

Abstract
Congestive heart failure (CHF) is one of the most common reasons for hospitalization in the United States. Despite multiple different beneficial medications for the treatment of chronic CHF, there are no therapies with a demonstrated mortality benefit in the treatment of acute decompensated heart failure. In fact, studies of inotropes used in this setting have demonstrated more harm than good. Arginine vasopressin has been shown to be up regulated in CHF. When bound to the V1a and/or V2 receptors, vasopressin causes vasoconstriction, left ventricular remodeling and free water reabsorption. Recently, two drugs have been approved for use that antagonize these receptors. Studies thus far have indicated that these medications, while effective at aquaresis (free water removal), are safe and not associated with increased morbidity such as renal failure and arrhythmias. Both conivaptan and tolvaptan have been approved for the treatment of euvolemic and hypervolemic hyponatremia. We review the results of these studies in patients with heart failure.
Keywords: Heart failure, Arginine vasopressin antagonist, Vaptan, Hyponatremia, Aquaresis, Vasopressin
Core tip: Beneficial therapies in the setting of acute decompensated heart failure are limited. When bound to the V1a and/or V2 receptors, vasopressin, which is upregulated in heart failure, causes vasoconstriction, left ventricular remodeling and free water reabsorption. Over recent years, vasopressin antagonists such as conivaptan and tolvaptan have been investigated and approved for use in the appropriate setting. We review the evidence and implications behind use of vaptans in the setting of heart failure.
INTRODUCTION
Congestive heart failure (CHF) is a growing problem, with high mortality, frequent hospitalizations and poor quality of life. CHF afflicts about 5 million people in the United States, with over half a million new diagnoses and 200000 deaths each year[1,2]. Despite advances in therapy such as the use of angiotensin converting enzyme (ACE) inhibitors and beta blockers, heart failure hospitalizations are on the rise, with over a million a year, partly due to patients living longer and surviving acute myocardial infarctions. One of the principle goals of therapy during a heart failure admission is to relieve excess volume in order to improve symptoms. This is primarily accomplished with the use of diuretics and vasodilators. Although these agents improve symptoms, they may be associated with an increase in mortality chronically[1,3,4]. Additionally, they are often associated with hyponatremia[5]. In an attempt to further advance heart failure treatment, several new medications have been studied including natriuretic peptides, adenosine antagonists and vasopressin antagonists. The purpose of this paper is to review the role of vasopressin antagonists for the therapy of acute heart failure exacerbations.
NEUROHORMONAL ACTIVATION IN ACUTE HEART FAILURE
Acute heart failure is associated with activation of several components of the neurohormonal system. In response to ventricular dysfunction and decreased perfusion, baroreceptors in the aorta, carotid body, and the kidney are activated. The immediate response is an increase in sympathetic nervous system outflow. Norepinephrine release results in tachycardia, arterial vasoconstriction, venoconstriction, and increased contractility. The renin-angiotensin-aldosterone system, which promotes the retention of sodium and subsequently water, is also activated. Additionally, arginine vasopressin (AVP), endothelin, atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), adenosine, and tumor necrosis factor are all released. These hormones have a variety of individual effects as outlined in Figure 1.
Figure 1.
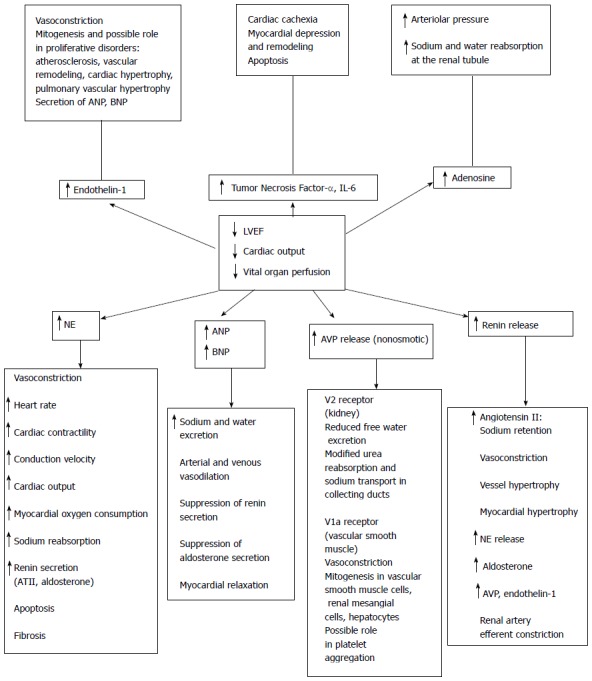
Summary of neurohormonal activation in heart failure. With injury to the left ventricle and subsequent decrease in left ventricular function, there is a decrease in cardiac output, subsequent decrease in perfusion of vital organs, and activation of the various neurohormonal systems. ANP: Atrial natriuretic peptide; AT II: Angiotensin II; AVP: Arginine vasopressin; BNP: Brain natriuretic peptide; IL-6: Interleukin 6; LVEF: Left ventricular ejection fraction; NE: Norepinephrine. From Russell et al[28] with permission from Springer.
Although the acute effects of these neurohormones are helpful to sustain life, chronically elevated levels may be quite detrimental. In both the Studies of Left Ventricular Dysfunction (SOLVD) Trial and the Vasodilator - Heart Failure Trial II (V-HeFT II), investigators demonstrated that plasma levels of norepinephrine, renin, ANP, and AVP are elevated in patients with left ventricular dysfunction when compared with healthy controls[6,7]. Furthermore, as New York Heart Association (NYHA) functional class worsens, the levels of these neurohormones are increased. Many of the beneficial effects of ACE inhibitors and beta blockers may be due to the blockade of these neurohormones. However, evidence for the use of these agents in reducing mortality is primarily in the chronic heart failure setting, and less is known about appropriate optimal management of acute heart failure.
Many studies have examined the effects of chronic diuretics on mortality in patients with heart failure. Cooper et al[3] performed a retrospective analysis of the SOLVD Trial and found that those using a diuretic at baseline were more likely to have an arrhythmic death than those not on a diuretic. Even after controlling for disease severity, comorbidities, and concomitant medications, the use of diuretics was associated with an increased risk of arrhythmic death. Similar results were found in a retrospective analysis of 1153 patients from the Prospective Randomized Amlodipine Survival Evaluation Trial, which examined the use of amlodipine in patients with NYHA functional class IIIb/IV heart failure[1]. High chronic doses of diuretics were associated with increased mortality, sudden death, and pump failure death. Although it is not surprising that higher diuretic doses are used in patients that have more advanced heart failure, a multivariate analysis controlling for disease severity revealed that high diuretic dose was still a predictor of mortality. This could possibly be explained by diuretic resistance, neurohormonal activation, or electrolyte changes rather than the dose itself. In fact, in the acute setting, higher-dose diuretic therapy has been shown to result in improved fluid loss and relief of congestive symptoms, lower adverse events, and despite acutely worsening renal function no difference in 60 d clinical outcomes when compared with lower-dose diuretic therapy[8].
Intravenous inotropes have also not improved outcomes in patients admitted with heart failure. In one of the first studies of chronic heart failure patients admitted with acute volume overload, the Outcomes of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure (OPTIME-CHF) investigators examined the use of the positive inotrope milrinone[9]. Nine hundred and fifty one patients admitted with chronic heart failure exacerbation were randomized to either milrinone or placebo. The primary endpoint of cumulative days of cardiovascular hospitalization in the first 60 d after randomization was similar between the two groups. Similarly, there was no difference in 60 d mortality, in-hospital mortality, or the composite of death or readmission. The use of milrinone was associated with more hypotension and new atrial arrhythmias. Perhaps more sobering, in this group of patients with NYHA functional class III and IV symptoms and a mean ejection fraction of 23%, the mean days of hospitalization for any cause within the first 60 d after discharge was 13.5 in the placebo group and 13.4 in the milrinone group. Additionally, after discharge from their initial hospitalization, 35.3% of the placebo group and 35.0% of the milrinone group were either readmitted to the hospital or dead within 60 d. Even after their admission and “optimization” of medical therapy by heart failure experts, 9.5% of the patients enrolled in this trial were dead within 2 mo of discharge.
Nesiritide is a B-type natriuretic peptide that has been associated with a decrease in pulmonary capillary wedge pressure via its vasodilation and natriuresis[10,11]. Despite only being demonstrated to be a vasodilator in clinical trials, many now, perhaps incorrectly, use nesiritide as a first line diuretic. Wang et al[12] demonstrated that this might not be the correct use for the drug. In a small trial of 15 patients hospitalized for heart failure with mild renal insufficiency (baseline creatinine of 1.8 mg/dL), they performed a double-blind, placebo-controlled, crossover study. Patients were randomized to receive either placebo or nesiritide for 24 h on consecutive days. There were no differences in glomerular filtration rate, renal plasma flow, urine output, or sodium excretion for the patients between the two agents. Sackner-Bernstein et al[13] also conducted a meta-analysis of three randomized controlled trials that suggests nesiritide may be associated with a higher risk of death compared to vasodilators and diuretics. Controversy still exists over nesiritide’s deleterious effects on renal function and short-term mortality. More recent trials have demonstrated similar safety endpoints, but no clear benefit to nesiritide therapy. The Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure Trial evaluated the utility and safety of nesiritide in a randomized controlled trial of 7141 patients. Though there was no significant difference in rate of all cause mortality or worsening renal function, there was also only a small, non-significant change in patient dyspnea and no effect on rehospitalization rate[14]. The recently published Renal Optimization Strategies Evaluation in Acute Heart Failure, which was also presented at the American Heart Association 2013 Annual Scientific Session Late Breaking Clinical Trials, also failed to show benefit of low dose nesiritide. This multicenter randomized trial showed no difference in 72 h urine volume, cystatin C levels changes, symptom relief or concomitant diuretic dose needs. Though there was no difference in renal function or death, there was increased incidence of hypotension in the nesiritide group[15].
Clearly, the currently available agents for the treatment of heart failure in the acute setting are not associated with satisfactory outcomes. The rest of this paper will review a newer class of agents, arginine vasopressin antagonists, for the therapy of this deadly syndrome.
ARGININE VASOPRESSIN: PATHOPHYSIOLOGY
AVP is a neurohypophyseal peptide that serves the roles of vasoconstrictor and body water regulator. Turner et al[16] were the first to isolate and synthesize vasopressin in 1951. Synthesized in the paraventricular and supraoptic nuclei of the hypothalamus and stored in the posterior pituitary gland, vasopressin is released in response to osmotic and non-osmotic forces. AVP’s release is sensitive to changes in osmolality. Osmoreceptors in the hypothalamus stimulate increased AVP secretion after sensing as little as a 1% increase in serum osmolality. A decrease in 5% to 10% of plasma volume is required for AVP release, stimulated via baroreceptors that sense a low volume state[17].
Three different vasopressin receptors have been isolated: V1a, V1b (also known as V3) and V2 receptors (Table 1). The V1b receptor is expressed in the anterior pituitary gland and pancreatic islet cells, and although it does not have a major role in CHF, it may mediate release of aldosterone via modulation of adrenocorticotropin hormone release[18]. The V1a receptor (V1aR) is present in blood vessels and the kidney, where stimulation is responsible for vascular constriction and possibly regulation of water reabsorption, respectively. V1aR is a Gq-protein coupled receptor and, via phosphotidylinositol hydrolysis, stimulates mobilization of intracellular calcium. V1aR knockout mice have a blunted response to AVP-induced vasoconstriction and decreased sympathetic activity[19]. Additionally, they have lower levels of aldosterone, renin and angiotensin II as well as higher urine output. V2 receptors are present in the thick ascending limb of the loop of Henle and collecting ducts of the renal tubular system. Via Gs-protein coupled receptor signaling and subsequent activation of adenylate cyclase, cyclic adenosine monophosphate levels increase and cause translocation of the water channel aquaporin-2 (AQP2), thereby increasing water permeability, reducing the rate of free water secretion and concentrating the urine[17,20]. This causes a decrease in urine production that has been found to be proportional to the concentration of plasma vasopressin.
Table 1.
| Receptor subtype | Location | Action | Cardiovascular end effects |
| V1A | Liver, vascular smooth muscle, platelets, adrenal cortex, kidney, spleen, adipocytes, reproductive organs, brain, lung | Vasoconstriction | Left ventricular hypertrophy and remodeling, increase in afterload, myocyte hypertrophy |
| V1B | Corticotroph cells, pancreas, adrenal medulla, possibly kidney | Release of adrenocorticotropic hormone | May mediate release of aldosterone |
| V2 | Renal collecting ducts | Antidiuresis via increased water permeability | Hyponatremia, edema, increase in preload, pulmonary vascular congestion and left sided filling pressures |
AVP IN HEART FAILURE AND HYPONATREMIA
AVP levels are elevated in congestive heart failure patients[21,22]. Investigators in the SOLVD trial found that AVP was significantly elevated in asymptomatic patients with left ventricular dysfunction (ejection fraction less than 35%) when compared to controls, and even more so elevated in symptomatic patients with left ventricular dysfunction[6]. When plasma osmolality increases in both control and CHF patients, there is a significant exaggerated AVP response in CHF patients[23]. Although known to primarily be produced in the hypothalamus, vasopressin has also been found in isolated rat hearts undergoing the stress of acute pressure overload or nitric oxide stimulation[24].
AVP leads to worsening heart failure by a variety of mechanisms. Activation of V1aR causes arteriolar vasoconstriction resulting in increased systemic vascular resistance and afterload. At higher physiologic AVP levels, V1aR also mediates coronary vasoconstriction, thus decreasing coronary blood flow and cardiac contractility[18,24]. Stimulation of rat cardiac fibroblasts with AVP leads to cellular hypertrophy and proliferation via activation of the V1aR[25,26]. AVP-stimulated rat myocytes also express increased levels of ANP, a marker of hypertrophy[25]. The end result of AVP binding to V1aR is left ventricular hypertrophy and remodeling via vasoconstriction, increase in afterload, and myocyte hypertrophy.
V2R stimulation primarily leads to free water retention, which in turn causes an increase in preload, pulmonary vascular congestion and left sided filling pressures. The low output state of heart failure results in V2R activity, with nonosmotic stimulation of vasopressin release predominating, despite hypotonicity. In experimental CHF induced in a rat model, Xu and colleagues found increased AVP levels and increased AQP2 channel expression in the apical membrane of collecting ducts when compared with controls. When the CHF rats were treated with a V2R vasopressin antagonist, OPC 31260, they had increased aquaresis and plasma osmolality as well as decreased AQP2 expression[27].
In addition to hemodynamic alteration, increased water permeability via AQP2 channels leads to edema and hyponatremia[22]. Hyponatremia is a marker for advanced disease and poor outcome in CHF. In a retrospective analysis of OPTIME-CHF, patients with sodium levels in the lowest quartile had higher 60 d mortality and rehospitalization rates when compared to patients with higher sodium levels[28]. The presence of hyponatremia also limits the use of diuretics, as these agents only exacerbate loss of sodium, and ACE inhibitors, since hyponatremia is an independent risk factor for decline in renal function during treatment with such agents[29]. Hyponatremia is treated primarily via difficult to adhere to free water restriction.
AVP ANTAGONISM IN ADHF
Recently, specific antagonists to vasopressin have been developed as potentially useful agents for patients with heart failure and hyponatremia. In theory, antagonism of the V1aR, V2R or both may be beneficial in patients with heart failure. There are many different vasopressin antagonists, and some have been evaluated in patients with heart failure as outlined in Table 2.
Table 2.
Summary of key studies of vasopressin antagonists
| Vasopressin antagonist | Study | Design | Endpoint | Results |
| Lixivaptan (VPA985) | Martin et al[30], 1999 | 21 NYHA II and III patients randomized to placebo vs one of four doses (30, 75, 150, 250 mg) | Urinary AQP-2 excretion | Decrease in urinary AQP-2 excretion, increased solute-free water clearance and urine output, decreased urinary osmolality |
| Wong et al[32], 2003 | 44 hyponatremic patients randomized to placebo vs one of three doses (25, 125, or 250 mg bid) over a 7-d inpatient stay | Correction of hyponatremia | Increased free water clearance and serum sodium | |
| Abraham et al[31], 2006 | 42 patients with mild to moderate CHF randomized to placebo vs ascending single-dose drug (10-400 mg) | 24 h urine volume and serum sodium | Increased urine volume at 4 h and 24 h, increased serum sodium at higher doses | |
| BALANCE[35] | 650 CHF patients randomized to placebo vs lixivaptan | Correction of hyponatremia | Increased serum sodium levels | |
| Conivaptan | Udelson et al[41], 2001 | 142 NYHA III and IV patients randomized to placebo vs single IV-dose (10, 20, 40 mg) | Effect on hemodynamic parameters | Reduced PCWP, RAP Increased urine output |
| Goldsmith et al[42], 2008 | Dose-ranging pilot study of IV conivaptan in 170 randomized patients with worsening CHF | Assessment of global and respiratory status Effect on urine output | No change in status Increased urine output | |
| Russell et al[43], 2003 | 143 patients randomized to placebo vs one of three po doses | Change in time to reach 70% of peak O2 consumption | No change in exercise endpoint | |
| Zeltser et al[45], 2007 | 84 euvolemic or hypervolemic hyponatremic patients randomized to placebo vs IV conivaptan for 4 d (40 or 80 mg/d) | Change in serum sodium, measured by area under the sodium-time curve | Increased serum sodium | |
| Annane et al[46], 2009 (The Conivaptan Study Group) | 83 euvolemic or hypervolemic hyponatremic patients randomized to placebo vs po conivaptan for 5 d (40 or 80 mg/d) | Change in serum sodium, measured by area under the sodium-time curve | Increased serum sodium | |
| Tolvaptan (OPC-41061) | Gheorghiade et al[47], 2003 | 254 patients randomized to placebo vs 30, 45 or 60 mg/d for 25 d | Change in body weight | Decreased body weight, increased urine output, increased serum sodium, decreased edema |
| Gheorghiade et al[48], 2004 (ACTIV CHF) | Phase II study in 319 patients randomized to placebo vs 30, 60, or 90 mg/d for 60 d | Change in body weight at 24 h Heart failure outcomes | Significant decrease in body weight No change in worsening heart failure at 60 d | |
| Gheorghiade et al[42,50], 2007 (EVEREST) | Large 4133 patient multi-center randomized study of short and long term effects of tolvaptan in ADHF | CHF symptoms Mortality and heart failure related morbidity | Improvement in some CHF symptoms No difference in long-term mortality or morbidity | |
| Udelson et al[54], 2007 (METEOR) | 240 patients, NYHA II or III, randomized to placebo vs tolvaptan | LVEDV | No change in LVEDV at one year | |
| Udelson et al[55], 2008 | 181 patients, NYHA III and IV, randomized | Hemodynamic effects | Decreased PCWP, RAP, PAP |
IV: Intravenous; PO: Oral; PCWP: Pulmonary capillary wedge pressure; RAP: Right atrial pressure; O2: Oxygen; CHF: Congestive heart failure; ADHF: Acute decompensated heart failure; LVEDV: Left ventricular end diastolic volume; PAP: Pulmonary artery pressure.
Lixivaptan
The first agent studied was lixivaptan (or VPA-985, Cardiokine Inc, Philadelphia, PA), an oral, V2R selective, vasopressin antagonist. Martin and colleagues reported administering this agent at four different doses to 21 chronic NYHA functional class II and III patients and found decreased urinary AQP2 secretion (a marker of AVP action), increased solute-free water clearance and urine output, and decreased urine osmolality[30]. These results were confirmed in a single-ascending-dose 42 patient study of safety, efficacy, and tolerability of lixivaptan by the same authors[31]. Wong et al[32] also found a dose-dependent increase in sodium concentrations amongst 44 hyponatremic patients (six of which had CHF) receiving VPA-985. Higher doses (250 mg) caused dehydration and increases in vasopressin levels.
Abraham and colleagues further studied the role of lixivaptan in three phase III clinical trials. The LIBRA and HARMONY trials demonstrated safety of initiation and efficacy of lixivaptan in patients with euvolemic hyponatremia in the inpatient and outpatient settings, respectively[33,34]. The BALANCE (Treatment of Hyponatremia BAsed on LixivAptan in NYHA Class III/IV Cardiac Patient Evaluation) Trial was specific to hospitalized heart failure patients. The BALANCE trial was a large, international, multicenter, randomized, placebo-controlled, double blind study of 650 hospitalized CHF patients with serum sodium < 135 mEq/L. The primary endpoint was correction of hyponatremia, with additional endpoints including dyspnea, cognitive function and days of hospital-free survival[35]. Patients were treated with 50-100 mg of lixivaptan a day, twice daily, for 60 d. Though results have not been published, they were presented at an Federal Drug Administration (FDA) advisory committee meeting in 2013[36]. At seven days, there was a significant increase in serum sodium in the lixivaptan versus placebo group (2.5 mEq/L vs 1.3 mEq/L, P = 0.001). There was a nonsignificant early increase in mortality in the lixivaptan group however overall death rates and hospitalization rates were no different from the placebo group. In an extension study, long-term safety of lixivaptan was studied in a 28-wk open label study, results of which have not been published[37,38]. Lixivaptan is not yet FDA approved.
Conivaptan
Binding of V1aR by vasopressin plays an important role in cardiac contractility and remodeling. Therefore, a dual receptor (V1a and V2) antagonist, conivaptan (YM087), has also been evaluated in heart failure. Early experimental studies in animals showed the utility of intravenous conivaptan. In a canine model of AVP-induced CHF, infusion of intravenous conivaptan corrected poor cardiac hemodynamics[39]. In rats with post myocardial infarction CHF, intravenous conivaptan not only significantly improved right ventricular systolic pressure, left ventricular end-diastolic pressure, lung/body weight and right atrial pressure, but also, when compared to a V2 selective antagonist, increased the first derivative of left ventricular pressure, a measure of cardiac contractility[40].
One hundred, forty-two patients were randomized to either placebo or an intravenous dose of conivaptan at one of 3 different doses[41]. These patients had NYHA III or IV functional symptoms, but were stable outpatients that were admitted for placement of a right heart catheter and infusion of study drug. The investigators found a significant reduction in pulmonary capillary wedge and right atrial pressure. Additionally, urine output increased by 176 ± 18 mL/h in the high dose conivaptan group, without affecting systemic blood pressure, heart rate or serum electrolytes, including serum sodium. The beneficial hemodynamic effects of conivaptan therefore may be generalizable to CHF patients, and not just those with hyponatremia. Goldsmith and colleagues studied the use of intravenous conivaptan in a pilot study of 170 hospitalized patients with acute decompensated heart failure[42]. Their randomized placebo-controlled multi-center trial administered conivaptan for 48 h (as opposed to 12 h in the former study) alongside loop diuretics and found an average 1.0 to 1.5 L/d increase in urine output. They did not measure hemodynamics, however they found no significant change in systemic blood pressure.
The oral formulation of conivaptan has also been investigated[43]. In a 12-wk study in patients with NYHA class II-IV symptoms, 343 patients were randomized to either placebo or one of three doses of oral conivaptan. Using the hypothesis that a dual vasopressin receptor blocker would cause pulmonary vasodilatation and aquaresis and a subsequent improvement in submaximal exercise, the primary endpoint of the study was a change in the time to reach 70% of peak oxygen consumption[44]. However, there were no differences in any exercise endpoints between the placebo arm and the three groups of patients on different doses of conivaptan.
Conivaptan has also proven efficacy in treatment of hyponatremia. Intravenous conivaptan administered to euvolemic and hypervolemic hyponatremic patients significantly increased serum sodium concentrations 9.4 ± 0.8 mEq/L at a conivaptan dose of 80 mg/d after four days of treatment[45]. Oral conivaptan showed similar results in a study of 84 hyponatremic patients (33% had CHF), with an increase in sodium of 9.1 ± 0.9 mEq/L at the end of five days of treatment with 80 mg/d of oral conivaptan[46].
Intravenous conivaptan (Vaprisol, Astellas Pharma US, Inc.) was the first Federal Drug Administration (FDA) approved vaptan for the treatment of euvolemic hyponatremia. It is currently approved for treatment of euvolemic and hypervolemic hyponatremia in hospitalized patients, including those with heart failure. Administration involves an additional loading dose of 20 mg IV over 30 min followed by a 20 mg continuous infusion over 24 h. Metabolism is via cytochrome P450 3A4. Conivaptan is not directly approved for the treatment of acute heart failure without hyponatremia. There are no planned future trials studying oral conivaptan.
Tolvaptan
Tolvaptan (OPC-41061), a V2 selective vasopressin receptor antagonist, has been the most studied drug of its class in patients with heart failure. Gheorghiade et al[47] reported the results of a study of 254 patients who were randomized to either placebo or three different doses of tolvaptan for 25 d. The primary endpoint was change in body weight. Additional endpoints included urine sodium excretion, urine volume, urine osmolality, and ankle edema measurements. A decrease in body weight of about 1 kg was found after the first day that was maintained throughout the study. There was also an increase in urine volume and a normalization of serum sodium with tolvaptan.
A second dose ranging phase II study, ACTIV in CHF (Acute and Chronic Therapeutic Effect of a Vasopressin Antagonist in Congestive Heart Failure), was performed with a primary endpoint of change in body weight at 24 h[48]. Additionally, heart failure outcomes including death, hospitalization, or unscheduled visits for heart failure at 60 d were collected. Body weight at 24 h after tolvaptan administration decreased by 1.8 kg, 2.1 kg, and 2.05 kg in the 30, 60, and 90 mg per day arms compared to a decrease of 0.6 kg in the placebo arm. This decrease occurred without a change in renal function or hypokalemia. There was no difference in the secondary outcome of worsening heart failure at 60 d. Additionally, there was an increase in serum sodium in the tolvaptan arms. Although the study was not powered for mortality, a post-hoc analysis of patients with high blood urea nitrogen levels or severe congestive symptoms demonstrated a statistically higher mortality rate in placebo vs tolvaptan treatment groups.
Study of Ascending Levels of Tolvaptan in Hyponatremia 1 and 2 (SALT-1 and SALT-2) were two simultaneous phase III trials published in 2006 by Schrier et al[49]. Patients with euvolemic and hypervolemic hyponatremia demonstrated an increase in serum sodium levels by day 4 and sustained at day 30. Hyponatremia recurred when the drug was discontinued after the 30-d treatment period.
The Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Trial was a large event-driven, randomized, double-blind, placebo-controlled study of 4133 patients hospitalized with acute heart failure. Patients were randomized to placebo or a minimum of 60 d of tolvaptan at 30 mg/d. Short-term clinical effects were examined in two identical trials of 2048 and 2085 patients[50]. There were statistically significant differences in endpoints for body weight and dyspnea in both trials’ tolvaptan arms when compared to placebo, significant decrease in edema in only one trial, and no change in global clinical status in either trial. There was also greater improvement in other physician-assessed CHF signs and symptoms such as dyspnea, edema, orthopnea and jugular venous distention (P < 0.05) as well as increased serum sodium levels in the tolvaptan group[51]. Patients receiving tolvaptan were discharged on lower doses of furosemide. Although the short-term trials from the EVEREST group showed improvement in congestion without significant adverse effects, the long-term outcome trial did not show any benefit on all-cause mortality or heart failure-related morbidity[52]. In a post-hoc analysis of the EVEREST trial studying the effect of QRS duration on heart failure outcomes, a prolonged QRS interval was associated with poorer outcomes however use of tolvaptan did not affect these endpoints[53].
Effects of tolvaptan on hemodynamics and left ventricular physiology have also been assessed. The METEOR (Multicenter Evaluation of Tolvaptan Effects on Left Ventricular Remodeling) Trial, which investigated tolvaptan versus placebo in 240 patients with NYHA class IIor III symptoms and left ventricular ejection fraction less than or equal to 30%, showed no difference in the primary end point of left ventricular end diastolic volume at the end of one year[54]. Although not powered for such outcomes, the study did show a decrease in morbidity and mortality in the tolvaptan-treated patients. In order to further substantiate the findings that tolvaptan improved congestion, Udelson and colleagues assessed the acute hemodynamic effects of tolvaptan in 181 patients with symptomatic heart failure (NYHA class III or IV)[55]. They found that tolvaptan effectively decreased pulmonary capillary wedge pressure, right atrial pressure and pulmonary arterial pressure.
Similar to other vasopressin antagonists, tolvaptan improves serum sodium concentrations in hyponatremic patients[49]. In a study of 448 patients with euvolemic or hypervolemic hyponatremia, tolvaptan significantly increased sodium levels at day 4 and day 30. Interestingly, though EVEREST was a study specifically of patients with acutely decompensated heart failure, only about 8% of patients had significant hyponatremia (< 134 mEq/L)[52]. In this subset of patients however, there was a significant rise in serum sodium seen as early as day 1.
Tolvaptan (Otsuka, Inc.) is the only oral vasopressin antagonist that is FDA approved[56]. Tolvaptan is approved specifically for treatment of euvolemic or hypervolemic hyponatremia (per FDA label, “serum sodium < 125 mEq/L or less marked hyponatremia that is symptomatic and has resisted correction with fluid restriction”). Starting dosage is typically 15 mg/d and can be increased to 30 mg/d at the second dose but should not exceed 60 mg/d. Initially approved for longer term treatment, due to liver failure observed in a study of patients with underlying cirrhosis, the FDA revised its label in April 2013 and limits treatment duration to 30 d[57]. It should be noted that in the SALTWATER Trial, an open-label extension of the SALT-1 and SALT-2 Trials, in which patients with hyponatremia were treated with tolvaptan for a mean duration of 701 d, six patients experienced drug-related adverse effects, all related to sodium levels[58]. Tolvaptan should be initiated or re-initiated only in hospitalized patients where serum sodium levels can be closely monitored. Serum sodium should not be too rapidly corrected (faster than 12 mEq/24 h) as this can lead to neurologic effects such as dysarthria, mutism, dysphagia, lethargy, affective changes, spastic quadriparesis, seizures, coma and death. Tolvaptan is primarily metabolized by cytochrome P450 3A4, and therefore attention should be paid to potential drug interactions. As with other vaptans, side effects include thirst, dry mouth and frequent urination.
CLINICAL IMPLICATIONS
Vasopressin antagonists are a new group of drugs that provide effective aquaresis without effecting morbidity or mortality. Thus far, the vaptans are approved only for use in treatment of hyponatremia (with or without hypervolemia). Additionally, most studies with CHF patients have included patients only with reduced, and not preserved, systolic function. Although not yet fully studied, these drugs may prove to be beneficial for the treatment of heart failure as a replacement for or in conjunction with diuretics. They do not cause hypokalemia and do not appear to be associated with upregulation of the neurohormonal system. Although these agents have not been shown to reduce mortality in the long term, perhaps their use will allow one to administer lower diuretic doses resulting in less electrolyte disturbance and improved patient safety. Currently, use of a vaptan may be considered in volume overloaded patients who either have or are developing hyponatremia. Routine use of vasopressin antagonists has currently not been shown to be beneficial.
When considering the initiation of a vaptan, it is important to think about duration of therapy, route of administration and whether these medications should be used in the inpatient or outpatient setting. Most studies have only used these medications for short durations and there is limited data to support long-term use of vaptans. Therefore, although the effective increase in serum sodium concentration has been shown to last through treatment duration, it must be emphasized that these medications have only short term effect in their current role, and do not by any means aim to cure the underlying disease process. Although vaptan use has been theorized to be beneficial in the short-term acute setting, many of the studies were done using stable outpatients. It is unclear how useful these drugs will be in acutely decompensated heart failure. The most thoroughly studied of these medications, tolvaptan, is an oral agent, and therefore absorption may be affected by gut edema. One possibility is for patients to acutely receive intravenous conivaptan as inpatients and then be transitioned over to oral tolvaptan, on which they may go home for a short duration such as 30 d. Lixivaptan is an additional promising oral agent that is not yet FDA approved. Vasopressin antagonists should only be initiated inpatient so that sodium levels can be closely monitored and rapid correction, which can be detrimental, can be avoided.
CONCLUSION
Further investigation of vasopressin antagonists is needed in patients with both preserved and reduced ejection fractions. Acute heart failure has been a challenge to treat thus far. Results from ongoing studies of vasopressin antagonists may change the treatment approach in both inpatient and outpatient heart failure patients. However, there is still work to be done to decrease long-term morbidity and mortality in this patient population, which the vaptans do not seem to promise.
Footnotes
P- Reviewer: Joseph J, Lin J S- Editor: Ji FF L- Editor: A E- Editor: Wu HL
References
- 1.Neuberg GW, Miller AB, O’Connor CM, Belkin RN, Carson PE, Cropp AB, Frid DJ, Nye RG, Pressler ML, Wertheimer JH, et al. Diuretic resistance predicts mortality in patients with advanced heart failure. Am Heart J. 2002;144:31–38. doi: 10.1067/mhj.2002.123144. [DOI] [PubMed] [Google Scholar]
- 2.Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, Jessup M, Konstam MA, Mancini DM, Michl K, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation. 2009;119:e391–e479. doi: 10.1161/CIRCULATIONAHA.109.192065. [DOI] [PubMed] [Google Scholar]
- 3.Cooper HA, Dries DL, Davis CE, Shen YL, Domanski MJ. Diuretics and risk of arrhythmic death in patients with left ventricular dysfunction. Circulation. 1999;100:1311–1315. doi: 10.1161/01.cir.100.12.1311. [DOI] [PubMed] [Google Scholar]
- 4.Bart BA, Goldsmith SR. Aggravated renal dysfunction and the acute management of advanced chronic heart failure. Am Heart J. 1999;138:200–202. doi: 10.1016/s0002-8703(99)70101-8. [DOI] [PubMed] [Google Scholar]
- 5.Liamis G, Milionis H, Elisaf M. A review of drug-induced hyponatremia. Am J Kidney Dis. 2008;52:144–153. doi: 10.1053/j.ajkd.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 6.Francis GS, Benedict C, Johnstone DE, Kirlin PC, Nicklas J, Liang CS, Kubo SH, Rudin-Toretsky E, Yusuf S. Comparison of neuroendocrine activation in patients with left ventricular dysfunction with and without congestive heart failure. A substudy of the Studies of Left Ventricular Dysfunction (SOLVD) Circulation. 1990;82:1724–1729. doi: 10.1161/01.cir.82.5.1724. [DOI] [PubMed] [Google Scholar]
- 7.Francis GS, Cohn JN, Johnson G, Rector TS, Goldman S, Simon A. Plasma norepinephrine, plasma renin activity, and congestive heart failure. Relations to survival and the effects of therapy in V-HeFT II. The V-HeFT VA Cooperative Studies Group. Circulation. 1993;87:VI40–VI48. [PubMed] [Google Scholar]
- 8.Felker GM, Lee KL, Bull DA, Redfield MM, Stevenson LW, Goldsmith SR, LeWinter MM, Deswal A, Rouleau JL, Ofili EO, et al. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med. 2011;364:797–805. doi: 10.1056/NEJMoa1005419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cuffe MS, Califf RM, Adams KF, Benza R, Bourge R, Colucci WS, Massie BM, O’Connor CM, Pina I, Quigg R, et al. Short-term intravenous milrinone for acute exacerbation of chronic heart failure: a randomized controlled trial. JAMA. 2002;287:1541–1547. doi: 10.1001/jama.287.12.1541. [DOI] [PubMed] [Google Scholar]
- 10.Colucci WS, Elkayam U, Horton DP, Abraham WT, Bourge RC, Johnson AD, Wagoner LE, Givertz MM, Liang CS, Neibaur M, et al. Intravenous nesiritide, a natriuretic peptide, in the treatment of decompensated congestive heart failure. Nesiritide Study Group. N Engl J Med. 2000;343:246–253. doi: 10.1056/NEJM200007273430403. [DOI] [PubMed] [Google Scholar]
- 11.Publication Committee for the VMAC Investigators (Vasodilatation in the Management of Acute CHF) Intravenous nesiritide vs nitroglycerin for treatment of decompensated congestive heart failure: a randomized controlled trial. JAMA. 2002;287:1531–1540. doi: 10.1001/jama.287.12.1531. [DOI] [PubMed] [Google Scholar]
- 12.Wang DJ, Dowling TC, Meadows D, Ayala T, Marshall J, Minshall S, Greenberg N, Thattassery E, Fisher ML, Rao K, et al. Nesiritide does not improve renal function in patients with chronic heart failure and worsening serum creatinine. Circulation. 2004;110:1620–1625. doi: 10.1161/01.CIR.0000141829.04031.25. [DOI] [PubMed] [Google Scholar]
- 13.Sackner-Bernstein JD, Kowalski M, Fox M, Aaronson K. Short-term risk of death after treatment with nesiritide for decompensated heart failure: a pooled analysis of randomized controlled trials. JAMA. 2005;293:1900–1905. doi: 10.1001/jama.293.15.1900. [DOI] [PubMed] [Google Scholar]
- 14.O’Connor CM, Starling RC, Hernandez AF, Armstrong PW, Dickstein K, Hasselblad V, Heizer GM, Komajda M, Massie BM, McMurray JJ, et al. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med. 2011;365:32–43. doi: 10.1056/NEJMoa1100171. [DOI] [PubMed] [Google Scholar]
- 15.Chen HH, Anstrom KJ, Givertz MM, Stevenson LW, Semigran MJ, Goldsmith SR, Bart BA, Bull DA, Stehlik J, LeWinter MM, et al. Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: the ROSE acute heart failure randomized trial. JAMA. 2013;310:2533–2543. doi: 10.1001/jama.2013.282190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turner RA, Pierce JG, du Vigneaud V. The purification and the amino acid content of vasopressin preparations. J Biol Chem. 1951;191:21–28. [PubMed] [Google Scholar]
- 17.Brown D, Nielsen S. Cell biology of vasopressin action. In: Brenner BM, ed , editors. Brenner and rector’s the kidney. 8th ed. Philadelphia, PA: Saunders; 2007. [Google Scholar]
- 18.Chatterjee K. Neurohormonal activation in congestive heart failure and the role of vasopressin. Am J Cardiol. 2005;95:8B–13B. doi: 10.1016/j.amjcard.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 19.Aoyagi T, Koshimizu TA, Tanoue A. Vasopressin regulation of blood pressure and volume: findings from V1a receptor-deficient mice. Kidney Int. 2009;76:1035–1039. doi: 10.1038/ki.2009.319. [DOI] [PubMed] [Google Scholar]
- 20.Russell SD, DeWald T. Vasopressin receptor antagonists. Therapeutic potential in the management of acute and chronic heart failure. Am J Cardiovasc Drugs. 2003;3:13–20. doi: 10.2165/00129784-200303010-00002. [DOI] [PubMed] [Google Scholar]
- 21.Cohn JN, Levine TB, Francis GS, Goldsmith S. Neurohumoral control mechanisms in congestive heart failure. Am Heart J. 1981;102:509–514. doi: 10.1016/0002-8703(81)90739-0. [DOI] [PubMed] [Google Scholar]
- 22.Goldsmith SR, Francis GS, Cowley AW, Levine TB, Cohn JN. Increased plasma arginine vasopressin levels in patients with congestive heart failure. J Am Coll Cardiol. 1983;1:1385–1390. doi: 10.1016/s0735-1097(83)80040-0. [DOI] [PubMed] [Google Scholar]
- 23.Uretsky BF, Verbalis JG, Generalovich T, Valdes A, Reddy PS. Plasma vasopressin response to osmotic and hemodynamic stimuli in heart failure. Am J Physiol. 1985;248:H396–H402. doi: 10.1152/ajpheart.1985.248.3.H396. [DOI] [PubMed] [Google Scholar]
- 24.Hupf H, Grimm D, Riegger GA, Schunkert H. Evidence for a vasopressin system in the rat heart. Circ Res. 1999;84:365–370. doi: 10.1161/01.res.84.3.365. [DOI] [PubMed] [Google Scholar]
- 25.Hiroyama M, Wang S, Aoyagi T, Oikawa R, Sanbe A, Takeo S, Tanoue A. Vasopressin promotes cardiomyocyte hypertrophy via the vasopressin V1A receptor in neonatal mice. Eur J Pharmacol. 2007;559:89–97. doi: 10.1016/j.ejphar.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 26.Yang XD, Zhao LY, Zheng QS, Li X. Effects of arginine vasopressin on growth of rat cardiac fibroblasts: role of V1 receptor. J Cardiovasc Pharmacol. 2003;42:132–135. doi: 10.1097/00005344-200307000-00020. [DOI] [PubMed] [Google Scholar]
- 27.Xu DL, Martin PY, Ohara M, St John J, Pattison T, Meng X, Morris K, Kim JK, Schrier RW. Upregulation of aquaporin-2 water channel expression in chronic heart failure rat. J Clin Invest. 1997;99:1500–1505. doi: 10.1172/JCI119312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klein L, O’Connor CM, Leimberger JD, Gattis-Stough W, Piña IL, Felker GM, Adams KF, Califf RM, Gheorghiade M. Lower serum sodium is associated with increased short-term mortality in hospitalized patients with worsening heart failure: results from the Outcomes of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure (OPTIME-CHF) study. Circulation. 2005;111:2454–2460. doi: 10.1161/01.CIR.0000165065.82609.3D. [DOI] [PubMed] [Google Scholar]
- 29.Packer M, Lee WH, Kessler PD, Medina N, Yushak M, Gottlieb SS. Identification of hyponatremia as a risk factor for the development of functional renal insufficiency during converting enzyme inhibition in severe chronic heart failure. J Am Coll Cardiol. 1987;10:837–844. doi: 10.1016/s0735-1097(87)80278-4. [DOI] [PubMed] [Google Scholar]
- 30.Martin PY, Abraham WT, Lieming X, Olson BR, Oren RM, Ohara M, Schrier RW. Selective V2-receptor vasopressin antagonism decreases urinary aquaporin-2 excretion in patients with chronic heart failure. J Am Soc Nephrol. 1999;10:2165–2170. doi: 10.1681/ASN.V10102165. [DOI] [PubMed] [Google Scholar]
- 31.Abraham WT, Shamshirsaz AA, McFann K, Oren RM, Schrier RW. Aquaretic effect of lixivaptan, an oral, non-peptide, selective V2 receptor vasopressin antagonist, in New York Heart Association functional class II and III chronic heart failure patients. J Am Coll Cardiol. 2006;47:1615–1621. doi: 10.1016/j.jacc.2005.11.071. [DOI] [PubMed] [Google Scholar]
- 32.Wong F, Blei AT, Blendis LM, Thuluvath PJ. A vasopressin receptor antagonist (VPA-985) improves serum sodium concentration in patients with hyponatremia: a multicenter, randomized, placebo-controlled trial. Hepatology. 2003;37:182–191. doi: 10.1053/jhep.2003.50021. [DOI] [PubMed] [Google Scholar]
- 33.Abraham WT, Hensen J, Gross PA, Bichet DG, Josiassen RC, Chafekar DS, Orlandi C. Lixivaptan safely and effectively corrects serum sodium concentrations in hospitalized patients with euvolemic hyponatremia. Kidney Int. 2012;82:1223–1230. doi: 10.1038/ki.2012.275. [DOI] [PubMed] [Google Scholar]
- 34.Abraham WT, Decaux G, Josiassen RC, Yagil Y, Kopyt N, Thacker HP, Mannelli M, Bichet DG, Orlandi C. Oral lixivaptan effectively increases serum sodium concentrations in outpatients with euvolemic hyponatremia. Kidney Int. 2012;82:1215–1222. doi: 10.1038/ki.2012.274. [DOI] [PubMed] [Google Scholar]
- 35.Abraham WT, Aranda JM, Boehmer JP, Elkayam U, Gilbert EM, Gottlieb SS, Hasenfuss G, Kukin M, Lowes BD, O’Connell JB, et al. Rationale and design of the treatment of hyponatremia based on lixivaptan in NYHA class III/IV cardiac patient evaluation (THE BALANCE) study. Clin Transl Sci. 2010;3:249–253. doi: 10.1111/j.1752-8062.2010.00217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.FDA Cardio-Renal Drugs Advisory Committee Meeting. Lixivaptan for the treatment of symptomatic euvolemic hyponatremia associated with syndrome of inappropriate antidiuretic hormone (SIADH) and symptomatic hypervolemic hyponatremia associated with heart failure (September 13, 2012) Available from: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/CardiovascularandRenalDrugsAdvisoryCommittee/UCM318869.pdf.
- 37.Zmily HD, Alani A, Ghali JK. Evaluation of lixivaptan in euvolemic and hypervolemic hyponatremia and heart failure treatment. Expert Opin Drug Metab Toxicol. 2013;9:645–655. doi: 10.1517/17425255.2013.783566. [DOI] [PubMed] [Google Scholar]
- 38.CardioKine Inc. international, multicenter study of a twenty-eight week, open-label, titrated oral Lixivaptan administration in patients with chronic hyponatremia: Extension to studies CK-LX3401, 3405, and 3430. NCT01056848 [updated 2011. accessed January 21] Available from: http://clinicaltrials.gov/ct2/show/NCT01056848?term=lixivaptan&rank=7.
- 39.Yatsu T, Kusayama T, Tomura Y, Arai Y, Aoki M, Tahara A, Wada K, Tsukada J. Effect of conivaptan, a combined vasopressin V(1a) and V(2) receptor antagonist, on vasopressin-induced cardiac and haemodynamic changes in anaesthetised dogs. Pharmacol Res. 2002;46:375–381. doi: 10.1016/s1043661802002062. [DOI] [PubMed] [Google Scholar]
- 40.Wada K, Fujimori A, Matsukawa U, Arai Y, Sudoh K, Yatsu T, Sasamata M, Miyata K. Intravenous administration of conivaptan hydrochloride improves cardiac hemodynamics in rats with myocardial infarction-induced congestive heart failure. Eur J Pharmacol. 2005;507:145–151. doi: 10.1016/j.ejphar.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 41.Udelson JE, Orlandi C, Ouyang J, Krasa H, Zimmer CA, Frivold G, Haught WH, Meymandi S, Macarie C, Raef D, et al. Acute hemodynamic effects of tolvaptan, a vasopressin V2 receptor blocker, in patients with symptomatic heart failure and systolic dysfunction: an international, multicenter, randomized, placebo-controlled trial. J Am Coll Cardiol. 2008;52:1540–1545. doi: 10.1016/j.jacc.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 42.Goldsmith SR, Elkayam U, Haught WH, Barve A, He W. Efficacy and safety of the vasopressin V1A/V2-receptor antagonist conivaptan in acute decompensated heart failure: a dose-ranging pilot study. J Card Fail. 2008;14:641–647. doi: 10.1016/j.cardfail.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 43.Russell SD, Adams KF, Shaw JP, Gattis WA, O’Connor CM. Results of twelve week double-blind, placebo-controlled, multicenter study of oral conivaptan to assess functional capacity in patients with class III chronic heart failure. J Card Fail. 2003;9 Suppl:S60. [Google Scholar]
- 44.Russell SD, Selaru P, Pyne DA, Ghazzi MM, Massey KD, Pressler M, Serikoff A, Coats AJ. Rationale for use of an exercise end point and design for the ADVANCE (A Dose evaluation of a Vasopressin ANtagonist in CHF patients undergoing Exercise) trial. Am Heart J. 2003;145:179–186. doi: 10.1067/mhj.2003.39. [DOI] [PubMed] [Google Scholar]
- 45.Zeltser D, Rosansky S, van Rensburg H, Verbalis JG, Smith N. Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia. Am J Nephrol. 2007;27:447–457. doi: 10.1159/000106456. [DOI] [PubMed] [Google Scholar]
- 46.Annane D, Decaux G, Smith N. Efficacy and safety of oral conivaptan, a vasopressin-receptor antagonist, evaluated in a randomized, controlled trial in patients with euvolemic or hypervolemic hyponatremia. Am J Med Sci. 2009;337:28–36. doi: 10.1097/MAJ.0b013e31817b8148. [DOI] [PubMed] [Google Scholar]
- 47.Gheorghiade M, Niazi I, Ouyang J, Czerwiec F, Kambayashi J, Zampino M, Orlandi C. Vasopressin V2-receptor blockade with tolvaptan in patients with chronic heart failure: results from a double-blind, randomized trial. Circulation. 2003;107:2690–2696. doi: 10.1161/01.CIR.0000070422.41439.04. [DOI] [PubMed] [Google Scholar]
- 48.Gheorghiade M, Gattis WA, O’Connor CM, Adams KF, Elkayam U, Barbagelata A, Ghali JK, Benza RL, McGrew FA, Klapholz M, et al. Effects of tolvaptan, a vasopressin antagonist, in patients hospitalized with worsening heart failure: a randomized controlled trial. JAMA. 2004;291:1963–1971. doi: 10.1001/jama.291.16.1963. [DOI] [PubMed] [Google Scholar]
- 49.Schrier RW, Gross P, Gheorghiade M, Berl T, Verbalis JG, Czerwiec FS, Orlandi C. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med. 2006;355:2099–2112. doi: 10.1056/NEJMoa065181. [DOI] [PubMed] [Google Scholar]
- 50.Gheorghiade M, Konstam MA, Burnett JC, Grinfeld L, Maggioni AP, Swedberg K, Udelson JE, Zannad F, Cook T, Ouyang J, et al. Short-term clinical effects of tolvaptan, an oral vasopressin antagonist, in patients hospitalized for heart failure: the EVEREST Clinical Status Trials. JAMA. 2007;297:1332–1343. doi: 10.1001/jama.297.12.1332. [DOI] [PubMed] [Google Scholar]
- 51.Pang PS, Gheorghiade M, Dihu J, Swedberg K, Khan S, Maggioni AP, Grinfeld L, Zannad F, Burnett JC, Ouyang J, et al. Effects of tolvaptan on physician-assessed symptoms and signs in patients hospitalized with acute heart failure syndromes: analysis from the efficacy of vasopressin antagonism in heart failure outcome study with tolvaptan (EVEREST) trials. Am Heart J. 2011;161:1067–1072. doi: 10.1016/j.ahj.2011.02.027. [DOI] [PubMed] [Google Scholar]
- 52.Konstam MA, Gheorghiade M, Burnett JC, Grinfeld L, Maggioni AP, Swedberg K, Udelson JE, Zannad F, Cook T, Ouyang J, et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA. 2007;297:1319–1331. doi: 10.1001/jama.297.12.1319. [DOI] [PubMed] [Google Scholar]
- 53.Wang NC, Maggioni AP, Konstam MA, Zannad F, Krasa HB, Burnett JC, Grinfeld L, Swedberg K, Udelson JE, Cook T, et al. Clinical implications of QRS duration in patients hospitalized with worsening heart failure and reduced left ventricular ejection fraction. JAMA. 2008;299:2656–2666. doi: 10.1001/jama.299.22.2656. [DOI] [PubMed] [Google Scholar]
- 54.Udelson JE, McGrew FA, Flores E, Ibrahim H, Katz S, Koshkarian G, O’Brien T, Kronenberg MW, Zimmer C, Orlandi C, et al. Multicenter, randomized, double-blind, placebo-controlled study on the effect of oral tolvaptan on left ventricular dilation and function in patients with heart failure and systolic dysfunction. J Am Coll Cardiol. 2007;49:2151–2159. doi: 10.1016/j.jacc.2007.01.091. [DOI] [PubMed] [Google Scholar]
- 55.Udelson JE, Smith WB, Hendrix GH, Painchaud CA, Ghazzi M, Thomas I, Ghali JK, Selaru P, Chanoine F, Pressler ML, et al. Acute hemodynamic effects of conivaptan, a dual V(1A) and V(2) vasopressin receptor antagonist, in patients with advanced heart failure. Circulation. 2001;104:2417–2423. doi: 10.1161/hc4501.099313. [DOI] [PubMed] [Google Scholar]
- 56.Thompson CA. FDA approves oral vasopressin antagonist. Am J Health Syst Pharm. 2009;66:1154. doi: 10.2146/news090054. [DOI] [PubMed] [Google Scholar]
- 57.Samsca (tolvaptan) Drug safety communication - FDA limits duration and usage due to possible liver injury leading to organ transplant or death [Updated 2013. Accessed January 21] Available from: http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm350185.htm.
- 58.Berl T, Quittnat-Pelletier F, Verbalis JG, Schrier RW, Bichet DG, Ouyang J, Czerwiec FS. Oral tolvaptan is safe and effective in chronic hyponatremia. J Am Soc Nephrol. 2010;21:705–712. doi: 10.1681/ASN.2009080857. [DOI] [PMC free article] [PubMed] [Google Scholar]