

Abstract
Myocardial infarction (MI) induces a sterile inflammatory response which contributes to adverse cardiac remodeling. The initiating mechanisms of this response remain incompletely defined. We found that necrotic cardiomyocytes released a heat-labile pro-inflammatory signal activating MAP kinases and NF-κB in cardiac fibroblasts, with secondary production of cytokines. This response was abolished in Myd88−/− fibroblasts, but was unaffected in nlrp3-deficient fibroblasts. Despite MyD88-dependency, the response was TLR-independent, as explored in TLR reporter cells, pointing to the implication of the IL-1 pathway. Indeed, necrotic cardiomyocytes released IL-1α, but not IL-1β, and the immune activation of cardiac fibroblasts was abrogated by an IL-1 receptor antagonist and an IL-1α blocking antibody. Moreover, immune responses triggered by necrotic Il1a−/− cardiomyocytes were markedly reduced. In vivo, mice exposed to MI released IL-1α in the plasma, and post-ischemic inflammation was attenuated in Il1a−/− mice. Thus, our findings identify IL-1α as a crucial early danger signal triggering post-MI inflammation.
INTRODUCTION
Myocardial infarction (MI) triggers an inflammatory response aimed at healing the infarct, but which may also foster the process of ventricular remodeling leading to cardiac dysfunction (1). It is acknowledged that such inflammation is triggered by danger signals (damage-associated molecular patterns, or DAMPs) released by necrotic myocardium and sensed by pattern receptors from the TLR and the NLR families in neighbor cells (2). Although some contributing role to these processes has been proposed for TLR2, TLR3, TLR4 (reviewed in (3)) and NLRP3 (4), as well as for DAMPs such as HMGB1 or S-100 proteins (5), the very proximal signal triggering inflammation in the ischemic heart has not been established. Therefore, the present study was designed to identify the nature of this signal and its sensing mechanisms.
MATERIAL AND METHODS
Animal experiments were approved by our institutional review board (authorizations 2477, 2484, 2664).
Mice, cells and treatments
Wild type mice in this study were C57BL6/J (Janvier Labs, Le Genest-Saint-Isle, France). Myd88−/− (a gift from Dr Thierry Roger, Lausanne University, Switzerland), Nlrp3−/− (obtained from Prof. Pascal Schneider, Lausanne University) and Il1a−/− (obtained from Prof. Yoichiro Iwakura, Tokyo University of Science) mice were backcrossed into C57BL6/J. Mouse neonatal ventricular cardiomyocytes (CMCs) and cardiac fibroblasts (CFs) were isolated by differential plating and cultured as described (6). Some experiments were done using cells isolated from adult mouse hearts immediately after differential plating and before culture, to separate CMCs from all non myoycte cells (containing CFs, vascular cells and some resident macrophages). For all experiments, cells were maintained in modified PBS-glucose buffer (PBS-G) (7). Necrosis of cells was done by 2 cycles of freeze/thaw lysis or by exposure to H2O2 (250 μM, 20 min), confirmed by propidium iodide staining and troponin release (troponin T immunoassay, Roche Diagnostics) (8). Conditioned media (2ml) were obtained by necrosis of 8×105 cells and cleared by centrifugation (10 min, 13000 rpm, 4 °C). Anakinra (Swedish Orphan Biovitrum AB) and IL-1α blocking antibody (eBioscience) were used at 0.01–10 μg/ml. Heat inactivation of conditioned media was done by heating 1h at 95 °C. DNase I (Roche) was used at 10 μg/ml. Salmonella LPS (Sigma) was used at 1 μg/ml.
RNA analyses by Real Time PCR
RNA was reversed transcribed and RT PCR was performed using standard procedures. Gene expressions were normalized to endogenous controls (Rpl19, Gapdh or Rps18). The list of oligonucleotides is given in table S1.
Cytokines, creatine kinase and troponin
CCL2/MCP1, IL-6 and IL-1α were measured by commercial ELISAs (R&D Systems and Biolegend). Plasma CK and troponin were assayed using a Cobas 8000 automated analyzer (Roche).
Western immunoblotting
Nuclear and cytosolic proteins were immunoblotted as described (7) using primary antibodies against ERK1/2, phospho-ERK1/2, p38, phospho-p38, JNK, phospho-JNK, phospho-cJUN (Cell Signaling Technology), NF-κB p65 (Santa Cruz Biotechnology), IL-1, TATA-binding protein (Abcam) and Tubulin (Sigma).
Cardiac myeloperoxidase (MPO) activity
MPO was measured in myocardial extracts using tetramethylbenzidine and H2O2, as described (9), and was expressed in mU/mg proteins.
Electromobility shift assay (EMSA)
Nuclear proteins (10 μg) were incubated with an [α-32P]dATP-labeled NF-κB probe (5′-GGCAGTTGAAGGGGACTTTCCCAGG-3′), and EMSA was performed as described (7).
TLR assay
HEK-293 cell lines overexpressing a given murine TLR protein and an inducible NF-κB reporter gene (HEK-Blue™ TLR cells assay, InVivoGen, Toulouse, France), were treated with conditioned media from necrotic or living CMCs for 4h. Specific TLR ligands were used as positive controls. TLR-negative, TNF-sensitive cells expressing the reporter gene only were used as negative controls.
Mouse myocardial infarction
Myocardial ischemia (30 min) and reperfusion (2h) was performed as described (8). At the end of reperfusion, blood and heart were obtained for measurement of cytokines. Area at risk (AAR in % of left ventricle, LV) and infarct size (in % of AAR or LV), were determined using standard Evan’s Blue/TTC staining (8).
Data analyses
Statistical analyses were done using GraphPad Prism 6.0 software. Student t-test was used for simple comparisons. Multiple comparisons were done with ANOVA followed by Dunnet’s or Bonferroni’s posttests. A p value <0.05 was considered statistically significant.
RESULTS AND DISCUSSION
In a first series of experiments, we determined whether dying cardiomyocytes (CMCs) release one or more factors able to trigger innate immune responses in cardiac fibroblasts (CFs), which may represent “sentinel cells” to detect CMCs damage (10). Mouse neonatal CMCs were killed by H2O2 (8), or by freeze-thaw lysis (11), and necrosis was confirmed by propidium iodide staining and troponin release (Fig. 1, A–B). CFs exposed to conditioned medium obtained from these necrotic CMCs displayed strong transcriptional activation and release of the chemokine CCL2 and the cytokine IL-6 (Fig. 1, C–E), associated with the activation of ERK, JNK, p38, NF-κB and AP-1 (Fig. 1, F–H). Since nucleic acids and proteins are two major families of DAMPs (5), we evaluated whether the pro-inflammatory activity released by CMCs would be altered by treatment with DNAse or by heating to denaturate proteins. The stimulated transcription of Ccl2 and Il6 was unaffected by DNAse, but was eliminated by heating (Fig. 2, A–B), supporting a protein origin of the DAMP in the medium. These findings indicate that one or more soluble, heat-sensitive, DAMPs released by necrotic CMCs promote an immediate, reflex, innate immune response in CFs, which could therefore represent a primary source of inflammatory cytokines during MI.
Figure 1. Conditioned medium from necrotic cardiomyocytes trigger innate immune responses in cardiac fibroblasts in vitro.
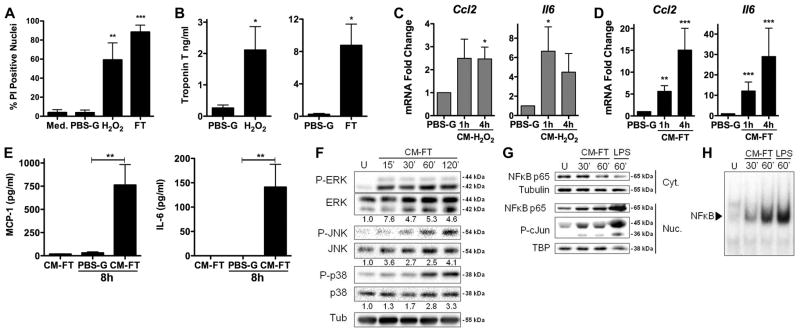
(A–B) Cardiomyocyte (CMCs) necrosis (A: nuclear PI staining, B: troponin T release) induced by H2O2 (250 μM, 20 min) or freeze/thaw (FT). CMCs in culture medium or in PBS-glucose (PBS-G) served as controls (N=3–5 per conditions). (C–D) mRNA of Ccl2 and Il6 in cardiac fibroblasts (CFs) exposed to conditioned medium from CMCs killed by H2O2 (CM-H2O2) or freeze/thaw (CM-FT) (N=3–7). CFs in PBS-G for 4 h served as controls. (E) MCP-1 and IL-6 release by CFs exposed (8 h) to CM-FT or PBS-G (N=4). (F) Phosphorylation of ERK, JNK and p38, (G) Cytoplasmic/nuclear NF-κB p65 and nuclear phospho-c-Jun, (H) NF-κB-DNA binding activity (EMSA) in CFs exposed to CM-FT. Tubulin and TBP (TATA-binding protein), loading control (F, G). LPS (1μg/ml, 60 min) was a positive control in G–H. U, untreated. Data representative of at least 3 independent experiments. * p<0.05. ** p<0.01. *** p<0.001.
Figure 2. Necrotic cardiomyocytes release a heat sensitive DAMP, triggering MyD88-dependent, NLRP3- and TLR-independent signaling pathways in cardiac fibroblasts.

(A–B) Ccl2 and Il6 mRNA in cardiac fibroblasts (CFs) exposed (4 h) to conditioned medium from necrotic cardiomyocytes (CM-FT), with or without DNaseI or heat inactivation (HI). CFs in PBS-glucose (PBS-G, 4 h) served as controls (N=3). (C–D) Ccl2 and Il6 mRNA in wild-type (WT) versus Myd88−/− (C) or Nlrp3−/− (D) CFs stimulated for (1 to 4 h) with CM-FT (N=3–4). (E) HEK-TLR assay in response to TLR agonists or to conditioned medium obtained from live (CM-Ctl) or necrotic (CM-FT) cardiomyocytes (N=2). * p<0.05. ** p<0.01. *** p<0.001.
Recent studies indicated that TLR2, TLR3, TLR4 (3) and NLRP3 (4) participate to the cardiac response to ischemia. To explore the role of TLRs or NLRP3 in our model, we examined the response to necrotic CMCs of CFs deficient either in the TLR adapter MyD88, or in Nlrp3. Transcriptional activation of Ccl2 and Il6 was suppressed in Myd88−/− CFs (Fig. 2, C), suggesting an instrumental role of a TLR-mediated process, consistent with previous findings implicating MyD88 in post-MI inflammation (12). In contrast, there was no significant influence of Nlrp3 deficiency (Fig. 2, D), arguing against a role of this receptor in early detection of necrotic CMCs by CFs. This is at variance with a recent study reporting NLRP3 activation in CFs 3 days after MI (4), suggesting that NLRP3 engagement is a delayed, but not immediate, event in post-MI immune response.
To explore further the role of TLRs, we examined the pro-inflammatory activity of conditioned medium in TLR-deficient HEK 293 reporter cells selectively transfected with several TLR family members (except TLR1 and TLR6, expressed at low levels in native HEK 293 cells). Unexpectedly, conditioned medium did not activate any of the TLR-transfected cells (Fig. 2, E), implicating the participation of an alternative MyD88-dependent signaling cascade in CFs. Besides TLRs, MyD88 is a crucial adapter downstream of the IL-1 receptor 1 (IL-R1), primarily activated by one of the two isoforms of IL-1, IL-1α and IL-1β (13). IL-1α is present in various cell types as a fully active cytoplasmic precursor, whereas IL-1β is present as a precursor activated upon proteolytic processing (13). In the heart, relative expression of the two ligands within the different cell populations is unknown. We found IL-1α strongly expressed in CMCs, while it was not detected in CFs, and only weakly detected in the whole population of non myocyte cells (NMCs) (Fig. 3, A–D), supporting that cardiac IL-1α expression is mainly restricted to CMCs. In contrast, both CMCs and CFs expressed IL-1β mRNA (Fig. 3, E), as well as the IL-1β precursor, but neither cell type displayed detectable levels of mature IL-1β, even when stimulated with LPS, which enhanced pro-IL-1β expression (Fig. 3, F–G). CFs were able to secrete IL-1β when co-stimulated with LPS and nigericin, a NLRP3 activator, but did not do so when exposed to conditioned medium from necrotic CMCs (Fig. 3, G), which is an additional indication that CFs do not detect CMCs-derived signals via NLRP3.
Figure 3. IL-1α is a major alarmin specifically released by necrotic cardiomyocytes.

(A) Il1a mRNA and (B) IL-1α protein in neonatal cardiomyocytes (CMCs, N=3) and cardiac fibroblasts (CFs, N=3). (C) IL-1α in conditioned media (CM-FT) obtained from necrotic CMCs or necrotic CFs (N=3). (D) IL-1α in adult cardiomyocytes (CMCs) and cardiac non myocyte cells (NMCs) (N=4). (E) Il1b mRNA in CMCs and CFs (N=3). (F) Pro-IL-1β and mature IL-1β (p17) in total cell lysates from CMCs either untreated (Ctrl) or stimulated with LPS (100 ng/ml, 18 h), and in conditioned medium from necrotic CMCs (CM-FT). Supernatants of bone-marrow macrophages (BMM) stimulated (18 h) with LPS (100 ng/ml) and nigericin (Nig, 5 μM) were used as positive control (N= 3). (G) Pro-IL-1β and mature IL-1β in total cell lysates from CFs, either left untreated (Ctrl) or stimulated (18 h) with LPS (100 ng/ml) or LPS and nigericin (Nig, 5 μM), and in the supernatants of CFs untreated (Ctrl) or exposed to CM-FT from necrotic CMCs (1 and 4 h). IL-1β in supernatants from CFs stimulated with LPS and nigericin (Nig) was used as a positive control (N= 3). (H) MCP-1 and IL-6 release by CFs stimulated (4 h) with CM-FT in presence or absence of Anakinra or IL-1α blocking antibody (0.01–10 μg/ml) (N=3). Levels of MCP-1 and IL-6 in CM-FT are shown as control. (I) Ccl2 and Il6 mRNA in CFs exposed (1 and 4 h) to conditioned media (CM-FT) from necrotic CMCs (CMCs→CFs) or necrotic CFs (CFs→CFs) (N=9). * p<0.05. ** p<0.01. *** p<0.001.
These findings prompted us to evaluate whether IL-1α was the DAMP activating CFs in our model. Both the IL-1R antagonist Anakinra (14) and an IL-1α blocking antibody almost suppressed the induced expression of CCL2/MCP-1 and IL-6 in CFs (Fig. 3, H), implicating IL-1α as a crucial pro-inflammatory danger signal released by CMCs. This is in line with two previous investigations reporting a role of IL-1α in the neutrophilic inflammation elicited in mice by the implantation of dead cells and extracts of necrotic tissue (15, 16). Such a role of IL-1α was further demonstrated by the striking attenuation of CFs immune activation upon stimulation with conditioned medium obtained from CFs (instead of CMCs) -which lack IL-1α- (Fig. 3, I), or from CMCs genetically deficient in IL-1α (Fig. 4, A).
Figure 4. Crucial role of IL-1α in the acute inflammatory response after myocardial infarction.

(A) MCP-1 and IL-6 release by cardiac fibroblasts (CFs) exposed 4 h to PBS-glucose (PBS-G) or to conditioned medium from wild-type (CM-FT WT) and Il1a−/− (CM-FT Il1a−/−) necrotic CMCs (N=3). MCP-1 and IL-6 in CM-FT WT and CM-FT Il1a−/− shown as controls. (B–C) IL-1α in myocardial tissue (B) and plasma (C) of mice in baseline conditions (base), and after myocardial ischemia (30 min) and reperfusion (2h, MIR) or sham ischemia (N=4–5/group). (D) Myocardial area at risk (AAR) and infarct size (IS), in % of AAR or left ventricle (LV) (N= 4/group). Plasma CK (E), troponin (F), IL-6 (G), Myocardial MCP-1(H), IL-6 (I) and MPO activity (J) in WT and Il1a−/− mice in baseline conditions (Base) and after MIR (N=5–6/group). *p<0.05. **p<0.01. ***p<0.001.
We next sought to determine whether a similar pro-inflammatory function of IL-1α would also take place during myocardial infarction in vivo, using a murine model of myocardial ischemia and reperfusion (MIR). The expression of IL-1α in the myocardium was detected at similar levels under physiological conditions, after sham surgery or after MIR (Fig. 4, B), implying that IL-1α is not up-regulated by MIR. In contrast, we found that IL-1α was detectable in plasma only after MIR, suggesting its passive release from the necrotic myocardium (Fig. 4, C). When MIR was induced in Il1a null mice, the degree of myocardial injury was not different from WT mice (comparable infarct size and circulating levels of CK and troponin, Fig. 4, D–F). Notwithstanding this comparable tissue damage, Il1a−/−mice developed significantly less inflammation following MIR, as shown by marked reduction of plasma and myocardial cytokines (Fig. 4, G–I), as well as suppressed myocardial neutrophil infiltration (evaluated by MPO activity, Fig. 4J).
Our results identify IL-1α, released by dead CMCs and sensed by CFs, as a crucial danger signal in the initiation of sterile inflammation in the infarcted heart. In contrast, our studies do not support a role for TLR- or NLR-dependent mechanisms in such initiating process, although it must be underscored that our experiments using TLR-expressing HEK 293 cells may be criticized, as this represents an artificial in vitro system which cannot rule out a role for potential TLR ligands in the in vivo setting. It is also worth to mention that our results do not exclude the involvement of additional signals which may be active in vivo, such as the autonomous nervous system, the renin-angiotensin system and possibly other yet to be identified signals. Taking these limitations into account, our observations have potential clinical implications, as they provide a conceptual framework for the development of novel pharmacological compounds with a highly specific target in patients with acute myocardial infarction. Such compounds could prevent adverse ventricular remodeling by interrupting a proximal trigger of excessive inflammatory responses, while leaving unaltered innate immune defenses essential for antimicrobial resistance.
Supplementary Material
Acknowledgments
Supported by the Swiss National Foundation for Scientific Research (grants 310030_135394/1 to LL, and 310030_132491 to NRV), the Muschamp Foundation (to LL) and Intramural Program of NIH/NIAAA (to PP). Breeding of Nlrp3−/− mice was supported by the Institute for Arthritis Research at Lausanne University.
The authors thank Yoichiro Iwakura (Tokyo University of Science, Chiba, Japan) for the agreement to use l1a−/− mice, and Alexander K. So (Lausanne University) for the gift of Anakinra.
References
- 1.Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. 2014;11:255–265. doi: 10.1038/nrcardio.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liaudet L, Rosenblatt-Velin N. Role of innate immunity in cardiac inflammation after myocardial infarction. Front Biosci. 2013;5S:86–104. doi: 10.2741/s359. [DOI] [PubMed] [Google Scholar]
- 3.Christia P, Frangogiannis NG. Targeting inflammatory pathways in myocardial infarction. Eur J Clin Invest. 2013;43:986–995. doi: 10.1111/eci.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sandanger O, Ranheim T, Vinge LE, Bliksoen M, Alfsnes K, Finsen AV, Dahl CP, Askevold ET, Florholmen G, Christensen G, Fitzgerald KA, Lien E, Valen G, Espevik T, Aukrust P, Yndestad A. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc Res. 2013;99:164–174. doi: 10.1093/cvr/cvt091. [DOI] [PubMed] [Google Scholar]
- 5.de Haan JJ, Smeets MB, Pasterkamp G, Arslan F. Danger signals in the initiation of the inflammatory response after myocardial infarction. Mediators Inflamm. 2013;2013:206039. doi: 10.1155/2013/206039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenblatt-Velin N, Lepore MG, Cartoni C, Beermann F, Pedrazzini T. FGF-2 controls the differentiation of resident cardiac precursors into functional cardiomyocytes. J Clin Invest. 2005;115:1724–1733. doi: 10.1172/JCI23418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loukili N, Rosenblatt-Velin N, Rolli J, Levrand S, Feihl F, Waeber B, Pacher P, Liaudet L. Oxidants positively or negatively regulate nuclear factor kappa B in a context-dependent manner. J Biol Chem. 2010;285:15746–15752. doi: 10.1074/jbc.M110.103259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loukili N, Rosenblatt-Velin N, Li J, Clerc S, Pacher P, Feihl F, Waeber B, Liaudet L. Peroxynitrite induces HMGB1 release by cardiac cells in vitro and HMGB1 upregulation in the infarcted myocardium in vivo. Cardiovasc Res. 2011;89:586–594. doi: 10.1093/cvr/cvq373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liaudet L, Yang Z, Al-Affar EB, Szabo C. Myocardial ischemic preconditioning in rodents is dependent on poly (ADP-ribose) synthetase. Mol Med. 2001;7:406–417. [PMC free article] [PubMed] [Google Scholar]
- 10.Shinde AV, Frangogiannis NG. Fibroblasts in myocardial infarction: A role in inflammation and repair. J Mol Cell Cardiol. 2013;70:74–82. doi: 10.1016/j.yjmcc.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, Eisenbarth SC, Florquin S, Flavell RA, Leemans JC, Sutterwala FS. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A. 2009;106:20388–20393. doi: 10.1073/pnas.0908698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng Y, Zhao H, Xu X, Buys ES, Raher MJ, Bopassa JC, Thibault H, Scherrer-Crosbie M, Schmidt U, Chao W. Innate immune adaptor MyD88 mediates neutrophil recruitment and myocardial injury after ischemia-reperfusion in mice. Am J Physiol Heart Circ Physiol. 2008;295:H1311–H1318. doi: 10.1152/ajpheart.00119.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity. 2013;39:1003–1018. doi: 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther. 2007;9:R28. doi: 10.1186/ar2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–856. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- 16.Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, White MR, Dinarello CA, Apte RN. IL-1alpha and IL-1beta recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol. 2011;187:4835–4843. doi: 10.4049/jimmunol.1102048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.