocardium and the right side of the interventricular septum[7]. This CMP has a predominant (but not necessarily exclusive) LV involvement, characterized by one or more of the following: LV wall motion abnormalities, chamber dilation, systolic impairment, and late gadolinium enhancement (LGE)[7]. Relevant clinical features of LDAC include ventricular arrhythmias of right bundle branch block (RBBB) morphology, and (infero)-lateral T-wave inversion at electrocardiogram (ECG). According to Sen-Chowdhry et al[6,7], LDAC can be considered one of the three possible patterns of the spectrum of ARVC, together with the “classical” form and the “biventricular” form, in consideration of the histopathologic and genetic similarities. However, as alternative hypothesis, LDAC could be considered as a novel distinct CMP.
Biventricular arrhythmogenic cardiomyopathy
The biventricular subtype of arrhythmogenic CMP is defined by early and parallel involvement of the RV and LV[6]. While milder cases typically demonstrate localized structural abnormalities on both sides; advanced disease is characterized by biventricular dilation and systolic impairment. The clinical picture is generally characterized by a composite of right-dominant and left-dominant features. Ventricular arrhythmias of both RBBB and LBBB configuration may occur, and at least 15% of cases show both morphologies of extrasystoles, underlining the presence of arrhythmogenic substrate in both ventricles. The ratio of RV to LV volume remains close to 1 throughout the disease course[6].
Finally, it must be remembered that, as noted above, during the progression of the disease an initial right or left-dominant pattern can evolve into a biventricular dysfunction[13,31].
Biventricular arrhythmogenic cardiomyopathy can mimic clinically and at imaging examinations a dilated CMP and be diagnosed only by pathologic examination at necropsy or of the explanted heart[32].
CRITERIA AND CHALLENGES IF DIAGNOSIS
As mentioned above, the clinical diagnosis of ARVC is often difficult because of the non-specific nature of the disease and the broad spectrum of phenotypic expressions. Consequently, ARVC is probably underestimated as milder cases frequently go unrecognized and non-classic sub-types are not incorporated. Furthermore, left-dominant and biventricular arrhythmogenic CMP are commonly misattributed to dilated CMP[32], hot phases to isolated viral myocarditis, and early disease to idiopathic ventricular tachycardia or benign ventricular ectopy[5,6]. The common thought that ARVC is a disease of the young and cannot present beyond middle age is probably an erroneous assumption, which becomes self-fulfilling as clinicians fail to consider it as a possibility in older patients. Raising clinicians’ awareness of the disease and its multiple presentations is critical to timely diagnosis and prevention of SCD.
There is no single gold-standard diagnostic test for ARVC, and the diagnosis relies on a scoring system with “major” and “minor” criteria based on the demonstration of a combination of defects in RV morphology and function, characteristic depolarization/repolarization ECG abnormalities, characteristic tissue pathology, typical arrhythmias, family history, and the results of genetic testing[33]. Definitive diagnosis, based on the Revised 2010 Task Force Criteria[33] (Table 1), requires 2 major criteria, 1 major and 2 minor criteria, or 4 minor criteria from different categories. Therefore, the initial evaluation of all patients suspected of having ARVC should include physical examination, clinical history, family history of arrhythmias or SCD, ECG (Figures 3 and 4), signal-averaged ECG, Holter ECG monitoring, and comprehensive noninvasive imaging tests focused on both ventricles, such as echocardiography (Figure 5). New tools for improving diagnostic accuracy have been introduced in the clinical practice. Among non-invasive investigations, cardiac magnetic resonance (CMR) gives accurate morpho-functional evaluation of both ventricles with quantitative assessment of ventricular volumes and ejection fractions, and can give information on myocardial tissue characterization (fatty infiltration, and fibrosis, at LGE study) (Figure 6)[34,35].
Table 1.
Revised arrhythmogenic right ventricular cardiomyopathy diagnostic criteria (modified from Marcus et al[33])
| Major criteria | Minor criteria | |
| RV systolic function and structure | By 2D echo: Regional RV akinesia, dyskinesia or aneurysm and one of the following (end diastole): PLAX RVOT ≥ 32 mm, PSAX RVOT ≥ 36 mm, Or fractional area change ≤ 33% | By 2D echo: Regional RV akinesia, dyskinesia or aneurysm and 1 of the following (end diastole): PLAX RVOT ≥ 29 to < 32 mm, PSAX RVOT ≥ 32 to < 36 mm, Or fractional area change > 33% to ≤ 40% |
| By MRI: Regional RV akinesia, dyskinesia or aneurysm or dyssynchronous RV contraction and 1 of following: Ratio of RV end-diastolic volume to BSA ≥ 110 mL/m2 or ≥ 100 mL/m2 (or RV EF ≤ 40%) | By MRI: Regional RV akinesia, dyskinesia or aneurysm or dyssynchronous RV contraction and 1 of the following: Ratio of RV end-diastolic volume to BSA ≥ 100 to < 110 mL/m2 (male) or ≥ 90 to < 100 mL/m2 (female) or RV > 40% to ≤ 45 % | |
| By RV angiography: Regional RV akinesia, dyskinesia or aneurysm | By RV angiography: Regional RV akinesia, dyskinesia or aneurysm | |
| Tissue characterization | Residual myocytes < 60% by morphometric analysis with fibrous replacement of the RV free wall myocardium in ≥ 1 sample, with or without fatty replacement of tissue on EMB | Residual myocytes 60% to 75% (or 50% to 65% if estimated), with fibrous replacement of the RV free wall myocardium in ≥ 1 sample, with or without fatty replacement of tissue on EMB |
| Repolarization abnormality | Inverted T waves in right precordial leads (V1-3) or beyond in individuals > 14 yr of age (in the absence of complete right bundle - branch block QRS ≥ 120 ms | Inverted T waves in leads V1 and V2 in individuals > 14 years of age (in the absence of complete right bundle branch block ) or in V4-6 or inverted T waves in leads V1-V4 individuals > 14 yr of age in the presence of complete right bundle branch block |
| Depolarization abnormality | Epsilon waves in the right precordial leads (V1-3) | Late potential by SAECG in ≥ 1 of 3 parameters in the absence of a QRS duration of ≥ 110 ms on the standard ECG; Filtered QRS duration ≥ 114 ms; Duration of terminal QRS < 40 mV or ≥ 38 μs; Root-mean-square voltage of terminal 40 ms ≤ 20 μV; Terminal activation duration of QRS ≥ 55 ms measured from the nadir of the S wave to the end of QRS |
| Arrhythmias | Nonsustained or sustained ventricular tachycardia of left bundle branch morphology with superior axis Frequent ventricular extrasystoles (> 1000 per 24 h) (Holter) | Nonsustained or sustained ventricular tachycardia of RV outflow configuration, left bundle branch morphology with inferior axis or > 500 ventricular extrasystoles per 24 h (Holter) |
| Familial history | ARVC confirmed pathologically in the first degree or identification of a pathogenic mutation categorized as associated or probably associated with ARVC | History of ARVC in a first degree relative or premature sudden death (< 35 yr of age) due to suspected ARVC or ARVC confirmed pathologically or by current Task Force Criteria in second-degree relative |
LV: Left ventricle; RV: Right ventricular; ARVC: Arrhythmogenic right ventricular cardiomyopathy; PLAX: Parasternal long axis; PSAX: Parasternal short axis; RVOT: Right ventricle outflow tract; ECG: Electrocardiogram; EMB: Endomyocardial biopsy; MRI; Magnetic resonance imaging; SAECG: Signal averaged ECG; BSA: body surface area.
Figure 3.
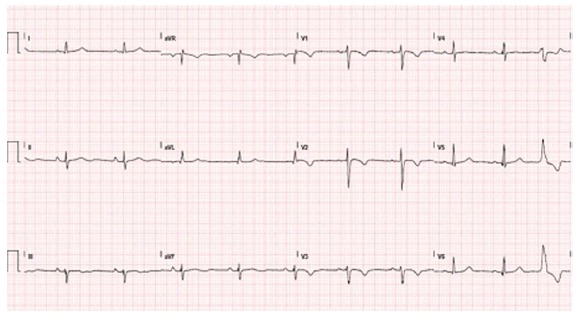
Typical electrocardiogram in a patient with classical arrhythmogenic right ventricular cardiomyopathy. Negative T waves in anterior precordial leads are present. A ventricular premature beat with left bundle branch block morphology is also observed.
Figure 4.
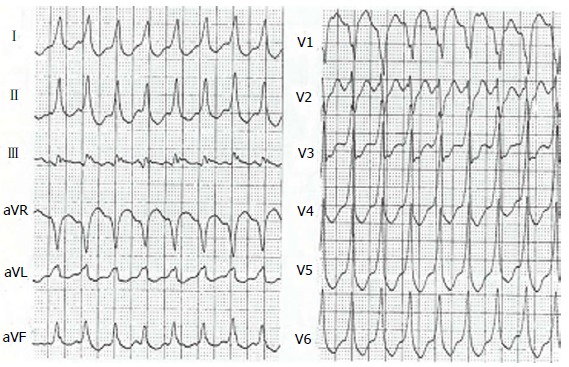
Ventricular tachycardia with left bundle branch block pattern in a patient with arrhythmogenic right ventricular cardiomyopathy.
Figure 5.
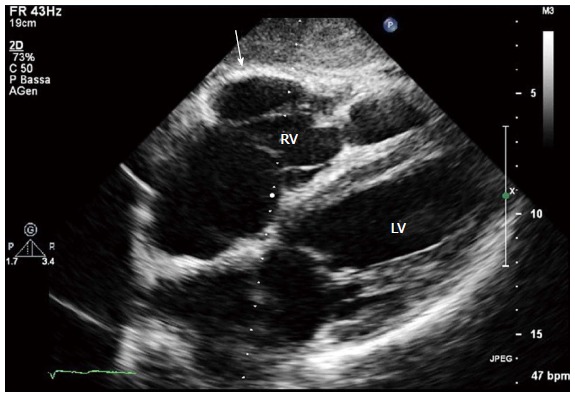
Two-dimensional echocardiogram, 4 chamber subcostal view, end diastolic frame, in an arrhythmogenic right ventricular cardiomyopathy patient. RV wall aneurysm at subtricuspid basal level is evident (arrow). LV: Left ventricle; RV: Right ventricle.
Figure 6.
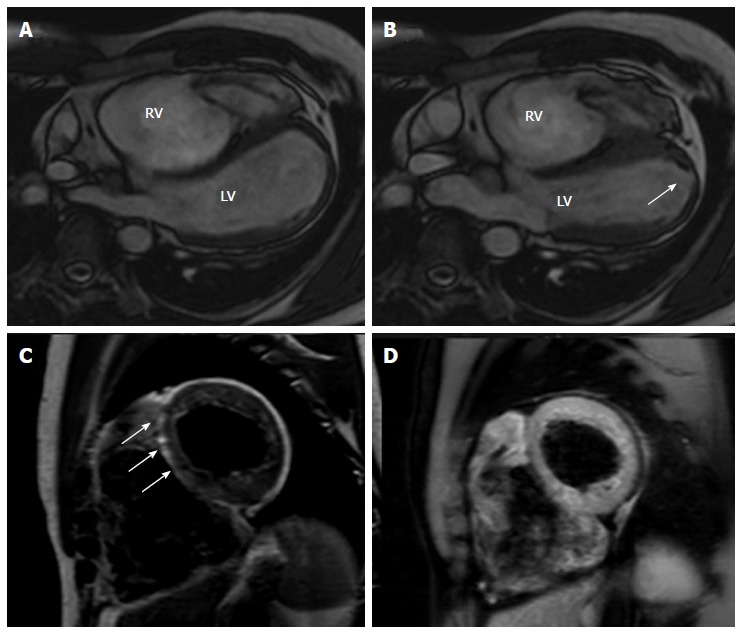
Cardiac magnetic resonance images of an arrhythmogenic right ventricular cardiomyopathy patient with mild left ventricular involvement. A, B: End-diastolic and end-systolic frames, off-axis 4 chamber view. Note the multiple bulging segments at right ventricular (RV) free wall. An apical hypokinesia of the left ventricle (LV) is also seen (arrow); C, D: Short axis T1-weighted dark blood imaging without (C) and with fat saturation (D) in the same patient with arrhythmogenic right ventricular cardiomyopathy. Note the hyperintense area in the interventricular (IV) septum in T1-weighted image (arrows) and corresponding hypointensity at T1-weighted dark blood imaging with fat-saturation indicating fatty infiltration in the IV septum.
In the new ARVC guidelines[33], “major” diagnostic criteria were selected because of their good sensitivity and specificity for the disease. It is important to note that imaging qualitative typical abnormalities of the disease, as aneurismal RV bulges are diagnostic only if associated with quantitative data as RV enlargement and/or depressed systolic function. Non-invasive tissue characterization by CMR was not considered because its poor specificity and reproducibility. Emerging major criteria is the demonstration of a typical genetic mutation, and genetic study is clinically useful particularly in borderline or possible ARVC[3-6]. If a noninvasive workup is suggestive but non diagnostic, further testing should be considered to establish the diagnosis, including electrophysiologic testing, RV angiography, electroanatomic mapping[36], and rarely also endomyocardial biopsy. This invasive procedure was frequently employed in the past and has been considered the “gold-standard”, but presently it is indicated only in very selected cases, with questionable diagnosis of ARVC despite thorough diagnostic assessment[6]. In fact, its sensitivity is not absolute, due to the frequent patchy distribution of the disease, and the procedure is not without serious risk of RV perforation because of the abnormally thin RV wall characteristic of the disease. If a biopsy is scheduled, it must be analyzed by optimal technique using quantitative morphometry[37], and the site of right ventricular puncture must be preferably chosen with echocardiographic, CMR or electroanatomic guidance[3]. Moreover, recent data suggest the diagnostic usefulness of immunohistochemical analysis of plakoglobin signal level at intercalated discs, diffusely reduced in myocardial tissue of ARVC[38].
DIFFERENTIAL DIAGNOSIS
Diagnosis of ARVC should be considered in any patient without a definite heart disease who presents with syncopal episodes, frequent ventricular extrasystoles or ventricular tachycardia. The main differential diagnoses include the following conditions:
Idiopathic RV outflow tract-ventricular tachycardia is a mostly benign condition not associated with structural heart disease. In early stage ARVC can be difficult to distinguish from this “idiopathic” type of ventricular arrhythmia in absence of structural changes[4]. A scoring system has been developed to identify a concealed ARVC in patients with apparently idiopathic VT[39]. Differential diagnosis is based on the fact that this arrhythmia is non-familial, and patients do not have the characteristic ECG/signal average ECG abnormalities of ARVC (inverted T waves in V1-V3, epsilon waves, QRS duration > 110 ms) Accurate imaging examination with CMR, and systematic follow-up reassessment can be useful to exclude RV abnormalities.
Brugada syndrome is an inherited cardiac condition that, similarly to ARVC, can be transmitted with an autosomal dominant pattern, and can lead to SCD from malignant ventricular arrhythmias. Differently from ARVC, it is characterized by a distinct typical ECG pattern with “J wave” in precordial leads, and absence of RV morpho-functional abnormalities at imaging.
Dilated cardiomyopathy may be difficult to distinguish from ARVC, especially in its advanced stage with severe biventricular involvement. In absence of classic ARVC hallmarks (RV aneurysms, bulging), the clinical distinction between these 2 CMP can be very difficult or impossible[32].
Myocarditis can mimic ARVC, especially when the RV is involved. Myocarditis can cause structural abnormalities, including microaneurysms, as well as the arrhythmic manifestations considered typical of ARVC. Moreover, myocardial inflammatory infiltrates, myocyte necrosis, replacement fibrosis and also fibro-fatty replacement of the RV myocardium can be observed also in myocarditis, resembling ARVC histologic features. New tools, such as 3-dimensional electro-anatomic mapping, applied to the standard endomyocardial biopsy, have been introduced to improve diagnostic accuracy in the clinical practice. Recently, in a provocative study Pieroni et al[40], found that 50% of patients with a noninvasive ARVC diagnosis fulfilled Dallas histological criteria of active myocarditis. These data would require confirmation in the future on large patient populations.
Sarcoidosis with cardiac involvement can mimic ARVC, making an accurate differential diagnosis is particularly challenging[6]. Cardiac sarcoidosis must be suspected in presence of concomitant mediastinal lymphadenopathy, extracardiac sarcoidosis, conduction defects with a high-grade atrio-ventricular block, and interventricular septal scar at imaging. A global RV hypokinesis or regional wall motion abnormalities can be present, due to the patchy nature of the granulomatous infiltration. The absence of myocardial fat infiltrates at CMR could be useful distinguishing feature to suspect cardiac sarcoidosis[41], although its diagnostic accuracy could vary, depending on the stage of the disease at which the CMR data were acquired. Endomyocardial biopsy may be indicated in selected cases with questionable diagnosis.
Other pathologies: (1) coronary artery disease and myocardial infarction can involve both ventricles and mimic aspects of ARVC; (2) pulmonary hypertension (RV pressure overload), and/or significant tricuspid regurgitation (RV volume overload) can cause RV dilation and dysfunction; (3) congenital heart diseases such as Uhl’s anomaly (a rare congenital heart disease with a total loss of the RV myocardial muscle and parchment appearance)[42] and repaired Tetralogy of Fallot have to be consider especially for their prevalent RV involvement; and (4) intracardiac left to-right shunts (e.g., atrial septal defects and anomalous pulmonary venous drainage) may cause RV volume overload. The diagnosis can be missed on standard echocardiogram, and transesophageal echocardiography and/or CMR can improve the diagnostic accuracy, in these selected cases.
PATIENT MANAGEMENT
Prevention of SCD is the most important management task for the patients affected by ARVC. Retrospective analysis of clinical and pathological series identified several risk factors, such as previous cardiac arrest, syncope, young age, malignant family history, participation in competitive sports, ventricular tachycardia, severe RV dysfunction, LV involvement, and QRS dispersion[43,44]. However, it has to be noted that the prognostic value of these single or combined risk factors has not been prospectively assessed. Two recent papers[45,46] tried to define the incidence and predictors of ICD therapy in patients with ARVC after placement of an ICD for primary prevention. Nearly one-half of the ARVC patients with primary prevention ICD implantation experienced appropriate ICD interventions. In one or both of these studies, proband status of patients, presence of unexplained syncopal episodes, inducibility of ventricular arrhythmias at electrophysiologic study and presence of non-sustained ventricular tachycardia at Holter monitoring resulted independent predictors of appropriate ICD discharge.
In summary, according to the International Guidelines and the consensus of the experts[47,48], the indications of ICD for prevention of SCD in ARVC patients are well established for high-risk patients with history of aborted SCD or episodes of sustained ventricular tachycardia (Level of Recommendation: IB), while in presence of unexplained syncope, non-sustained ventricular tachycardia, familial history of sudden death, extensive disease including those with LV involvement and are considered as possible indications for ICD at intermediate risk of SCD (Class of Recommendation IIa, Level of evidence: C). Additionally, the rare patients with genotypes of ARVC associated with a high genetic risk for SCD (e.g., ARVC 5)[49] may be considered as possible candidates for ICD therapy. It is currently recommended that asymptomatic patients have to be managed in a case-by-case basis.
The role of the electrophysiology with programmed ventricular stimulation remains controversial in the specific setting of ARVC. In fact, contrary to the above mentioned study by Bhonsale et al[45], in the Darvin II study[46], this test showed poor accuracy in predicting appropriate ICD interventions.
The impact of ICD implantation in ARVC has been evaluated in a recent meta-analysis[50]: a total of 610 patients were collected from 18 cohorts with ICD for either primary or secondary prevention and the annualized rate of appropriate ICD therapies resulted 9.5%.
The clinical relevance of ICD implantation in improving survival in patients with ARVC was clearly demonstrated by Corrado et al[46], by comparing the actual survival curve of their implanted patients with the ventricular fibrillation/flutter-free survival (estimated mortality reduction at 48 mo of 23%).
In ARVC patients, pharmacologic treatment as well as radiofrequency ablation (RFA) must not be considered a definitive therapy for ventricular arrhythmias, and they are not an equivalent alternative to ICD therapy in patients at high risk of SCD. RFA can be appropriate in selected patients who are not candidates for an ICD, or in those with an ICD who have frequent episodes of VT and ICD shocks despite antiarrhythmic drugs. Multiple recent studies suggest that simultaneous epicardial and endocardial approaches for VT mapping and ablation are feasible - although technically more demanding - and might even result in suppression of recurrent VT. This could be explained by the preferential epicardial infiltration characteristic of the disease[51].
Antiarrhythmic medications have been used for symptomatic control in ARVC. The combination of beta-blockers and amiodarone has a proved beneficial effect in suppression of non-sustained VT, in reduction of sustained VT arrhythmias and rate, preventing syncope and favoring anti-tachycardia pacing termination rather than shock therapy. Hence, sotalol and amiodarone have been proposed as effective treatment of sustained VT or VF as adjunctive therapy to ICD or in patients with ARVC that are not candidates for ICD implantation (Class of Recommendation IIa, Level of evidence: C)[48,52].
Furthermore, the North American ARVC Registry has demonstrated that amiodarone alone showed greatest efficacy at preventing sustained ventricular tachycardia or ICD discharge[53]. Conversely, a study from our group[54] reported that the treatment with amiodarone was independently related with increased mortality, presumably because the treated cases were those with higher arrhythmic risk.
Beta-blockers and angiotensin-converting enzyme inhibitors can be also used in ARVC patients, particularly in those with biventricular dysfunction and HF, due to their proven benefit in reducing mortality and slowing disease progression in other CMP, although no studies are presently available specifically on the response of ARVC patients to these medications[6].
General life education measures are also important in ARVC patients. Particular caution must be addressed to avoid competitive sport activities and strong physical efforts[6], which could increase the phenotypic expression and the arrhythmic risk[55,56].
Cardiac transplantation is indicated in patients with severe intractable heart failure, generally with end-stage disease and severe biventricular involvement, and in selected cases with intractable incessant ventricular arrhythmias[57].
UNSOLVED PROBLEMS AND FUTURE PERSPECTIVES
Despite considerable improvement in knowledge, ARVC has still several unsolved problems that deserve further research. In our opinion, the main future challenges would answer to the following questions: (1) what is the clinical role of genetic testing[6,17,58,59]? (2) how to improve the identification of affected cases, particularly in the concealed phase and in disease variants (LDAC, atypical forms)? and (3) how to improve the risk stratification of patients?
The use of genetic testing is growing very rapidly in recent years in CMPs[17], and its role is changing from a research tool to a clinically useful exam. In our opinion, based on the present knowledge, its clinical role in ARVC is not well defined. In fact, a pathogenic mutation can be recognized only in approximately a half of probands, and the possibility of multiple mutations or a non pathogenic benign mutation, encountered also in healthy individuals can cause considerable diagnostic problems[59-61]. A genetic study is considered clinically useful in equivocal cases, because the demonstration of a pathogenic mutation is a major criteria in the revised ARVC diagnostic criteria[33], and with a “cascade” analysis, in relatives of ARVC patients with identified mutation[17-60]. Appropriate genetic counseling and clinical management are very important particularly in genetic positive apparently healthy familial subjects (regular follow-up visits, possible caution about competitive sports). A clinically oriented approach which considers the presence of diagnostic “red flags” is preferable, in order to help in the proper selection of candidate genetic mutations[58].
New methods for early and precise identification of ARVC in initial phases are presently under active research. Advanced echocardiographic analyses can be helpful, particularly the study of myocardial deformation using speckle tracking analysis[62-64]. The modification of diagnostic ARVC criteria by the recent revision[33] significantly improved the diagnostic power of available methods, increasing both sensitivity and specificity[20,62,65]. However it has to be observed that the diagnostic criteria of LDAC were not considered in the last task force revision, and that would be advisable in the near future[7].
Accurate risk stratification is problematic in patients with ARVC, particularly for patients without history of severe life threatening arrhythmias (primary prevention of SCD)[44].
Additional potentially useful prognostic data recently were demonstrated by cardiovascular imaging, such as echocardiography[54,66], CMR[67,68], and electroanatomic mapping[69], thus reinforcing the importance of identifying the pathologic substrate of arrhythmias in the disease (areas of myocardial scarring and fibro-fatty infiltration with a probable reentry mechanism). Bhonsale et al[70] recently proposed a strategy for risk stratification for ARVC associated desmosomal mutation carriers based on pedigree evaluation, ECG and Holter information.
ACKNOWLEDGMENTS
We thank Prof R. Bussani (Department of Cardiovascular Pathology and Morbid Anatomy, University of Trieste) for gross specimens and histopathologic images and Dr. G. Vitrella (Cardiovascular Department, University of Trieste) for cardiac magnetic resonance images.
Footnotes
P- Reviewer: Amiya E, Li XP S- Editor: Song XX L- Editor: A E- Editor: Wu HL
References
- 1.Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, Grosgogeat Y. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65:384–398. doi: 10.1161/01.cir.65.2.384. [DOI] [PubMed] [Google Scholar]
- 2.Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129–133. doi: 10.1056/NEJM198801213180301. [DOI] [PubMed] [Google Scholar]
- 3.Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289–1300. doi: 10.1016/S0140-6736(09)60256-7. [DOI] [PubMed] [Google Scholar]
- 4.James CA, Calkins H. Update on Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/C) Curr Treat Options Cardiovasc Med. 2013;15:476–487. doi: 10.1007/s11936-013-0251-8. [DOI] [PubMed] [Google Scholar]
- 5.Sen-Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E, McKenna WJ. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007;115:1710–1720. doi: 10.1161/CIRCULATIONAHA.106.660241. [DOI] [PubMed] [Google Scholar]
- 6.Sen-Chowdhry S, Morgan RD, Chambers JC, McKenna WJ. Arrhythmogenic cardiomyopathy: etiology, diagnosis, and treatment. Annu Rev Med. 2010;61:233–253. doi: 10.1146/annurev.med.052208.130419. [DOI] [PubMed] [Google Scholar]
- 7.Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, Pennell DJ, McKenna WJ. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol. 2008;52:2175–2187. doi: 10.1016/j.jacc.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 8.Corrado D, Thiene G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: clinical impact of molecular genetic studies. Circulation. 2006;113:1634–1637. doi: 10.1161/CIRCULATIONAHA.105.616490. [DOI] [PubMed] [Google Scholar]
- 9.Basso C, Corrado D, Thiene G. Cardiovascular causes of sudden death in young individuals including athletes. Cardiol Rev. 1999;7:127–135. doi: 10.1097/00045415-199905000-00009. [DOI] [PubMed] [Google Scholar]
- 10.Tabib A, Loire R, Chalabreysse L, Meyronnet D, Miras A, Malicier D, Thivolet F, Chevalier P, Bouvagnet P. Circumstances of death and gross and microscopic observations in a series of 200 cases of sudden death associated with arrhythmogenic right ventricular cardiomyopathy and/or dysplasia. Circulation. 2003;108:3000–3005. doi: 10.1161/01.CIR.0000108396.65446.21. [DOI] [PubMed] [Google Scholar]
- 11.Corrado D, Thiene G, Nava A, Rossi L, Pennelli N. Sudden death in young competitive athletes: clinicopathologic correlations in 22 cases. Am J Med. 1990;89:588–596. doi: 10.1016/0002-9343(90)90176-e. [DOI] [PubMed] [Google Scholar]
- 12.Dalal D, Nasir K, Bomma C, Prakasa K, Tandri H, Piccini J, Roguin A, Tichnell C, James C, Russell SD, et al. Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation. 2005;112:3823–3832. doi: 10.1161/CIRCULATIONAHA.105.542266. [DOI] [PubMed] [Google Scholar]
- 13.Pinamonti B, Di Lenarda A, Sinagra G, Silvestri F, Bussani R, Camerini F. Long-term evolution of right ventricular dysplasia-cardiomyopathy. The Heart Muscle Disease Study Group. Am Heart J. 1995;129:412–415. doi: 10.1016/0002-8703(95)90029-2. [DOI] [PubMed] [Google Scholar]
- 14.Sinagra G, Mestroni L, Camerini F. The Role of Clinical Observation: Red Flag 5—Right Ventricular Involvement, Arrhythmogenic Right Ventricular Cardiomyopathy and Associated Phenotypes in Genetic Cardiomyopathies. Milan: Springer; 2013. pp. 61–69. [Google Scholar]
- 15.Pinamonti B, Sinagra G, Salvi A, Di Lenarda A, Morgera T, Silvestri F, Bussani R, Camerini F. Left ventricular involvement in right ventricular dysplasia. Am Heart J. 1992;123:711–724. doi: 10.1016/0002-8703(92)90511-s. [DOI] [PubMed] [Google Scholar]
- 16.Protonotarios N, Tsatsopoulou A. Naxos disease and Carvajal syndrome: cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Pathol. 2004;13:185–194. doi: 10.1016/j.carpath.2004.03.609. [DOI] [PubMed] [Google Scholar]
- 17.Marcus FI, Edson S, Towbin JA. Genetics of arrhythmogenic right ventricular cardiomyopathy: a practical guide for physicians. J Am Coll Cardiol. 2013;61:1945–1948. doi: 10.1016/j.jacc.2013.01.073. [DOI] [PubMed] [Google Scholar]
- 18.Campuzano O, Alcalde M, Allegue C, Iglesias A, García-Pavía P, Partemi S, Oliva A, Pascali VL, Berne P, Sarquella-Brugada G, et al. Genetics of arrhythmogenic right ventricular cardiomyopathy. J Med Genet. 2013;50:280–289. doi: 10.1136/jmedgenet-2013-101523. [DOI] [PubMed] [Google Scholar]
- 19.Fressart V, Duthoit G, Donal E, Probst V, Deharo JC, Chevalier P, Klug D, Dubourg O, Delacretaz E, Cosnay P, et al. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. Europace. 2010;12:861–868. doi: 10.1093/europace/euq104. [DOI] [PubMed] [Google Scholar]
- 20.Quarta G, Muir A, Pantazis A, Syrris P, Gehmlich K, Garcia-Pavia P, Ward D, Sen-Chowdhry S, Elliott PM, McKenna WJ. Familial evaluation in arrhythmogenic right ventricular cardiomyopathy: impact of genetics and revised task force criteria. Circulation. 2011;123:2701–2709. doi: 10.1161/CIRCULATIONAHA.110.976936. [DOI] [PubMed] [Google Scholar]
- 21.Sheikh F, Ross RS, Chen J. Cell-cell connection to cardiac disease. Trends Cardiovasc Med. 2009;19:182–190. doi: 10.1016/j.tcm.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peters NS, Green CR, Poole-Wilson PA, Severs NJ. Reduced content of connexin43 gap junctions in ventricular myocardium from hypertrophied and ischemic human hearts. Circulation. 1993;88:864–875. doi: 10.1161/01.cir.88.3.864. [DOI] [PubMed] [Google Scholar]
- 23.Smith JH, Green CR, Peters NS, Rothery S, Severs NJ. Altered patterns of gap junction distribution in ischemic heart disease. An immunohistochemical study of human myocardium using laser scanning confocal microscopy. Am J Pathol. 1991;139:801–821. [PMC free article] [PubMed] [Google Scholar]
- 24.Azaouagh A, Churzidse S, Konorza T, Erbel R. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: a review and update. Clin Res Cardiol. 2011;100:383–394. doi: 10.1007/s00392-011-0295-2. [DOI] [PubMed] [Google Scholar]
- 25.Sen-Chowdhry S, Syrris P, McKenna WJ. Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2007;50:1813–1821. doi: 10.1016/j.jacc.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 26.Iyer VR, Chin AJ. Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) Am J Med Genet C Semin Med Genet. 2013;163C:185–197. doi: 10.1002/ajmg.c.31368. [DOI] [PubMed] [Google Scholar]
- 27.Taylor M, Graw S, Sinagra G, Barnes C, Slavov D, Brun F, Pinamonti B, Salcedo EE, Sauer W, Pyxaras S, et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation. 2011;124:876–885. doi: 10.1161/CIRCULATIONAHA.110.005405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.LeWinter MM, Granzier HL. Titin is a major human disease gene. Circulation. 2013;127:938–944. doi: 10.1161/CIRCULATIONAHA.112.139717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chung CS, Hutchinson KR, Methawasin M, Saripalli C, Smith JE, Hidalgo CG, Luo X, Labeit S, Guo C, Granzier HL. Shortening of the elastic tandem immunoglobulin segment of titin leads to diastolic dysfunction. Circulation. 2013;128:19–28. doi: 10.1161/CIRCULATIONAHA.112.001268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anderson BR, Granzier HL. Titin-based tension in the cardiac sarcomere: molecular origin and physiological adaptations. Prog Biophys Mol Biol. 2012;110:204–217. doi: 10.1016/j.pbiomolbio.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, Nava A, Silvestri F, Blomstrom-Lundqvist C, Wlodarska EK, et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol. 1997;30:1512–1520. doi: 10.1016/s0735-1097(97)00332-x. [DOI] [PubMed] [Google Scholar]
- 32.Nemec J, Edwards BS, Osborn MJ, Edwards WD. Arrhythmogenic right ventricular dysplasia masquerading as dilated cardiomyopathy. Am J Cardiol. 1999;84:237–239, A9. doi: 10.1016/s0002-9149(99)00244-1. [DOI] [PubMed] [Google Scholar]
- 33.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533–1541. doi: 10.1161/CIRCULATIONAHA.108.840827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tandri H, Saranathan M, Rodriguez ER, Martinez C, Bomma C, Nasir K, Rosen B, Lima JA, Calkins H, Bluemke DA. Noninvasive detection of myocardial fibrosis in arrhythmogenic right ventricular cardiomyopathy using delayed-enhancement magnetic resonance imaging. J Am Coll Cardiol. 2005;45:98–103. doi: 10.1016/j.jacc.2004.09.053. [DOI] [PubMed] [Google Scholar]
- 35.Sen-Chowdhry S, Prasad SK, Syrris P, Wage R, Ward D, Merrifield R, Smith GC, Firmin DN, Pennell DJ, McKenna WJ. Cardiovascular magnetic resonance in arrhythmogenic right ventricular cardiomyopathy revisited: comparison with task force criteria and genotype. J Am Coll Cardiol. 2006;48:2132–2140. doi: 10.1016/j.jacc.2006.07.045. [DOI] [PubMed] [Google Scholar]
- 36.Corrado D, Basso C, Leoni L, Tokajuk B, Bauce B, Frigo G, Tarantini G, Napodano M, Turrini P, Ramondo A, et al. Three-dimensional electroanatomic voltage mapping increases accuracy of diagnosing arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2005;111:3042–3050. doi: 10.1161/CIRCULATIONAHA.104.486977. [DOI] [PubMed] [Google Scholar]
- 37.Basso C, Ronco F, Marcus F, Abudureheman A, Rizzo S, Frigo AC, Bauce B, Maddalena F, Nava A, Corrado D, et al. Quantitative assessment of endomyocardial biopsy in arrhythmogenic right ventricular cardiomyopathy/dysplasia: an in vitro validation of diagnostic criteria. Eur Heart J. 2008;29:2760–2771. doi: 10.1093/eurheartj/ehn415. [DOI] [PubMed] [Google Scholar]
- 38.Asimaki A, Tandri H, Huang H, Halushka MK, Gautam S, Basso C, Thiene G, Tsatsopoulou A, Protonotarios N, McKenna WJ, et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360:1075–1084. doi: 10.1056/NEJMoa0808138. [DOI] [PubMed] [Google Scholar]
- 39.Hoffmayer KS, Machado ON, Marcus GM, Yang Y, Johnson CJ, Ermakov S, Vittinghoff E, Pandurangi U, Calkins H, Cannom D, Gear KC, Tichnell C, Park Y, Zareba W, Marcus FI, Scheinman MM. Electrocardiographic comparison of ventricular arrhythmias in patients with arrhythmogenic right ventricular cardiomyopathy and right ventricular outflow tract tachycardia. J Am Coll Cardiol. 2011;58:831–838. doi: 10.1016/j.jacc.2011.05.017. [DOI] [PubMed] [Google Scholar]
- 40.Pieroni M, Dello Russo A, Marzo F, Pelargonio G, Casella M, Bellocci F, Crea F. High prevalence of myocarditis mimicking arrhythmogenic right ventricular cardiomyopathy differential diagnosis by electroanatomic mapping-guided endomyocardial biopsy. J Am Coll Cardiol. 2009;53:681–689. doi: 10.1016/j.jacc.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 41.Steckman DA, Schneider PM, Schuller JL, Aleong RG, Nguyen DT, Sinagra G, Vitrella G, Brun F, Cova MA, Pagnan L, et al. Utility of cardiac magnetic resonance imaging to differentiate cardiac sarcoidosis from arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2012;110:575–579. doi: 10.1016/j.amjcard.2012.04.029. [DOI] [PubMed] [Google Scholar]
- 42.Gerlis LM, Schmidt-Ott SC, Ho SY, Anderson RH. Dysplastic conditions of the right ventricular myocardium: Uhl’s anomaly vs arrhythmogenic right ventricular dysplasia. Br Heart J. 1993;69:142–150. doi: 10.1136/hrt.69.2.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Corrado D, Leoni L, Link MS, Della Bella P, Gaita F, Curnis A, Salerno JU, Igidbashian D, Raviele A, Disertori M, et al. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2003;108:3084–3091. doi: 10.1161/01.CIR.0000103130.33451.D2. [DOI] [PubMed] [Google Scholar]
- 44.Silvano M, Corrado D, Köbe J, Mönnig G, Basso C, Thiene G, Eckardt L. Risk stratification in arrhythmogenic right ventricular cardiomyopathy. Herzschrittmacherther Elektrophysiol. 2013;24:202–208. doi: 10.1007/s00399-013-0291-5. [DOI] [PubMed] [Google Scholar]
- 45.Bhonsale A, James CA, Tichnell C, Murray B, Gagarin D, Philips B, Dalal D, Tedford R, Russell SD, Abraham T, et al. Incidence and predictors of implantable cardioverter-defibrillator therapy in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy undergoing implantable cardioverter-defibrillator implantation for primary prevention. J Am Coll Cardiol. 2011;58:1485–1496. doi: 10.1016/j.jacc.2011.06.043. [DOI] [PubMed] [Google Scholar]
- 46.Corrado D, Calkins H, Link MS, Leoni L, Favale S, Bevilacqua M, Basso C, Ward D, Boriani G, Ricci R, et al. Prophylactic implantable defibrillator in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia and no prior ventricular fibrillation or sustained ventricular tachycardia. Circulation. 2010;122:1144–1152. doi: 10.1161/CIRCULATIONAHA.109.913871. [DOI] [PubMed] [Google Scholar]
- 47.Epstein AE, DiMarco JP, Ellenbogen KA, Estes NAM, Freedman RA, Gettes LS, Gillinov AM, Gregoratos G, Hammill SC, Hayes DL, et al. ACC/AHA/HRS 2008 Guidelines for Device-Based Therapy of Cardiac Rhythm Abnormalities: Executive Summary A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices): Developed in Collaboration With the American Association for Thoracic Surgery and Society of Thoracic Surgeons. Circulation. 2008;117:2820–2840. [Google Scholar]
- 48.Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, Gregoratos G, Klein G, Moss AJ, Myerburg RJ, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death) J Am Coll Cardiol. 2006;48:e247–e346. doi: 10.1016/j.jacc.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 49.Merner ND, Hodgkinson KA, Haywood AF, Connors S, French VM, Drenckhahn JD, Kupprion C, Ramadanova K, Thierfelder L, McKenna W, et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82:809–821. doi: 10.1016/j.ajhg.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schinkel AF. Implantable cardioverter defibrillators in arrhythmogenic right ventricular dysplasia/cardiomyopathy: patient outcomes, incidence of appropriate and inappropriate interventions, and complications. Circ Arrhythm Electrophysiol. 2013;6:562–568. doi: 10.1161/CIRCEP.113.000392. [DOI] [PubMed] [Google Scholar]
- 51.Garcia FC, Bazan V, Zado ES, Ren JF, Marchlinski FE. Epicardial substrate and outcome with epicardial ablation of ventricular tachycardia in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2009;120:366–375. doi: 10.1161/CIRCULATIONAHA.108.834903. [DOI] [PubMed] [Google Scholar]
- 52.Wichter T, Borggrefe M, Haverkamp W, Chen X, Breithardt G. Efficacy of antiarrhythmic drugs in patients with arrhythmogenic right ventricular disease. Results in patients with inducible and noninducible ventricular tachycardia. Circulation. 1992;86:29–37. doi: 10.1161/01.cir.86.1.29. [DOI] [PubMed] [Google Scholar]
- 53.Marcus GM, Glidden DV, Polonsky B, Zareba W, Smith LM, Cannom DS, Estes NA, Marcus F, Scheinman MM. Efficacy of antiarrhythmic drugs in arrhythmogenic right ventricular cardiomyopathy: a report from the North American ARVC Registry. J Am Coll Cardiol. 2009;54:609–615. doi: 10.1016/j.jacc.2009.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pinamonti B, Dragos AM, Pyxaras SA, Merlo M, Pivetta A, Barbati G, Di Lenarda A, Morgera T, Mestroni L, Sinagra G. Prognostic predictors in arrhythmogenic right ventricular cardiomyopathy: results from a 10-year registry. Eur Heart J. 2011;32:1105–1113. doi: 10.1093/eurheartj/ehr040. [DOI] [PubMed] [Google Scholar]
- 55.Corrado D, Basso C, Rizzoli G, Schiavon M, Thiene G. Does sports activity enhance the risk of sudden death in adolescents and young adults? J Am Coll Cardiol. 2003;42:1959–1963. doi: 10.1016/j.jacc.2003.03.002. [DOI] [PubMed] [Google Scholar]
- 56.James CA, Bhonsale A, Tichnell C, Murray B, Russell SD, Tandri H, Tedford RJ, Judge DP, Calkins H. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62:1290–1297. doi: 10.1016/j.jacc.2013.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tedford RJ, James C, Judge DP, Tichnell C, Murray B, Bhonsale A, Philips B, Abraham T, Dalal D, Halushka MK, et al. Cardiac transplantation in arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2012;59:289–290. doi: 10.1016/j.jacc.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lopes LR, Elliott PM. New approaches to the clinical diagnosis of inherited heart muscle disease. Heart. 2013;99:1451–1461. doi: 10.1136/heartjnl-2012-301995. [DOI] [PubMed] [Google Scholar]
- 59.Murray B. Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C): a review of molecular and clinical literature. J Genet Couns. 2012;21:494–504. doi: 10.1007/s10897-012-9497-7. [DOI] [PubMed] [Google Scholar]
- 60.Raman SV, Basso C, Tandri H, Taylor MR. Imaging phenotype vs genotype in nonhypertrophic heritable cardiomyopathies: dilated cardiomyopathy and arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Imaging. 2010;3:753–765. doi: 10.1161/CIRCIMAGING.110.957563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kapplinger JD, Landstrom AP, Salisbury BA, Callis TE, Pollevick GD, Tester DJ, Cox MG, Bhuiyan Z, Bikker H, Wiesfeld AC, et al. Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J Am Coll Cardiol. 2011;57:2317–2327. doi: 10.1016/j.jacc.2010.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guttmann OP, Mohiddin SA, Elliott PM. Almanac 2014: cardiomyopathies. Heart. 2014;100:756–764. doi: 10.1136/heartjnl-2013-305420. [DOI] [PubMed] [Google Scholar]
- 63.Teske AJ, Cox MG, Te Riele AS, De Boeck BW, Doevendans PA, Hauer RN, Cramer MJ. Early detection of regional functional abnormalities in asymptomatic ARVD/C gene carriers. J Am Soc Echocardiogr. 2012;25:997–1006. doi: 10.1016/j.echo.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 64.Atsumi A, Ishizu T, Kameda Y, Yamamoto M, Harimura Y, Machino-Ohtsuka T, Kawamura R, Enomoto M, Seo Y, Aonuma K. Application of 3-dimensional speckle tracking imaging to the assessment of right ventricular regional deformation. Circ J. 2013;77:1760–1768. doi: 10.1253/circj.cj-12-1445. [DOI] [PubMed] [Google Scholar]
- 65.Cox MG, van der Smagt JJ, Noorman M, Wiesfeld AC, Volders PG, van Langen IM, Atsma DE, Dooijes D, Houweling AC, Loh P, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy diagnostic task force criteria: impact of new task force criteria. Circ Arrhythm Electrophysiol. 2010;3:126–133. doi: 10.1161/CIRCEP.109.927202. [DOI] [PubMed] [Google Scholar]
- 66.Saguner AM, Vecchiati A, Baldinger SH, Rüeger S, Medeiros-Domingo A, Mueller-Burri AS, Haegeli LM, Biaggi P, Manka R, Lüscher TF, et al. Different prognostic value of functional right ventricular parameters in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ Cardiovasc Imaging. 2014;7:230–239. doi: 10.1161/CIRCIMAGING.113.000210. [DOI] [PubMed] [Google Scholar]
- 67.Deac M, Alpendurada F, Fanaie F, Vimal R, Carpenter JP, Dawson A, Miller C, Roussin I, di Pietro E, Ismail TF, et al. Prognostic value of cardiovascular magnetic resonance in patients with suspected arrhythmogenic right ventricular cardiomyopathy. Int J Cardiol. 2013;168:3514–3521. doi: 10.1016/j.ijcard.2013.04.208. [DOI] [PubMed] [Google Scholar]
- 68.te Riele AS, Bhonsale A, James CA, Rastegar N, Murray B, Burt JR, Tichnell C, Madhavan S, Judge DP, Bluemke DA, et al. Incremental value of cardiac magnetic resonance imaging in arrhythmic risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62:1761–1769. doi: 10.1016/j.jacc.2012.11.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Migliore F, Zorzi A, Silvano M, Bevilacqua M, Leoni L, Marra MP, Elmaghawry M, Brugnaro L, Dal Lin C, Bauce B, et al. Prognostic value of endocardial voltage mapping in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ Arrhythm Electrophysiol. 2013;6:167–176. doi: 10.1161/CIRCEP.111.974881. [DOI] [PubMed] [Google Scholar]
- 70.Bhonsale A, James CA, Tichnell C, Murray B, Madhavan S, Philips B, Russell SD, Abraham T, Tandri H, Judge DP, et al. Risk stratification in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. Circ Arrhythm Electrophysiol. 2013;6:569–578. doi: 10.1161/CIRCEP.113.000233. [DOI] [PubMed] [Google Scholar]