

Abstract
Hemolytic uremic syndrome (HUS) is a disease characterized by hemolysis, thrombocytopenia, and acute kidney injury, although other organs may be involved. Most cases are due to infection with Shiga toxin-producing Escherichia coli (STEC). Early identification and initiation of best supportive care, with microbiological input to identify the pathogen, result in a favorable outcome in most patients. The remaining 10% of HUS cases are classed together as atypical HUS and have a diverse etiology. The majority are due to inherited or acquired abnormalities that lead to a failure to control complement activation. Atypical HUS occurring in other situations (for example, related to pregnancy or kidney transplantation) may also involve excessive complement activation. Plasma therapies can reverse defective complement control, and it is now possible to specifically target complement activation. This has led to improved outcomes in patients with atypical forms of HUS. We will review our current understanding of the pathogenesis of HUS and how this has led to advances in patient care.
Introduction
The thrombotic microangiopathies (TMAs) are a group of diseases that are characterized by a microangiopathic hemolytic anemia, thrombocytopenia, and the occlusion of small vessels by thrombi (Figure 1). Endothelial injury and an inability to control coagulation are common features of all of these diseases. The clinical manifestations of a TMA depend on the site of vascular involvement and, on the basis of clinical presentation, were classically divided into two main types: HUS and thrombotic thrombocytopenic purpura (TTP). HUS typically affects the microvasculature of the kidney causing acute kidney injury (AKI), whereas in TTP neurological disease is more common. However, it is clear that disease classification based purely on clinical presentation can be misleading, as HUS can cause neurological disease (as well as affecting many other organs) and TTP can affect the kidney. In addition, it is clear that HUS does not represent a single disease with a common pathogenesis. Instead, HUS is a group of diseases that are due to different environmental or genetic factors and that have a similar clinical presentation.
Figure 1. Glomerular pathology in hemolytic uremic syndrome.
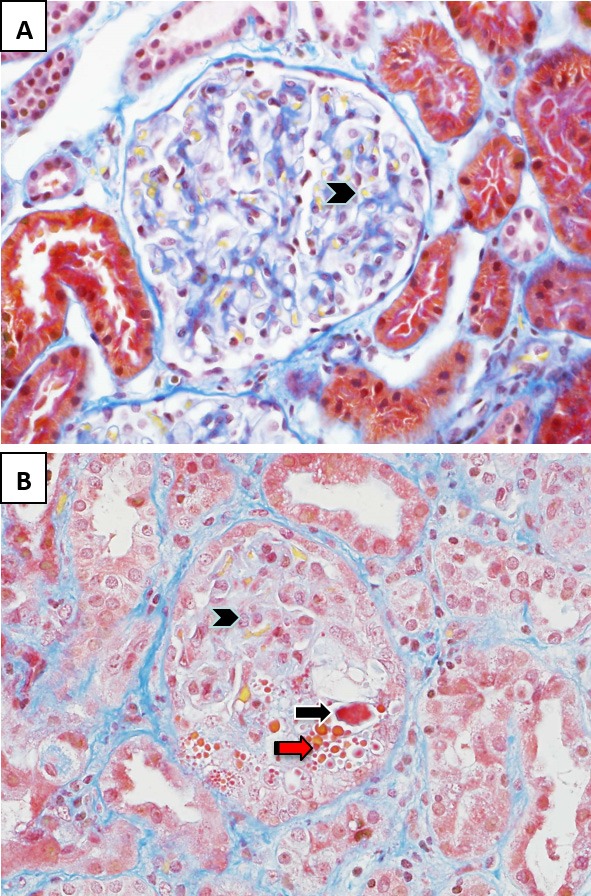
(A) In the normal glomerulus, patent capillary lumina containing erythrocytes stain yellow (arrowhead). (B) In hemolytic uremic syndrome, the glomerular capillary loops contain fibrin thrombi and microthrombi that stain bright red (black arrow). There is endothelial cell swelling with obliteration of some of the capillary lumina (arrowhead) and red cell fragmentation (red arrow). Stain: Martius scarlet blue trichrome; magnification: 400×.
As our understanding of the molecular basis of these diseases increases, it is now possible to classify HUS and TTP on the basis of etiology. Most cases of HUS follow infection with Shiga toxin-producing enteric pathogens or excessive activation of the complement system. TTP is due to diminished activity of ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13), an enzyme responsible for the breakdown of multimeric von Willebrand factor, a pro-thrombotic protein produced by the endothelium, megakaryocytes, and vascular connective tissue [1]. It is evident that a disruption of endothelium function is common to all TMAs and therefore it is possible that there are links between the pathways that lead to specific diseases.
Differentiating between types of TMA on the basis of clinical parameters is a vital part of patient care. However, this is not always possible, and diagnostic accuracy requires identification of the underlying abnormality. Accurate and early diagnosis is increasingly important as new treatments become available that are specific for different types of TMA and with increasing evidence that early initiation of treatment improves patient outcomes. This review will discuss the pathogenic mechanisms that cause the different types of HUS, clinical and laboratory diagnosis of HUS, and the available treatment options.
Pathogenesis of hemolytic uremic syndrome
Although HUS is primarily a disease of the kidney, neurological involvement is common, potentially life-threatening, and an important determinant of mortality [2]. The incidence of HUS, usually as a consequence of infection, is higher in children; The overall incidence in the general population, including adults, is approximately 1 to 2 in 100,000 [3,4]. It can cause dialysis-requiring AKI, severe multi-organ failure, and death (<5%); however, in most cases, complete recovery occurs (75%), and the remaining patients have a reduced glomerular filtration rate, hypertension, or proteinuria [5]. Neurological involvement can present with irritability, confusion, focal deficit, seizures, or coma, and involvement of the heart, gastrointestinal tract, and pancreas has been described.
Most cases of HUS (90%) occur following infection with STEC, typically serotype O157. Other serotypes of Escherichia coli can produce toxin, and the largest recorded outbreak of STEC HUS occurred in Northern Europe in 2011 because of infection with serotype O104 [6]. Infection is acquired by ingestion of contaminated food and is followed about 3 days later, although not invariably [7], by abdominal pain and diarrhea that progresses to a colitic illness with bloody diarrhea (hence its previous name: diarrhea-positive HUS). Features of TMA and AKI develop over the following 3 to 4 days in 5% to 15% of patients with STEC infection [8]. Why only a small proportion of patients develop HUS following infection is not known. A genetic predisposition, perhaps involving genes associated with other forms of TMA, is possible, and complement gene mutations have been described in patients with STEC HUS and may predispose them to a poor outcome [9].
Pathogenic STEC strains express adhesins, which are thought to promote adherence to the intestinal epithelium following which there is translocation of toxin (usually Shiga toxin type 2) through the epithelium. It has been proposed that the toxin could then bind to circulating leukocytes [10–12], although the evidence for this is conflicting [13]. The Shiga toxin receptor is a glycosphingolipid: globotriaosylceramide (Gb3). When Shiga toxin binds Gb3, the complex is internalized and transported to the endoplasmic reticulum, where the complex is cleaved to release a protease into the cytoplasm. This protease inhibits ribosomal function and protein synthesis, leading to cell death. It can also activate signaling pathways, inducing an inflammatory response in affected cells [14]. Although Gb3 is found in other organs, the kidney is most susceptible to injury. This may be due to higher levels of Gb3 on renal cells, particularly the glomerular endothelium (but also podocytes, mesangial, and tubular epithelial cells [15]), the high blood flow to the kidney, or a greater susceptibility of renal cells to the effects of the toxin [16]. The expression of Gb3 at other sites (for example, within the central nervous system) explains the extra-renal manifestations of STEC HUS.
Atypical hemolytic uremic syndrome
Atypical HUS (aHUS) accounts for most of the remaining 10% of cases of HUS. With aHUS, as with STEC HUS, the kidneys are most frequently involved, but any organ can be affected. It is difficult to distinguish aHUS from STEC HUS or TTP on clinical grounds alone. A diagnosis of aHUS should be considered once STEC infection or reduced ADAMTS13 activity has been excluded. Although it is more common in childhood, aHUS can occur at any age and has a worse prognosis than STEC HUS, with a 3-year patient or kidney survival of 50% [17].
Genetic or acquired defects in control of complement activation are commonly found in patients with aHUS. Complement is a complex system of enzymes, effector proteins, and inhibitors and forms part of the innate immune system. Low levels of circulating complement protein C3, suggestive of consumption, were first described in 1973 [18]. Twenty-five years later, loss-of-function mutations in the complement regulatory protein complement factor H (CFH) were described [19]. Since that first report, loss-of-function mutations in other complement regulators (complement factor I [20] and membrane cofactor protein (CD46) [21]) or gain-of-function mutations in complement activators (C3 [22] and complement factor B [23]), gene rearrangement in CFH and its related proteins [24], autoantibodies affecting regulator function [25–27], and “at risk” haplotypes [28,29] have been described in patients with aHUS. An abnormality in complement control can be identified in approximately 65% of patients with HUS and is also described in other glomerular [30–32] and non-renal [33] diseases. aHUS is usually inherited as an autosomal dominant trait, and there is incomplete penetrance with approximately 50% of people with a mutation developing disease [34]. The incomplete penetrance may be explained by the need for an environmental trigger, which can include pregnancy [35], infection (including a diarrheal illness which can occur in 23% of patients [36]), or drugs [37]. The presence of more than one mutation or susceptible haplotype may also increase the risk of disease [36]. Complement activation, primarily via the alternative pathway, leads to endothelial damage and activation, leading to small-vessel thrombosis in target organs (Figure 2).
Figure 2. Algorithm for the diagnosis of the main types of thrombotic microangiopathy.

Abbreviations: ADAMTS13, a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13; HUS, hemolytic uremic syndrome; LDH, lactate dehydrogenase; PCR, polymerase chain reaction; STEC, Shiga toxin-producing Escherichia coli; TTP, thrombotic thrombocytopenic purpura.
Not all cases of aHUS are due to defects in proteins traditionally regarded as important in complement control. Genetic variants in thrombomodulin, a protein with anti-fibrinolytic and anti-inflammatory properties, have been associated with aHUS [38,39]. Thrombomodulin also accelerates the factor I-mediated cleavage of C3b, and loss of this effect may predispose to aHUS [38]. However, these variants can occur in the general population and may occur in combination with other disease-associated mutations [39,40]. In early childhood, recessive mutations in diacylglycerol kinase epsilon (DGKE) cause an HUS-like TMA that frequently progresses to renal failure [41,42]. How DGKE mutations cause TMA is not known, but defective DGKE signaling appears to promote a pro-thrombotic state.
Other forms of hemolytic uremic syndrome
Thrombotic microangiopathic diseases with clinical, hematological, and histological features of HUS can occur in response to other triggers. Infection with neuraminidase-producing Streptococcus pneumoniae account for 5% of childhood HUS, inducing a severe TMA with high mortality [43]. One hypothesis for the link between infection and HUS is that neuraminidase strips N-acetylneuraminic acid from the glycocalyx of cells exposing epitopes on endothelial cells, erythrocytes, and platelets that are recognized by naturally occurring antibodies, leading to a pro-thrombotic state. Treatment is supportive with eradication of Streptococcal infection.
An autosomal recessive defect in vitamin B12 metabolism leads in early childhood to severe TMA, which can be controlled with daily vitamin B12 treatment [44,45]. The use of quinine is associated with a TMA [46,47] as are other drugs, in particular calcineurin inhibitors [48,49]. Infections (for example, HIV [50] and malignancy or its treatment [51]) are also associated with TMA and renal involvement. In some of these cases, particularly those related to pregnancy, the association may reflect a triggering event in patients carrying a genetic defect in complement control. In a series of patients with pregnancy-induced HUS, over 80% had a complement gene mutation [35].
Diagnosis and treatment of patients with STEC hemolytic uremic syndrome
A diagnosis of STEC HUS should be considered in any patient developing a TMA, particularly if it follows a diarrheal illness and is associated with AKI. However, atypical forms of HUS also may be associated with diarrhea and therefore STEC infection should be confirmed (Figure 3). Stool samples should be sent for STEC culture, but this may be negative, particularly if sample collection is delayed [52]. Diarrhea may have stopped by the time of presentation, but STEC can still be cultured from feces or rectal swab. Samples should also be assessed for the presence of Shiga toxin [53]. Shiga toxin can be detected directly from feces, but the results are variable [54], and this is best performed after overnight culture. Polymerase chain reaction-based assays can be used to detect STEC Shiga toxin genes [55]. Serology can also be useful indicator of STEC infection [56].
Figure 3. Complement activation in atypical hemolytic uremic syndrome.

The complement system, through the alternative pathway, is in a state of continuous low-level activation with generation of a fluid phase C3 convertase (C3H2OBb). This generates the fragment of C3, C3b, which can bind to cell membranes, which leads to cell-bound C3 convertase. If unchecked, this process leads to rapid amplification of complement activation and generation of the effector proteins of the system: C5b-9, C3a, C5a, and more C3b. To prevent this, there are cell surface (or membrane co-factor protein [MCP]) and fluid phase—complement factor H and complement factor I (CFI)—inhibitors of complement activation. Failure to adequately control activation leads to endothelial injury, with thrombus formation, red cell fragmentation, and platelet consumption.
Best supportive care (BSC), with particular attention to fluid replacement, renal support, and treatment of neurological manifestations of the disease, is the mainstay of treatment for patients with HUS [57,58]. The improved survival in patients with STEC HUS in recent years probably reflects better supportive care. Evidence for the addition of plasma-based therapy to BSC for the treatment for STEC HUS is limited and it probably has no role [59,60]. There was a report from the 1990s suggesting a reduced mortality in older patients treated with plasma exchange [61]. However, several studies have found no benefit or worse outcomes following treatment with plasma therapies [59,60], including a prospective study of 619 children [62] and analysis of outcomes from the German outbreak [63,64].
The use of antibiotics is controversial. It is recommended not to use antibiotics to treat STEC infection (E. coli O157) because of either no benefit or potential increase in the risk of developing HUS [65–68]. There is evidence that antibiotic use leads to an increase in toxin production [69] or release [70]. A study from Japan reported a lower rate of HUS in patients with STEC infection treated with fosfomycin [71]. However, this was evident only on subgroup analysis of the fosfomycin-treated cohort, and patients in the control cohort may have received other antibiotics. A beneficial effect of fosfomycin is therefore unproven. A subsequent meta-analysis failed to show a harmful or beneficial effect of antibiotics but included the subgroup analysis from the Japanese study [72,73]. Data from the 2011 German outbreak with E. coli O104 support the use of antibiotics in patients with established STEC HUS, with a reduction in seizures, deaths, and requirement for abdominal surgery when combined antibiotics were used [63]. Antibiotics also eliminate STEC from the gut more rapidly [74] but will have no effect on the individual patient. It may be that the results from the German outbreak due to E. coli O104 may not be generalizable to infections with other serotypes as E. coli O104, in contrast to O157, does not release increased quantities of Shiga toxin in response to therapeutic concentrations of antibiotics [75].
There is evidence from a pre-clinical rodent model of Shiga toxin-mediated injury that complement activation may augment the effects of the toxin on cells [76,77], although no evidence of complement activation was seen in a primate model [78]. There is also evidence of complement activation in the circulation of patients with STEC HUS [79], and a report of three children with severe neurological involvement suggested a response to treatment with the anti-C5 monoclonal antibody, eculizumab [80]. Eculizumab was used extensively during the German outbreak with no reported benefit in an analysis of 67 patients [63]. The rate of resolution of disease was equivalent in eculizumab-treated and untreated case controls. A report in 2014 of nine cases of STEC O104 HUS in France suggested a benefit with eculizumab. However, in that report, unlike the report from Germany, there were no control cases [81]. The use of eculizumab in STEC HUS requires further evaluation before its use can be recommended.
Several other treatments for STEC HUS, including oral administration of Shiga toxin-binding agents [82], corticosteroids, anti-platelet agents, and heparin, have been suggested but have not proven beneficial [58]. Although other treatments have been associated with good outcomes, it is difficult to assess their benefit in a disease that in most cases is self-limiting. These treatments include immunoadsorption to treat an immunoglobulin-mediated component of the neurological disease [83] and recombinant thrombomodulin [84,85].
Diagnosis and treatment of patients with atypical hemolytic uremic syndrome
aHUS should be considered in any patient presenting with a TMA, particularly if the preceding history does not suggest STEC infection. Clinical features may suggest a diagnosis of aHUS but alone are insufficient to confidently diagnose aHUS. Evidence of STEC infection should be sought, and reduced ADAMTS13 activity should be excluded [86]. Serum levels of C3, C4, factor H, and factor I should be measured before plasma exchange is started. Low C3 concentrations suggest but are not diagnostic of aHUS [87]. Genetic screening and testing for autoantibodies should be performed on all suspected cases. Though important to elucidate the cause of aHUS, these tests take time and do not form part of the initial diagnostic algorithm.
Plasma exchange (PE) until recently has been considered the most effective treatment [88,89]. PE replaces functionally defective proteins and removes overactive proteins. It is also the treatment for TTP and therefore is appropriate while laboratory investigations are being performed to differentiate between aHUS and TTP. PE should be started as soon as the diagnosis is suspected, replacing 1 to 2 plasma volumes in adults and 50 to 100 mL/kg in children with each treatment. The volume exchanged and the frequency of treatments should be increased until the TMA is controlled. In those patients who respond, PE can be gradually withdrawn, although a significant proportion will require continued PE to maintain remission.
The effective use of eculizumab in aHUS was first described in 2009 [90], and since then there have been many reports of its use [91–94]. Two trials of eculizumab in adolescents and adults with plasma therapy-resistant or -dependent aHUS have recently been reported. In total, 37 patients were treated in these open-label, uncontrolled trials. Eculizumab was effective in more than 80% of patients, controlling hemolysis, improving renal function, and allowing the withdrawal of plasma therapy [95]. Treatment of aHUS should be started early (as soon as other causes of TMA have been excluded on the basis of clinical and laboratory assessment) since early treatment is associated with a better renal outcome [96]. There are still unanswered questions about eculizumab treatment of aHUS, in particular how long to continue treatment. Withdrawal of eculizumab is associated with relapse, but it can be withdrawn in some patients, with a recent report describing successful withdrawal in 7 out of 10 patients [97]. Because of the risk of Neisseria meningitides infection, all patients receiving eculizumab should be vaccinated and long-term antibiotics should be considered [98].
The rationale for anti-complement treatment in patients without a defined defect in complement control or with a non-complement mutation is less clear. Nevertheless, patients without an identified mutation responded similarly to treatment with eculizumab [95], which may reflect a role for complement in the development of aHUS, even if the causative mutation lies outside the complement system. There are also links between thrombomodulin and complement activation. The exception is patients with DGKE mutations who appear resistant to treatment targeting complement activation [41].
Transplantation in patients with hemolytic uremic syndrome
Although STEC frequently causes AKI, recovery of renal function usually occurs. Recovery of function may not be complete, and in some cases maintenance dialysis is required. In these cases, transplantation is possible provided that there is no coexisting defect in complement control [9]. In contrast, aHUS frequently recurs after transplantation. Overall, 60% of patients develop recurrent disease after transplantation, 90% of whom will lose their graft [99]. The risk of recurrence depends on the underlying abnormality, with high risk in patients with factor H, C3, and B mutations but lower risk with CD46 mutations or if no abnormality is detected [99–101]. Recurrent disease after transplantation can be treated successfully with eculizumab, and 40% of patients in the recent trails developed disease in a transplanted kidney [95,102]. Eculizumab can also be used prophylactically to prevent recurrence in patients at high risk of recurrence who are undergoing transplantation [96].
As most complement proteins are synthesized in the liver (with the exception of CD46), combined liver and kidney transplantation reverses the complement abnormality, controls disease, and allows successful kidney transplantation [103,104]. Initial outcomes of combined transplantation were poor [105]. Aggressive plasma exchange or eculizumab to correct the complement abnormality prior to transplantation allows successful transplantation and this option should be discussed with patients [106].
If the predisposing mutation occurs in other pathways, aHUS can recur after transplantation. Recurrence after transplantation has been reported in patients with thrombomodulin mutations [38,107]. Transplantation has been performed in patients with aHUS due to DGKE mutations without evidence of recurrent aHUS [41].
The etiology of de novo HUS after transplantation is complex, and ischemia reperfusion, drugs, and humoral immunity are potential precipitants. Complement may also be involved. Thirty percent of patients who develop de novo HUS have a complement mutation [108], and even in the absence of a mutation eculizumab can be effective [109].
Conclusions
Most commonly, HUS is due to STEC infection. This can be a severe, life-threatening disease but in most cases is self-limiting and will resolve completely. Treatment is with BSC, and although other treatments, including plasma therapy, eculizumab, and toxin binders, have been proposed, there is very little evidence to support their use outside of clinical trials. aHUS is, in most cases, due to impaired control of complement activation. It can be treated effectively with complement inhibition, but this may not be successful if treatment is started late or the disease is caused by a problem in another pathway.
Undoubtedly, progress has been made in the treatment of HUS. However, there are very few high-quality clinical trials to guide treatment. HUS is a rare, etiologically heterogeneous disease and there is a need for multicenter collaborative studies to test the efficacy of new treatments and inform best practice in the treatment of this disease.
Abbreviations
- ADAMTS13
a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13
- aHUS
atypical hemolytic uremic syndrome
- AKI
acute kidney injury
- BSC
best supportive care
- CFH
complement factor H
- DGKE
diacylglycerol kinase epsilon
- Gb3
globotriaosylceramide
- HUS
hemolytic uremic syndrome
- PE
plasma exchange
- STEC
Shiga toxin-producing Escherichia coli
- TMA
thrombotic microangiopathy
- TTP
thrombotic thrombocytopenic purpura
Disclosures
David Kavanagh and Neil Sheerin have received support to attend meetings and honoraria for presentations from Alexion Pharmaceuticals (Cheshire, CT, USA). Shreya Raman declares that she has no disclosures.
The electronic version of this article is the complete one and can be found at: http://f1000.com/prime/reports/m/6/119
References
- 1.Furlan M, Robles R, Solenthaler M, Wassmer M, Sandoz P, Lammle B. Deficient activity of von Willebrand factor-cleaving protease in chronic relapsing thrombotic thrombocytopenic purpura. Blood. 1997;89:3097–103. [PubMed] [Google Scholar]
- 2.Bale JF, Jr., Brasher C, Siegler RL. CNS manifestations of the hemolytic-uremic syndrome. Relationship to metabolic alterations and prognosis. Am J Dis Child. 1980;134:869–72. doi: 10.1001/archpedi.1980.02130210053014. [DOI] [PubMed] [Google Scholar]
- 3.Trachtman H, Austin C, Lewinski M, Stahl RA. Renal and neurological involvement in typical Shiga toxin-associated HUS. Nat Rev Nephrol. 2012;8:658–69. doi: 10.1038/nrneph.2012.196. [DOI] [PubMed] [Google Scholar]
- 4.Lynn RM, O’Brien SJ, Taylor CM, Adak GK, Chart H, Cheasty T, Coia JE, Gillespie IA, Locking ME, Reilly WJ, Smith HR, Waters A, Willshaw GA. Childhood hemolytic uremic syndrome, United Kingdom and Ireland. Emerg Infect Dis. 2005;11:590–6. doi: 10.3201/eid1104.040833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garg AX, Suri RS, Barrowman N, Rehman F, Matsell D, Rosas-Arellano MP, Salvadori M, Haynes RB, Clark WF. Long-term renal prognosis of diarrhea-associated hemolytic uremic syndrome: a systematic review, meta-analysis, and meta-regression. Jama. 2003;290:1360–70. doi: 10.1001/jama.290.10.1360. [DOI] [PubMed] [Google Scholar]
- 6.Frank C, Werber D, Cramer JP, Askar M, Faber M, an der Heiden M, Bernard H, Fruth A, Prager R, Spode A, Wadl M, Zoufaly A, Jordan S, Kemper MJ, Follin P, Müller L, King LA, Rosner B, Buchholz U, Stark K, Krause G, HUS Investigation Team Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N Engl J Med. 2011;365:1771–80. doi: 10.1056/NEJMoa1106483. [DOI] [PubMed] [Google Scholar]
- 7.Miceli S, Jure MA, de Saab OA, de Castillo MC, Rojas S, de Holgado AP, de Nader OM. A clinical and bacteriological study of children suffering from haemolytic uraemic syndrome in Tucuman, Argentina. Jpn J Infect Dis. 1999;52:33–7. [PubMed] [Google Scholar]
- 8.Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365:1073–86. doi: 10.1016/S0140-6736(05)71144-2. [DOI] [PubMed] [Google Scholar]
- 9.Alberti M, Valoti E, Piras R, Bresin E, Galbusera M, Tripodo C, Thaiss F, Remuzzi G, Noris M. Two patients with history of STEC-HUS, posttransplant recurrence and complement gene mutations. Am J Transplant. 2013;13:2201–6. doi: 10.1111/ajt.12297. [DOI] [PubMed] [Google Scholar]
- 10.Brigotti M, Caprioli A, Tozzi AE, Tazzari PL, Ricci F, Conte R, Carnicelli D, Procaccino MA, Minelli F, Ferretti AV, Paglialonga F, Edefonti A, Rizzoni G. Shiga toxins present in the gut and in the polymorphonuclear leukocytes circulating in the blood of children with hemolytic-uremic syndrome. J Clin Microbiol. 2006;44:313–7. doi: 10.1128/JCM.44.2.313-317.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brigotti M, Tazzari PL, Ravanelli E, Carnicelli D, Rocchi L, Arfilli V, Scavia G, Minelli F, Ricci F, Pagliaro P, Ferretti AV, Pecoraro C, Paglialonga F, Edefonti A, Procaccino MA, Tozzi AE, Caprioli A. Clinical relevance of shiga toxin concentrations in the blood of patients with hemolytic uremic syndrome. Pediatr Infect Dis J. 2011;30:486–90. doi: 10.1097/INF.0b013e3182074d22. [DOI] [PubMed] [Google Scholar]
- 12.Brigotti M, Carnicelli D, Ravanelli E, Barbieri S, Ricci F, Bontadini A, Tozzi AE, Scavia G, Caprioli A, Tazzari PL. Interactions between Shiga toxins and human polymorphonuclear leukocytes. J Leukoc Biol. 2008;84:1019–27. doi: 10.1189/jlb.0308157. [DOI] [PubMed] [Google Scholar]
- 13.Geelen JM, van der Velden TJ, Te Loo DM, Boerman OC, van den Heuvel LP, Monnens LA. Lack of specific binding of Shiga-like toxin (verocytotoxin) and non-specific interaction of Shiga-like toxin 2 antibody with human polymorphonuclear leucocytes. Nephrol Dial Transplant. 2007;22:749–55. doi: 10.1093/ndt/gfl688. [DOI] [PubMed] [Google Scholar]
- 14.Obrig TG. Escherichia coli Shiga Toxin Mechanisms of Action in Renal Disease. Toxins (Basel) 2010;2:2769–94. doi: 10.3390/toxins2122769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chaisri U, Nagata M, Kurazono H, Horie H, Tongtawe P, Hayashi H, Watanabe T, Tapchaisri P, Chongsa-nguan M, Chaicumpa W. Localization of Shiga toxins of enterohaemorrhagic Escherichia coli in kidneys of paediatric and geriatric patients with fatal haemolytic uraemic syndrome. Microb Pathog. 2001;31:59–67. doi: 10.1006/mpat.2001.0447. [DOI] [PubMed] [Google Scholar]
- 16.Obrig TG, Louise CB, Lingwood CA, Boyd B, Barley-Maloney L, Daniel TO. Endothelial heterogeneity in Shiga toxin receptors and responses. J Biol Chem. 1993;268:15484–8. [PubMed] [Google Scholar]
- 17.Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, Daina E, Fenili C, Castelletti F, Sorosina A, Piras R, Donadelli R, Maranta R, van der Meer I, Conway EM, Zipfel PF, Goodship TH, Remuzzi G. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5:1844–59. doi: 10.2215/CJN.02210310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cameron JS, Vick R. Letter: Plasma-C3 in haemolytic-uraemic syndrome and thrombotic thrombocytopenic purpura. Lancet. 1973;2:975. doi: 10.1016/S0140-6736(73)92645-7. [DOI] [PubMed] [Google Scholar]
- 19.Warwicker P, Goodship TH, Donne RL, Pirson Y, Nicholls A, Ward RM, Turnpenny P, Goodship JA. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. 1998;53:836–44. doi: 10.1111/j.1523-1755.1998.00824.x. [DOI] [PubMed] [Google Scholar]
- 20.Fremeaux-Bacchi V, Dragon-Durey MA, Blouin J, Vigneau C, Kuypers D, Boudailliez B, Loirat C, Rondeau E, Fridman WH. Complement factor I: a susceptibility gene for atypical haemolytic uraemic syndrome. J Med Genet. 2004;41:e84. doi: 10.1136/jmg.2004.019083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richards A, Kemp EJ, Liszewski MK, Goodship JA, Lampe AK, Decorte R, Muslumanoglu MH, Kavukcu S, Filler G, Pirson Y, Wen LS, Atkinson JP, Goodship TH. Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci USA. 2003;100:12966–71. doi: 10.1073/pnas.2135497100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fremeaux-Bacchi V, Miller EC, Liszewski MK, Strain L, Blouin J, Brown AL, Moghal N, Kaplan BS, Weiss RA, Lhotta K, Kapur G, Mattoo T, Nivet H, Wong W, Gie S, Hurault de Ligny B, Fischbach M, Gupta R, Hauhart R, Meunier V, Loirat C, Dragon-Durey MA, Fridman WH, Janssen BJ, Goodship TH, Atkinson JP. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood. 2008;112:4948–52. doi: 10.1182/blood-2008-01-133702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, Carreras L, Arranz EA, Garrido CA, Lopez-Trascasa M, Sanchez-Corral P, Morgan BP, Rodriguez de Cordoba S. Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci USA. 2007;104:240–5. doi: 10.1073/pnas.0603420103. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1058916
- 24.Venables JP, Strain L, Routledge D, Bourn D, Powell HM, Warwicker P, Diaz-Torres ML, Sampson A, Mead P, Webb M, Pirson Y, Jackson MS, Hughes A, Wood KM, Goodship JA, Goodship TH. Atypical haemolytic uraemic syndrome associated with a hybrid complement gene. PLoS Med. 2006;3:e431. doi: 10.1371/journal.pmed.0030431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kavanagh D, Pappworth IY, Anderson H, Hayes CM, Moore I, Hunze EM, Bennaceur K, Roversi P, Lea S, Strain L, Ward R, Plant N, Nailescu C, Goodship TH, Marchbank KJ. Factor I autoantibodies in patients with atypical hemolytic uremic syndrome: disease-associated or an epiphenomenon? Clin J Am Soc Nephrol. 2012;7:417–26. doi: 10.2215/CJN.05750611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dragon-Durey MA, Sethi SK, Bagga A, Blanc C, Blouin J, Ranchin B, Andre JL, Takagi N, Cheong HI, Hari P, Le Quintrec M, Niaudet P, Loirat C, Fridman WH, Frémeaux-Bacchi V. Clinical features of anti-factor H autoantibody-associated hemolytic uremic syndrome. J Am Soc Nephrol. 2010;21:2180–7. doi: 10.1681/ASN.2010030315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moore I, Strain L, Pappworth I, Kavanagh D, Barlow PN, Herbert AP, Schmidt CQ, Staniforth SJ, Holmes LV, Ward R, Morgan L, Goodship TH, Marchbank KJ. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood. 2010;115:379–87. doi: 10.1182/blood-2009-05-221549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Esparza-Gordillo J, Goicoechea de Jorge E, Buil A, Carreras Berges L, Lopez-Trascasa M, Sanchez-Corral P, Rodriguez de Cordoba S. Predisposition to atypical hemolytic uremic syndrome involves the concurrence of different susceptibility alleles in the regulators of complement activation gene cluster in 1q32. Hum Mol Genet. 2005;14:703–12. doi: 10.1093/hmg/ddi066. [DOI] [PubMed] [Google Scholar]
- 29.Fremeaux-Bacchi V, Kemp EJ, Goodship JA, Dragon-Durey MA, Strain L, Loirat C, Deng HW, Goodship TH. The development of atypical haemolytic-uraemic syndrome is influenced by susceptibility factors in factor H and membrane cofactor protein: evidence from two independent cohorts. J Med Genet. 2005;42:852–6. doi: 10.1136/jmg.2005.030783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gale DP, de Jorge EG, Cook HT, Martinez-Barricarte R, Hadjisavvas A, McLean AG, Pusey CD, Pierides A, Kyriacou K, Athanasiou Y, Voskarides K, Deltas C, Palmer A, Frémeaux-Bacchi V, de Cordoba SR, Maxwell PH, Pickering MC. Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet. 2010;376:794–801. doi: 10.1016/S0140-6736(10)60670-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/5851956
- 31.Chen Q, Wiesener M, Eberhardt HU, Hartmann A, Uzonyi B, Kirschfink M, Amann K, Buettner M, Goodship T, Hugo C, Skerka C, Zipfel PF. Complement factor H-related hybrid protein deregulates complement in dense deposit disease. J Clin Invest. 2014;124:145–55. doi: 10.1172/JCI71866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Servais A, Noel LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, Macher MA, Zuber J, Karras A, Provot F, Moulin B, Grünfeld JP, Niaudet P, Lesavre P, Frémeaux-Bacchi V. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012;82:454–64. doi: 10.1038/ki.2012.63. [DOI] [PubMed] [Google Scholar]
- 33.Wang X, Geng P, Zhang Y, Zhang M. Association between complement factor H Val62Ile polymorphism and age-related macular degeneration susceptibility: a meta-analysis. Gene. 2014;538:306–12. doi: 10.1016/j.gene.2014.01.032. [DOI] [PubMed] [Google Scholar]
- 34.Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, Mele C, Bresin E, Cassis L, Gamba S, Porrati F, Bucchioni S, Monteferrante G, Fang CJ, Liszewski MK, Kavanagh D, Atkinson JP, Remuzzi G, International Registry of Recurrent and Familial HUS/TTP Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108:1267–79. doi: 10.1182/blood-2005-10-007252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fakhouri F, Roumenina L, Provot F, Sallee M, Caillard S, Couzi L, Essig M, Ribes D, Dragon-Durey MA, Bridoux F, Rondeau E, Frémeaux-Bacchi V. Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J Am Soc Nephrol. 2010;21:859–67. doi: 10.1681/ASN.2009070706. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/4022956
- 36.Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, Macher MA, Niaudet P, Guest G, Boudailliez B, Bouissou F, Deschenes G, Gie S, Tsimaratos M, Fischbach M, Morin D, Nivet H, Alberti C. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2007;18:2392–400. doi: 10.1681/ASN.2006080811. [DOI] [PubMed] [Google Scholar]
- 37.Kavanagh D, Goodship TH, Richards A. Atypical hemolytic uremic syndrome. Semin Nephrol. 2013;33:508–30. doi: 10.1016/j.semnephrol.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delvaeye M, Noris M, De Vriese A, Esmon CT, Esmon NL, Ferrell G, Del-Favero J, Plaisance S, Claes B, Lambrechts D, Zoja C, Remuzzi G, Conway EM. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:345–57. doi: 10.1056/NEJMoa0810739. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1163595
- 39.Maga TK, Nishimura CJ, Weaver AE, Frees KL, Smith RJ. Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum Mutat. 2010;31:E1445–60. doi: 10.1002/humu.21256. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718839449
- 40.Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaime F, Dragon-Durey MA, Ngo S, Moulin B, Servais A, Provot F, Rostaing L, Burtey S, Niaudet P, Deschênes G, Lebranchu Y, Zuber J, Loirat C. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. 2013;8:554–62. doi: 10.2215/CJN.04760512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lemaire M, Fremeaux-Bacchi V, Schaefer F, Choi M, Tang WH, Le Quintrec M, Fakhouri F, Taque S, Nobili F, Martinez F, Ji W, Overton JD, Mane SM, Nürnberg G, Altmüller J, Thiele H, Morin D, Deschenes G, Baudouin V, Llanas B, Collard L, Majid MA, Simkova E, Nürnberg P, Rioux-Leclerc N, Moeckel GW, Gubler MC, Hwa J, Loirat C, Lifton RP. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet. 2013;45:531–6. doi: 10.1038/ng.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/717995801
- 42.Ozaltin F, Li B, Rauhauser A, An SW, Soylemezoglu O, Gonul II, Taskiran EZ, Ibsirlioglu T, Korkmaz E, Bilginer Y, Duzova A, Ozen S, Topaloglu R, Besbas N, Ashraf S, Du Y, Liang C, Chen P, Lu D, Vadnagara K, Arbuckle S, Lewis D, Wakeland B, Quigg RJ, Ransom RF, Wakeland EK, Topham MK, Bazan NG, Mohan C, Hildebrandt F, Bakkaloglu A, Huang CL, Attanasio M. DGKE variants cause a glomerular microangiopathy that mimics membranoproliferative GN. J Am Soc Nephrol. 2013;24:377–84. doi: 10.1681/ASN.2012090903. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/717973260
- 43.Copelovitch L, Kaplan BS. Streptococcus pneumoniae-associated hemolytic uremic syndrome. Pediatr Nephrol. 2008;23:1951–6. doi: 10.1007/s00467-007-0518-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/722941704
- 44.Kind T, Levy J, Lee M, Kaicker S, Nicholson JF, Kane SA. Cobalamin C disease presenting as hemolytic-uremic syndrome in the neonatal period. J Pediatr Hematol Oncol. 2002;24:327–9. doi: 10.1097/00043426-200205000-00023. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/721738242
- 45.Sharma AP, Greenberg CR, Prasad AN, Prasad C. Hemolytic uremic syndrome (HUS) secondary to cobalamin C (cblC) disorder. Pediatr Nephrol. 2007;22:2097–103. doi: 10.1007/s00467-007-0604-1. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/722941649
- 46.Gottschall JL, Elliot W, Lianos E, McFarland JG, Wolfmeyer K, Aster RH. Quinine-induced immune thrombocytopenia associated with hemolytic uremic syndrome: a new clinical entity. Blood. 1991;77:306–10. [PubMed] [Google Scholar]
- 47.Gottschall JL, Neahring B, McFarland JG, Wu GG, Weitekamp LA, Aster RH. Quinine-induced immune thrombocytopenia with hemolytic uremic syndrome: clinical and serological findings in nine patients and review of literature. Am J Hematol. 1994;47:283–9. doi: 10.1002/ajh.2830470407. [DOI] [PubMed] [Google Scholar]
- 48.Van Buren D, Van Buren CT, Flechner SM, Maddox AM, Verani R, Kahan BD. De novo hemolytic uremic syndrome in renal transplant recipients immunosuppressed with cyclosporine. Surgery. 1985;98:54–62. [PubMed] [Google Scholar]
- 49.Medina PJ, Sipols JM, George JN. Drug-associated thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Curr Opin Hematol. 2001;8:286–93. doi: 10.1097/00062752-200109000-00004. [DOI] [PubMed] [Google Scholar]
- 50.Turner ME, Kher K, Rakusan T, D’Angelo L, Kapur S, Selby D, Ray PE. A typical hemolytic uremic syndrome in human immunodeficiency virus-1-infected children. Pediatr Nephrol. 1997;11:161–3. doi: 10.1007/s004670050249. [DOI] [PubMed] [Google Scholar]
- 51.Lesesne JB, Rothschild N, Erickson B, Korec S, Sisk R, Keller J, Arbus M, Woolley PV, Chiazze L, Schein PS, et al. Cancer-associated hemolytic-uremic syndrome: analysis of 85 cases from a national registry. J Clin Oncol. 1989;7:781–9. doi: 10.1200/JCO.1989.7.6.781. [DOI] [PubMed] [Google Scholar]
- 52.Mody RK, Luna-Gierke RE, Jones TF, Comstock N, Hurd S, Scheftel J, Lathrop S, Smith G, Palmer A, Strockbine N, Talkington D, Mahon BE, Hoekstra RM, Griffin PM. Infections in pediatric postdiarrheal hemolytic uremic syndrome: factors associated with identifying shiga toxin-producing Escherichia coli. Arch Pediatr Adolesc Med. 2012;166:902–9. doi: 10.1001/archpediatrics.2012.471. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/720432355
- 53.Klein EJ, Stapp JR, Clausen CR, Boster DR, Wells JG, Qin X, Swerdlow DL, Tarr PI. Shiga toxin-producing Escherichia coli in children with diarrhea: a prospective point-of-care study. J Pediatr. 2002;141:172–7. doi: 10.1067/mpd.2002.125908. [DOI] [PubMed] [Google Scholar]
- 54.Cornick NA, Jelacic S, Ciol MA, Tarr PI. Escherichia coli O157:H7 infections: discordance between filterable fecal shiga toxin and disease outcome. J Infect Dis. 2002;186:57–63. doi: 10.1086/341295. [DOI] [PubMed] [Google Scholar]
- 55.Gerritzen A, Wittke JW, Wolff D. Rapid and sensitive detection of Shiga toxin-producing Escherichia coli directly from stool samples by real-time PCR in comparison to culture, enzyme immunoassay and Vero cell cytotoxicity assay. Clin Lab. 2011;57:993–8. [PubMed] [Google Scholar]; http://f1000.com/prime/725237529
- 56.Chart H, Cheasty T. Human infections with verocytotoxin-producing Escherichia coli O157--10 years of E. coli O157 serodiagnosis. J Med Microbiol. 2008;57:1389–93. doi: 10.1099/jmm.0.2008/003632-0. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/725237530
- 57.Michael M, Elliott EJ, Craig JC, Ridley G, Hodson EM. Interventions for hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: a systematic review of randomized controlled trials. Am J Kidney Dis. 2009;53:259–72. doi: 10.1053/j.ajkd.2008.07.038. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/721895840
- 58.Michael M, Elliott EJ, Ridley GF, Hodson EM, Craig JC. Interventions for haemolytic uraemic syndrome and thrombotic thrombocytopenic purpura. Cochrane Database Syst Rev. 2009:CD003595. doi: 10.1002/14651858.CD003595.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/721156369
- 59.Rizzoni G, Claris-Appiani A, Edefonti A, Facchin P, Franchini F, Gusmano R, Imbasciati E, Pavanello L, Perfumo F, Remuzzi G. Plasma infusion for hemolytic-uremic syndrome in children: results of a multicenter controlled trial. J Pediatr. 1988;112:284–90. doi: 10.1016/S0022-3476(88)80071-4. [DOI] [PubMed] [Google Scholar]
- 60.Loos S, Ahlenstiel T, Kranz B, Staude H, Pape L, Hartel C, Vester U, Buchtala L, Benz K, Hoppe B, Beringer O, Krause M, Müller D, Pohl M, Lemke J, Hillebrand G, Kreuzer M, König J, Wigger M, Konrad M, Haffner D, Oh J, Kemper MJ. An outbreak of Shiga toxin-producing Escherichia coli O104:H4 hemolytic uremic syndrome in Germany: presentation and short-term outcome in children. Clin Infect Dis. 2012;55:753–9. doi: 10.1093/cid/cis531. [DOI] [PubMed] [Google Scholar]
- 61.Dundas S, Murphy J, Soutar RL, Jones GA, Hutchinson SJ, Todd WT. Effectiveness of therapeutic plasma exchange in the 1996 Lanarkshire Escherichia coli O157:H7 outbreak. Lancet. 1999;354:1327–30. doi: 10.1016/S0140-6736(99)01251-9. [DOI] [PubMed] [Google Scholar]
- 62.Rosales A, Hofer J, Zimmerhackl LB, Jungraithmayr TC, Riedl M, Giner T, Strasak A, Orth-Holler D, Wurzner R, Karch H, German-Austrian HUSSG Need for long-term follow-up in enterohemorrhagic Escherichia coli-associated hemolytic uremic syndrome due to late-emerging sequelae. Clin Infect Dis. 2012;54:1413–21. doi: 10.1093/cid/cis196. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/724518921
- 63.Menne J, Nitschke M, Stingele R, Abu-Tair M, Beneke J, Bramstedt J, Bremer JP, Brunkhorst R, Busch V, Dengler R, Deuschl G, Fellermann K, Fickenscher H, Gerigk C, Goettsche A, Greeve J, Hafer C, Hagenmüller F, Haller H, Herget-Rosenthal S, Hertenstein B, Hofmann C, Lang M, Kielstein JT, Klostermeier UC, Knobloch J, Kuehbacher M, Kunzendorf U, Lehnert H, Manns MP. Validation of treatment strategies for enterohaemorrhagic Escherichia coli O104:H4 induced haemolytic uraemic syndrome: case-control study. BMJ. 2012;345:e4565. doi: 10.1136/bmj.e4565. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718131010
- 64.Kielstein JT, Beutel G, Fleig S, Steinhoff J, Meyer TN, Hafer C, Kuhlmann U, Bramstedt J, Panzer U, Vischedyk M, Busch V, Ries W, Mitzner S, Mees S, Stracke S, Nürnberger J, Gerke P, Wiesner M, Sucke B, Abu-Tair M, Kribben A, Klause N, Schindler R, Merkel F, Schnatter S, Dorresteijn EM, Samuelsson O, Brunkhorst R, Collaborators of the DGfN STEC-HUS registry Best supportive care and therapeutic plasma exchange with or without eculizumab in Shiga-toxin-producing E. coli O104:H4 induced haemolytic-uraemic syndrome: an analysis of the German STEC-HUS registry. Nephrol Dial Transplant. 2012;27:3807–15. doi: 10.1093/ndt/gfs394. [DOI] [PubMed] [Google Scholar]
- 65.Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med. 2000;342:1930–6. doi: 10.1056/NEJM200006293422601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bell BP, Griffin PM, Lozano P, Christie DL, Kobayashi JM, Tarr PI. Predictors of hemolytic uremic syndrome in children during a large outbreak of Escherichia coli O157:H7 infections. Pediatrics. 1997;100:E12. doi: 10.1542/peds.100.1.e12. [DOI] [PubMed] [Google Scholar]
- 67.Wong CS, Mooney JC, Brandt JR, Staples AO, Jelacic S, Boster DR, Watkins SL, Tarr PI. Risk factors for the hemolytic uremic syndrome in children infected with Escherichia coli O157:H7: a multivariable analysis. Clin Infect Dis. 2012;55:33–41. doi: 10.1093/cid/cis299. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/717952621
- 68.Smith KE, Wilker PR, Reiter PL, Hedican EB, Bender JB, Hedberg CW. Antibiotic treatment of Escherichia coli O157 infection and the risk of hemolytic uremic syndrome, Minnesota. Pediatr Infect Dis J. 2012;31:37–41. doi: 10.1097/INF.0b013e31823096a8. [DOI] [PubMed] [Google Scholar]
- 69.Kimmitt PT, Harwood CR, Barer MR. Toxin gene expression by shiga toxin-producing Escherichia coli: the role of antibiotics and the bacterial SOS response. Emerg Infect Dis. 2000;6:458–65. doi: 10.3201/eid0605.000503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grif K, Dierich MP, Karch H, Allerberger F. Strain-specific differences in the amount of Shiga toxin released from enterohemorrhagic Escherichia coli O157 following exposure to subinhibitory concentrations of antimicrobial agents. Eur J Clin Microbiol Infect Dis. 1998;17:761–6. doi: 10.1007/s100960050181. [DOI] [PubMed] [Google Scholar]
- 71.Ikeda K, Ida O, Kimoto K, Takatorige T, Nakanishi N, Tatara K. Effect of early fosfomycin treatment on prevention of hemolytic uremic syndrome accompanying Escherichia coli O157:H7 infection. Clin Nephrol. 1999;52:357–62. [PubMed] [Google Scholar]
- 72.Safdar N, Said A, Gangnon RE, Maki DG. Risk of hemolytic uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 enteritis: a meta-analysis. JAMA. 2002;288:996–1001. doi: 10.1001/jama.288.8.996. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/723496466
- 73.Wong CS, Brandt JR. Risk of hemolytic uremic syndrome from antibiotic treatment of Escherichia coli O157:H7 colitis. JAMA. 2002;288:3111. doi: 10.1001/jama.288.24.3111-JLT1225-2-2. author reply 3112. [DOI] [PubMed] [Google Scholar]
- 74.Nitschke M, Sayk F, Hartel C, Roseland RT, Hauswaldt S, Steinhoff J, Fellermann K, Derad I, Wellhoner P, Büning J, Tiemer B, Katalinic A, Rupp J, Lehnert H, Solbach W, Knobloch JK. Association between azithromycin therapy and duration of bacterial shedding among patients with Shiga toxin-producing enteroaggregative Escherichia coli O104:H4. JAMA. 2012;307:1046–52. doi: 10.1001/jama.2012.264. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/14249959
- 75.Corogeanu D, Willmes R, Wolke M, Plum G, Utermohlen O, Kronke M. Therapeutic concentrations of antibiotics inhibit Shiga toxin release from enterohemorrhagic E. coli O104:H4 from the 2011 German outbreak. BMC Microbiol. 2012;12:160. doi: 10.1186/1471-2180-12-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Orth D, Khan AB, Naim A, Grif K, Brockmeyer J, Karch H, Joannidis M, Clark SJ, Day AJ, Fidanzi S, Stoiber H, Dierich MP, Zimmerhackl LB, Würzner R. Shiga toxin activates complement and binds factor H: evidence for an active role of complement in hemolytic uremic syndrome. J Immunol. 2009;182:6394–400. doi: 10.4049/jimmunol.0900151. [DOI] [PubMed] [Google Scholar]
- 77.Morigi M, Galbusera M, Gastoldi S, Locatelli M, Buelli S, Pezzotta A, Pagani C, Noris M, Gobbi M, Stravalaci M, Rottoli D, Tedesco F, Remuzzi G, Zoja C. Alternative pathway activation of complement by Shiga toxin promotes exuberant C3a formation that triggers microvascular thrombosis. J Immunol. 2011;187:172–80. doi: 10.4049/jimmunol.1100491. [DOI] [PubMed] [Google Scholar]
- 78.Lee BC, Mayer CL, Leibowitz CS, Stearns-Kurosawa DJ, Kurosawa S. Quiescent complement in nonhuman primates during E coli Shiga toxin-induced hemolytic uremic syndrome and thrombotic microangiopathy. Blood. 2013;122:803–6. doi: 10.1182/blood-2013-03-490060. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718016835
- 79.Thurman JM, Marians R, Emlen W, Wood S, Smith C, Akana H, Holers VM, Lesser M, Kline M, Hoffman C, Christen E, Trachtman H. Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2009;4:1920–4. doi: 10.2215/CJN.02730409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lapeyraque AL, Malina M, Fremeaux-Bacchi V, Boppel T, Kirschfink M, Oualha M, Proulx F, Clermont MJ, Le Deist F, Niaudet P, Schaefer F. Eculizumab in severe Shiga-toxin-associated HUS. N Engl J Med. 2011;364:2561–3. doi: 10.1056/NEJMc1100859. [DOI] [PubMed] [Google Scholar]
- 81.Delmas Y, Vendrely B, Clouzeau B, Bachir H, Bui HN, Lacraz A, Helou S, Bordes C, Reffet A, Llanas B, Skopinski S, Rolland P, Gruson D, Combe C. Outbreak of Escherichia coli O104:H4 haemolytic uraemic syndrome in France: outcome with eculizumab. Nephrol Dial Transplant. 2014;29:565–72. doi: 10.1093/ndt/gft470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Trachtman H, Cnaan A, Christen E, Gibbs K, Zhao S, Acheson DW, Weiss R, Kaskel FJ, Spitzer A, Hirschman GH. Effect of an oral Shiga toxin-binding agent on diarrhea-associated hemolytic uremic syndrome in children: a randomized controlled trial. JAMA. 2003;290:1337–44. doi: 10.1001/jama.290.10.1337. [DOI] [PubMed] [Google Scholar]
- 83.Greinacher A, Friesecke S, Abel P, Dressel A, Stracke S, Fiene M, Ernst F, Selleng K, Weissenborn K, Schmidt BM, Schiffer M, Felix SB, Lerch MM, Kielstein JT, Mayerle J. Treatment of severe neurological deficits with IgG depletion through immunoadsorption in patients with Escherichia coli O104:H4-associated haemolytic uraemic syndrome: a prospective trial. Lancet. 2011;378:1166–73. doi: 10.1016/S0140-6736(11)61253-1. [DOI] [PubMed] [Google Scholar]
- 84.Kawasaki Y, Suyama K, Ono A, Oikawa T, Ohara S, Suzuki Y, Sakai N, Hosoya M. Efficacy of recombinant human soluble thrombomodulin for childhood hemolytic uremic syndrome. Pediatr Int. 2013;55:e139–42. doi: 10.1111/ped.12165. [DOI] [PubMed] [Google Scholar]
- 85.Honda T, Ogata S, Mineo E, Nagamori Y, Nakamura S, Bando Y, Ishii M. A novel strategy for hemolytic uremic syndrome: successful treatment with thrombomodulin alpha. Pediatrics. 2013;131:e928–33. doi: 10.1542/peds.2012-1466. [DOI] [PubMed] [Google Scholar]
- 86.Loirat C, Fremeaux-Bacchi V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis. 2011;6:60. doi: 10.1186/1750-1172-6-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kavanagh D, Richards A, Fremeaux-Bacchi V, Noris M, Goodship T, Remuzzi G, Atkinson JP. Screening for complement system abnormalities in patients with atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2007;2:591–6. doi: 10.2215/CJN.03270906. [DOI] [PubMed] [Google Scholar]
- 88.Ariceta G, Besbas N, Johnson S, Karpman D, Landau D, Licht C, Loirat C, Pecoraro C, Taylor CM, Van de Kar N, Vandewalle J. Guideline for the investigation and initial therapy of diarrhea-negative hemolytic uremic syndrome. Pediatr Nephrol. 2009;24:687–96. doi: 10.1007/s00467-008-0964-1. [DOI] [PubMed] [Google Scholar]
- 89.Taylor CM, Machin S, Wigmore SJ, Goodship TH. Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br J Haematol. 2010;148:37–47. doi: 10.1111/j.1365-2141.2009.07916.x. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1872960
- 90.Gruppo RA, Rother RP. Eculizumab for congenital atypical hemolytic-uremic syndrome. N Engl J Med. 2009;360:544–6. doi: 10.1056/NEJMc0809959. [DOI] [PubMed] [Google Scholar]
- 91.Ariceta G, Arrizabalaga B, Aguirre M, Morteruel E, Lopez-Trascasa M. Eculizumab in the treatment of atypical hemolytic uremic syndrome in infants. Am J Kidney Dis. 2012;59:707–10. doi: 10.1053/j.ajkd.2011.11.027. [DOI] [PubMed] [Google Scholar]
- 92.Lapeyraque AL, Fremeaux-Bacchi V, Robitaille P. Efficacy of eculizumab in a patient with factor-H-associated atypical hemolytic uremic syndrome. Pediatr Nephrol. 2011;26:621–4. doi: 10.1007/s00467-010-1719-3. [DOI] [PubMed] [Google Scholar]
- 93.Prescott HC, Wu HM, Cataland SR, Baiocchi RA. Eculizumab therapy in an adult with plasma exchange-refractory atypical hemolytic uremic syndrome. Am J Hematol. 2010;85:976–7. doi: 10.1002/ajh.21862. [DOI] [PubMed] [Google Scholar]
- 94.Tschumi S, Gugger M, Bucher BS, Riedl M, Simonetti GD. Eculizumab in atypical hemolytic uremic syndrome: long-term clinical course and histological findings. Pediatr Nephrol. 2011;26:2085–8. doi: 10.1007/s00467-011-1989-4. [DOI] [PubMed] [Google Scholar]
- 95.Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, Bingham C, Cohen DJ, Delmas Y, Douglas K, Eitner F, Feldkamp T, Fouque D, Furman RR, Gaber O, Herthelius M, Hourmant M, Karpman D, Lebranchu Y, Mariat C, Menne J, Moulin B, Nürnberger J, Ogawa M, Remuzzi G, Richard T, Sberro-Soussan R, Severino B, Sheerin NS, Trivelli A, Zimmerhackl LB, Goodship T, Loirat C. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368:2169–81. doi: 10.1056/NEJMoa1208981. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718017565
- 96.Zuber J, Fakhouri F, Roumenina LT, Loirat C, Fremeaux-Bacchi V. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol. 2012;8:643–57. doi: 10.1038/nrneph.2012.214. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/725237531
- 97.Ardissino G, Testa S, Possenti I, Tel F, Paglialonga F, Salardi S, Tedeschi S, Belingheri M, Cugno M. Discontinuation of Eculizumab Maintenance Treatment for Atypical Hemolytic Uremic Syndrome: A Report of 10 Cases. Am J Kidney Dis. 2014;64:633–7. doi: 10.1053/j.ajkd.2014.01.434. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718321887
- 98.Wong EK, Goodship TH, Kavanagh D. Complement therapy in atypical haemolytic uraemic syndrome (aHUS) Mol Immunol. 2013;56:199–212. doi: 10.1016/j.molimm.2013.05.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bresin E, Daina E, Noris M, Castelletti F, Stefanov R, Hill P, Goodship TH, Remuzzi G. Outcome of renal transplantation in patients with non-Shiga toxin-associated hemolytic uremic syndrome: prognostic significance of genetic background. Clin J Am Soc Nephrol. 2006;1:88–99. doi: 10.2215/CJN.00050505. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/722837078
- 100.Noris M, Remuzzi G. Thrombotic microangiopathy after kidney transplantation. Am J Transplant. 2010;10:1517–23. doi: 10.1111/j.1600-6143.2010.03156.x. [DOI] [PubMed] [Google Scholar]
- 101.Le Quintrec M, Zuber J, Moulin B, Kamar N, Jablonski M, Lionet A, Chatelet V, Mousson C, Mourad G, Bridoux F, Cassuto E, Loirat C, Rondeau E, Delahousse M, Frémeaux-Bacchi V. Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome. Am J Transplant. 2013;13:663–75. doi: 10.1111/ajt.12077. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/717997982
- 102.Zuber J, Le Quintrec M, Krid S, Bertoye C, Gueutin V, Lahoche A, Heyne N, Ardissino G, Chatelet V, Noël LH, Hourmant M, Niaudet P, Frémeaux-Bacchi V, Rondeau E, Legendre C, Loirat C, French Study Group for Atypical HUS Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. Am J Transplant. 2012;12:3337–54. doi: 10.1111/j.1600-6143.2012.04252.x. [DOI] [PubMed] [Google Scholar]
- 103.Saland JM, Ruggenenti P, Remuzzi G. Liver-kidney transplantation to cure atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2009;20:940–9. doi: 10.1681/ASN.2008080906. [DOI] [PubMed] [Google Scholar]
- 104.Saland JM, Emre SH, Shneider BL, Benchimol C, Ames S, Bromberg JS, Remuzzi G, Strain L, Goodship TH. Favorable long-term outcome after liver-kidney transplant for recurrent hemolytic uremic syndrome associated with a factor H mutation. Am J Transplant. 2006;6:1948–52. doi: 10.1111/j.1600-6143.2006.01375.x. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/720798095
- 105.Remuzzi G, Ruggenenti P, Colledan M, Gridelli B, Bertani A, Bettinaglio P, Bucchioni S, Sonzogni A, Bonanomi E, Sonzogni V, Bonanomi E, Sonzogni V, Platt JL, Perico N, Noris M. Hemolytic uremic syndrome: a fatal outcome after kidney and liver transplantation performed to correct factor h gene mutation. Am J Transplant. 2005;5:1146–50. doi: 10.1111/j.1600-6143.2005.00783.x. [DOI] [PubMed] [Google Scholar]
- 106.Saland J. Liver-kidney transplantation to cure atypical HUS: still an option post-eculizumab? Pediatr Nephrol. 2014;29:329–32. doi: 10.1007/s00467-013-2722-2. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/725237532
- 107.Sinibaldi S, Guzzo I, Piras R, Bresin E, Emma F, Dello Strologo L. Post-transplant recurrence of atypical hemolytic uremic syndrome in a patient with thrombomodulin mutation. Pediatr Transplant. 2013;17:E177–81. doi: 10.1111/petr.12151. [DOI] [PubMed] [Google Scholar]
- 108.Le Quintrec M, Lionet A, Kamar N, Karras A, Barbier S, Buchler M, Fakhouri F, Provost F, Fridman WH, Thervet E, Legendre C, Zuber J, Frémeaux-Bacchi V. Complement mutation-associated de novo thrombotic microangiopathy following kidney transplantation. Am J Transplant. 2008;8:1694–701. doi: 10.1111/j.1600-6143.2008.02297.x. [DOI] [PubMed] [Google Scholar]
- 109.Wilson CH, Brown AL, White SA, Goodship TH, Sheerin NS, Manas DM. Successful treatment of de novo posttransplant thrombotic microangiopathy with eculizumab. Transplantation. 2011;92:e42–3. doi: 10.1097/TP.0b013e318230c0bd. [DOI] [PubMed] [Google Scholar]