

Abstract
Three new asperentin-type compounds, 6-O-α-d-ribosylasperentin (1) and 6-O-α-d-ribosyl-8-O-methylasperentin (2) and 5-hydroxyl-6-O-methylasperentin (3), along with asperentin (4) and its known analogues (5–9), were isolated from a halotolerant Aspergillus sp. strain F00785, an endotrophic fungus from marine alga. Their structures were determined using extensive NMR and HRESIMS spectroscopic analysis, including the X-ray crystallographic data for the assignment of the absolute configurations of compound 9. Compound 4 exhibited highly potent inhibitory activity against crop pathogens, Colletotrichum gleosporioides Penz. and Colletotrichum gleosporioides (Penz.) Sacc.
Keywords: asperentin, endophytic fungus, crop pathogen, α-d-ribofuranose
1. Introduction
Marine microorganisms inhabiting relatively uninvestigated and extreme environments have been the focus of attractive sources for novel and bioactive secondary metabolites [1,2]. Marine natural products with broad spectra of bioactivities such as anti-tumor, anti-microtubule, photoprotective, antibiotic and anti-infective [3,4,5], are exceptionally interesting high value products for applications in the pharmaceutical industry. At the moment there are eight Food and Drug Administration (FDA) or European Medicines Agency (EMEA) approved drugs [6], for example trabectedin [7,8], an anti-tumor agent, and brentuximab vedotin [9], an antibody-drug conjugate. Moreover, several compounds are in different phases of the clinical trials [10,11]. Halotolerant microorganisms are microbes that are able to grow well in varied saline conditions, such as marine, salt lake, salt field, etc. [12,13]. It is believed that the high-salt environment might activate some silent genes and induce unique biosynthesis pathway in these microbes [14,15]. In the course of our ongoing search for marine-originated bioactive microbial metabolites, a halotolerant endogenic fungal Aspergillus sp. F00785 was isolated from the marine alga collected in Jinjiang Saltern, Fujian province, China. A solvent partition followed by repeated chromatographic purifications of the fermentation extracts afforded three new asperentin derivatives, 6-O-α-d-ribosylasperentin (1), 6-O-α-d-ribosyl-8-O-methylasperentin (2) and 5-hydroxyl-6-O-methylasperentin (3), along with (−)-asperentin (4), and five asperentin derivatives, 5′-hydroxyasperentin (5), 4′-hydroxyasperentin (6), asperentin-8-methyl ether (7), 5′-hydroxyasperentin-8-methyl ether (8) and 4′-hydroxyasperentin-6-methyl ether (9) (Figure 1). Here, we report the isolation, structure determination and biological activity of asperentin and its derivatives (1−9). The absolute configurations of stereocentres, C-2′, C-6′ and C-3 in 9, were determined for the first time by X-ray crystallography. Compound 4 exhibited potent activity against Colletotrichum gleosporioides Penz. (C. gleosporioides Penz.) and C. gleosporioides (Penz.) Sacc.
Figure 1.

Structures of new asperentin analogs (1–3), (−)-asperentin (4) and its derivatives (5–9).
2. Results and Discussion
2.1. Structure Elucidation
6-O-α-d-Ribosylasperentin (1) was isolated as pale yellow oil, which is soluble in acetone, chloroform and methanol. The Rf value was 0.3 on TLC over silica gel developed with CHCl3/MeOH (v/v, 10:1). The molecular formula of 1 was deduced as C21H28O9 based on the HRESIMS (m/z 447.1632 [M + Na]+, calculated for C21H28O9Na, 447.1631). The IR absorptions at 3364 and 1667 cm−1 suggested the presence of hydroxyl and carbonyl groups. The 1H- and 13C-NMR spectra of 1 in CDCl3 displayed signals for one methyl, six aliphatic methylenes, seven aliphatic methines, two meta-coupled aromatic methines, four aromatic quaternary carbons, and one ester carbonyl carbon (Table 1). The structure of fragment 1a was established on the basis of HMBC correlations from H-4 to C-1′, C-3, C-8a, C-4a and C-5, H-7 to C-6, C-8 and C-8a, H-3′ to C-4′, and H-4′ to C-5′; and 1H-1H COSY correlations (H-1′/H-2′, H-2′/H-3′, H3-6′/H-6′, and H-6′/H-5′) (Figure 2). Its 1H and 13C NMR data are compatible with those reported for asperentin [16]. Fragment 1b was further determined on the basis of HMBC correlations from H-3″ to C-1″, H-1″ to C-3″ and C-4″, and the 1H-1HCOSY correlations from H-5″ to H-4″, H-2″ to H-1″. Finally, fragments 1a and 1b were linked together via a glycosidic bond indicated by the H-1″−C-6 three-bond correlation in the HMBC spectrum.
Table 1.
1H and 13CNMR data for 1–4 in CDCl3.
| No. | 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|
| δH a (J in Hz) | δC b | δH a (J in Hz) | δC b | δH a (J in Hz) | δC b | δH a (J in Hz) | δC b | |
| 1 | 169.6, C | 161.6, C | 170.0, C | 169.9, C | ||||
| 3 | 4.72, m | 76.5, CH | 4.60, m | 74.6, CH | 4.63, m | 76.3, CH | 4.72, m | 76.4, CH |
| 4 | 2.87, m | 33.6, CH2 | 2.92, dd, (16.2, 11.5) | 35.4, CH2 | 3.10, dd, (16.8, 3.4) | 27.3, CH2 | 2.86, m | 33.7, CH2 |
| 2.83, dd, (16.2, 2.9) | 2.62, dd, (16.8, 11.5) | |||||||
| 4a | 141.5, C | 144.0, C | 122.6, C | 141.8, C | ||||
| 5 | 6.42, s | 107.6, CH | 6.56, d, (2.0) | 106.8, CH | 134.3, C | 6.32, s | 106.7, CH | |
| OH-5 | 5.20, s | |||||||
| 6 | 162.5, C | 162.6, C | 153.1, C | 162.9, C | ||||
| OCH3-6 | 3.86, s | 56.2, CH3 | ||||||
| 7 | 6.56, s | 102.9, CH | 6.61, d, (2.0) | 100.1, CH | 6.34, s | 97.8, CH | 6.19, s | 102.0, CH |
| 8 | 163.8, C | 162.9, C | 157.8, C | 164.3, C | ||||
| OCH3-8 | 3.94, s | 56.3, CH3 | ||||||
| 8a | 103.2, C | 106.8, C | 100.7, C | 101.7, C | ||||
| 1′ | 1.98, m; 1.82, m | 39.2, CH2 | 1.92, m; 1.83, m | 39.5, CH2 | 1.95, m; 1.78, m | 39.5, CH2 | 1.99, m; 1.87, m | 39.4, CH2 |
| 2′ | 4.10, m | 66.4, CH | 4.10, m | 66.2, CH | 4.05, m | 66.3, CH | 4.13, m | 66.5, CH |
| 3′ | 1.69, m; 1.35, m | 30.8, CH2 | 1.36, m | 30.9, CH2 | 1.64, m; 1.29, m | 30.8, CH2 | 1.73, m; 1.38, m | 30.9, CH2 |
| 4′ | 1.70, m; 1.62, m | 18.2, CH2 | 1.73, m; 1.64, m | 18.3, CH2 | 1.65, m; 1.56, m | 18.3, CH2 | 1.74, m; 1.65, m | 18.2, CH2 |
| 5′ | 1.68, m; 1.32, m | 31.3, CH2 | 1.72, m | 31.0, CH2 | 1.63, m; 1.26, m | 31.1, CH2 | 1.72, m; 1.36, m | 31.0, CH2 |
| 6′ | 3.94, m | 67.6, CH | 3.96, m | 67.7, CH | 3.89, m | 67.4, CH | 4.02, m | 67.9, CH |
| CH3-6′ | 1.21, d, (4.1) | 19.1, CH3 | 1.22, d, (6.5) | 18.9, CH3 | 1.14, d, (6.5) | 19.2, CH3 | 1.25, d, (6.5) | 18.9, CH3 |
| 1″ | 5.69, d, (3.5) | 100.1, CH | 5.77, d, (4.5) | 100.5, CH | ||||
| 2″ | 4.29, brs | 72.1, CH | 4.34, m | 72.2, CH | ||||
| 3″ | 4.19, brs | 70.1, CH | 4.24, dd, (6.2, 2.7) | 70.4, CH | ||||
| 4″ | 4.21, brs | 86.4, CH | 4.28, dd, (6.2, 2.7) | 86.5, CH | ||||
| 5″ | 3.78, d, (11.6); 3.74, d, (11.6) | 62.3, CH2 | 3.89, dd, (12.1, 3.1); 3.81, dd, (12.1, 3.1) | 62.5, CH2 | ||||
| 8-OH | 11.13, s | 10.86, s | 11.10, s | |||||
a 600 MHz; b 150 MHz.
Figure 2.
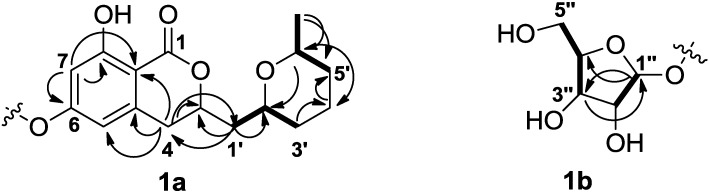
The structures of fragments 1a and 1b of 6-O-α-d-ribosylasperentin (1) and selected HMBCs (H→C) and 1H-1HCOSY correlations (bold line).
Acid hydrolysis of 1 afforded a pentofuranose and (−)-asperentin (4) ( = −23°, c = 0.83, EtOH) [17]. The latter was also known as (−)-cladosporin [18], its absolute configuration of (R)-3,4-dihydro-3-(((2′R,6′S)tetrahydro-6′-methyl-2H-pyran-2-yl)-methyl)-6,8-dihydroxy-isochromen-1-one was confirmed by comparing its NMR and physiochemical data with literature data [17,18]. The pentofuranose was determined to be ribose on the basis of 1H- and 13C-NMR data [19]. The sugar moiety was determined as d-ribose by comparison of the optical rotation of its tetra-acetate derivative ( = −17°, c = 0.68, MeOH) with the reported data [20,21]. Additionally, the stereochemistry of the anomeric carbon of the d-ribofuranose moiety was determined as α-configuration on the basis of the chemical shift and coupling constant of C-1″ (δH 5.69 (d, J = 3.5 Hz), δC 100.1) that is consistent with the reported value [21].
The two hydrolysates of 1 further validated the structures of fragments 1a and 1b. With all the obtained data, the structure of 6-O-α-d-ribosylasperentin (1) was deduced as (R)-3,4-dihydro-3-(((2′R, 6′S) tetrahydro-6′-methyl-2H-pyran-2′-yl)-methyl)-6-O-(α-d-ribofuranosyl)-8-hydroxyl-isochromen-1-one, an asperentin-6-O-riboside.
6-O-α-d-Ribosyl-8-O-methylasperentin (2) was isolated as pale yellow oil, which is soluble in acetone, chloroform and methanol. The Rf value was 0.35 eluting with the mixed solvent of CHCl3/MeOH (v/v, 10:1). The molecular formula of 2 was deduced as C22H30O9 based on the HRESIMS (m/z 439.1975 [M + H]+, calculated for C22H31O9, 439.1968). Analysis of the IR spectrum indicated the presence of hydroxyl and carbonyl functionalities with IR absorption at 3445 and 1700 cm−1, respectively.
The structure of 2 was determined as 8-methoxyl analogue of 1 on the basis of the similar NMR data of both compounds with the exception of the absence of a hydroxyl group and the presence of a methoxyl at C-8 (δH-OMe 3.94, δc-OMe56.3) (Table 1). That the methoxyl substituent on C-8 was further confirmed by HMBC correlation from OCH3 (δH 3.94) to C-8 (δC-8 162.9). Thus, 2 was 8-methoxyasperentin-6-O-riboside (Figure 1).
5-Hydroxyl-6-O-methylasperentin (3), another structurally similar metabolite, was isolated as white needle, which is soluble in acetone, chloroform and methanol. The molecular formula of 3 was deduced as C17H22O6 based on the HRESIMS (m/z 345.1308 [M + Na]+, calculated for C17H22O6Na, 345.1314). The IR absorptions at 3319 and 1657 cm−1 suggested the presence of hydroxyl and carbonyl groups. The NMR spectra were closely related to those of fragment 1a, except that the signals (δH-5 6.42, δC-5 107.6) of 1a was replaced with an aromatic oxygenated quaternary carbon (δc 134.3) which indicated a hydroxyl-substitution at C-5 (Table 1). Additionally, HMBC correlations from phenol hydrogen (δH5.20) at C-5 to C-4a (δC-4a 122.6), C-5 (δC-5 134.3) and C-6 (δC-6 153.1), and from OCH3 (δH 3.86) to C-6 (δC-6 153.1) further confirmed that 3 was 5-hydroxyasperentin-6-methyl ether.
Compounds 4−9 were isolated along with 6-O-α-d-ribosylasperentin (1), 6-O-α-d-ribosyl-8-O-methylasperentin (2) and 5-hydroxyl-6-O-methylasperentin (3). By analyses and comparisons of their NMR spectra, MS data and specific rotations with those reported in the literatures [17,18,22], compounds 4−9 were identified as (−)-asperentin (4) [17,18], 5′(S)-hydroxy-asperentin (5) [22], 4′(R)-hydroxyasperentin (6) [22], asperentin-8-methyl ether (7) [22], 5′(S)-hydroxy-asperentin-8-methyl ether (8) [22] and 4′(R)-hydroxy-asperentin-6-methyl ether (9) [22], respectively. The absolute configuration of stereocentres, C-2′, C-6′ and C-3 in 9, was determined as (R)-8-hydroxy-3-(((2′R,4′R,6′S)-4-hydroxy-6-methyltetrahydro-2H-pyran-2-yl)methyl)-6-methoxy-isochroman-1-one by the results of X-ray single-crystal diffraction using Flack parameters (Figure 3 and please see Supplementary Information). An intramolecular hydrogen bond was observed, which is consistent with the NMR data of δ8-OH (11.20), downfield shift compared with the free phenol hydrogen.
Figure 3.

X-ray crystal structure of 9.
2.2. Bioactivity Results
The antifungal activity of compounds 1–9 against three crop pathogens, C. gleosporioides Penz, C. gleosporioides (Penz) Sacc. and Botrytis cinerea Pers, were evaluated by filter-paper disk method using amphotericin B as positive control. The results showed that only (−)-asperentin (4) exhibited strong inhibitory activity and no activity were observed for the other compounds. At a concentration of 5 mg/mL, the inhibition zone of 4 to C. gleosporioides Penz. was 19.7 ± 0.58 mm, while that of amphotericin B was 15.7 ± 1.25 mm (Table 2).
Table 2.
Antimicrobial activity of (−) asperentin (4).
| Compounds | Inhibition Zone (mm) | ||
|---|---|---|---|
| C. gleosporioides Penz | C. gleosporioides (Penz) Sacc. | B. cinerea Pers | |
| (−)-asperentin (4) | 19.7 ± 0.58 | 13.3 ± 3.40 | 1.67 ± 1.11 |
| amphotericin B | 15.7 ± 1.25 | 16.0 ±1.41 | 11.0 ± 0.82 |
3. Experimental Section
3.1. General Experimental Procedures
Optical rotations were measured using a Perkin-Elmer 341 polarimeter (PerkinElmer Inc., Waltham, MA, USA). UV spectra were recorded on Jasco V-530 spectrophotometer (JASCO International Co., Tokyo, Japan). IR spectra were obtained on Perkin-Elmer 552 spectrophotometer. NMR spectra were recorded on a Bruker Avance-600 spectrometer (600 MHz) (Bruker Co., Bremen, Germany) using TMS as the internal standard. ESI-MS was measured on a Thermo-Finnigan LCQ Advantage mass spectrometer (Thermo Fisher Scientific Inc, San Jose, CA, USA). HR-ESI-MS was obtained on a Bruker LC-QTOF mass spectrometer. Semi-preparative high pressure liquid chromatography (HPLC) was performed on Agilent 1200 using XDB C18 column (10 × 250 mm, 5 μm, flow = 2 mL/min) (Agilent Technologies Inc., Santa Clara, CA, USA). TLC detection was carried out using precoated silica gel GF254 plates (10–40 μm, Qingdao Marine Chemical Plant, Qingdao, China). Column chromatography was performed with silica gel (200–300 mesh, Qingdao Marine Chemical Plant, Qingdao, China), reverse phase RP-18 (40–63 μm, Merck, Darmstadt, Germany), and Sephadex LH-20 (Amersham Biosciences, Sweden). All solvents were of analytical grade.
3.2. Fungi Materials
The marine-derived endophytic fungus Aspergillums sp. strain F00785 was identified by morphological characteristics. It was isolated from marine alga, Enteromorpha prolifera, collected in Jinjiang Saltern, Fujian province, China, in 2006, and preserved in liquid paraffin at 4 °C until its use for fermentation.
3.3. Extraction and Purification
The marine-derived fungal strain F00785 was statically cultured on potato-dextrose-agar (PDA) medium at 28 °C for 18 days. The fermented material was diced and extracted with the mixed solvent of EtOAc/MeOH/AcOH (v/v/v, 80/15/5) (3 × 5 L). The organic solution was combined and concentrated in vacuum at 40 °C to yield crude syrup (49.2 g). The crude syrup was suspended in EtOAc and washed with H2O, then the EtOAc layer was concentrated and resuspended in MeOH and petroleum ether. The MeOH layer was concentrated to give the crude extract.
The crude extract (12.87 g) was fractionated by RP-18 column (160 g) using MeOH–H2O gradient (30%, 40%, 50%, and 100%) as the eluent to yield four fractions (Fraction I-IV). Fraction I (840.0 mg) was sequentially subjected to a Sephadex LH-20 (160 g) using MeOH as the mobile phase and a Sephadex LH-20 (160 g) using acetone as the eluent to yield two portions, part A (32.5 mg) and part B (78.9 mg). Part A was subjected to preparative TLC to yield 6 (4.0 mg). Part B was purified on silica column chromatography using PE (petroleum ether)/EtOAc (v/v, 3/1) as the eluent to obtain 8 (3.5 mg). Fraction II (339.0 mg) was subjected to Sephadex LH-20 (160 g) eluting with CH2Cl2/MeOH (v/v, 1/2) and then silica gel column eluting with PE/EtOAc (v/v, 1/2) to afford 2 (7.0 mg). Fraction IV (1.56 g) was sequentially subjected to a RP-18 column (80 g) eluting with a MeOH–H2O gradient (30%, 40%, 50%, and 100%), Sephadex LH-20 (160 g) eluting with CH2Cl2/MeOH (v/v, 1/2), Sephadex LH-20 (80 g) eluting with acetone, and preparative TLC to afford 1 (28.9 mg), 4 (10.0 mg), 5 (8.0 mg),7 (38.0 mg), and 9 (4.5 mg). Compound 3 (2 mg) was obtained by further purification using semi-preparative HPLC (XDB C18 column, 10 × 250 mm, 5 μm, flow = 2 mL/min). The Rf values of compounds 4–9 on TLC over silica gel developed with CHCl3/MeOH (v/v, 10:1) are 0.85, 0.40, 0.40, 0.65, 0.35, and 0.35, respectively.
6-O-α-d-Ribosylasperentin (1). Pale yellow oil; = +122 (c = 0.7, MeOH), UV (MeOH) λmax 265.9 and 302.0 nm; IR (KBr) νmax 3364 and 1667 cm−1; 1H and 13C NMR, see Table 1; HR-ESI-MS m/z 447.1632 [M + Na]+ (calcd for C21H28O9Na, 447.1631).
6-O-α-d-Ribosyl-8-O-methylasperentin (2). Pale yellow oil; = +96 (c = 0.44, MeOH), UV (MeOH) λmax 260.0 and 302.0 nm; IR (KBr) νmax 3445 and 1700 cm−1; 1H and 13C NMR, see Table 1; HR-ESI-MS m/z 439.1975 [M + H]+ (calcd for C22H31O9, 439.1968).
5-Hydroxyl-6-O-methylasperentin (3). White needle; = −12 (c = 0.18, MeOH), m.p. 125.7 °C, UV (MeOH) λmax 268.9 and 333.0 nm; IR (KBr) νmax 3319 and 1657 cm−1; 1H and 13C NMR, see Table 1; HR-ESI-MS m/z 345.1308 [M + Na]+, (calcd for C17H22O6Na, 345.1314).
3.4. Acid Hydrolysis and Stereochemistry Determination of the Ribofuranose of 6-O-α-d-Ribosyl Asperentin (1)
A solution of 1 (90.0 mg) in 3 N HCl (60 mL) was stirred at 80 °C for 1 h. After the hydrolysis reaction was completed, the reaction mixture was concentrated under vacuum. The resulting residue was suspended in H2O (40 mL) and extracted with CH2Cl2 (3 × 40 mL). Then the aqueous layer was concentrated and dried under vacuum. The resulting residue was acetylated with Ac2O/Py at room temperature to yield an acetate product that was purified by Sephadex LH-20 (80 g) eluting with acetone/MeOH (v/v, 4/1) to afford a tetra-acetate of ribofuranose (4.1 mg). The specific rotation of this tetra-acetate product, = −17 (c = 0.68, MeOH), is consistent with the reported value of d-ribose [20]. 1H-NMR (600 MHz, CDCl3) δ 6.06 (1H, d, J = 4.9 Hz, H-1), 5.06 (1H, d, J = 3.6 Hz, H-2), 5.51 (1H, brs, H-3), 5.18 (1H, d, J = 3.2 Hz, H-4), 3.94 (1H, dd, J = 12.5, 5.9 Hz, Ha-5), 4.05 (1H, dd, J = 12.5, 3.0 Hz, Hb-5), 2.16 (3H, s, Ac), 2.15 (3H, s, Ac), 2.13 (3H, s, Ac), 2.12 (3H, s, Ac). 13C-NMR (150 MHz, CDCl3) δ 90.9 (C-1), 67.3 (C-2), 66.2 (C-3), 66.3 (C-4), 62.7 (C-5), 169.9 (CH3CO), 169.8 (CH3CO), 169.5 (CH3CO), 168.8 (CH3CO), 20.9 (CH3CO), 20.8 (CH3CO), 20.7 (CH3CO), 20.7 (CH3CO).
3.5. Anti-Crop Pathogens Test
Anti-crop pathogen activity against C. gleosporioides Penz, C. gleosporioides (Penz) Sacc., and B. cinerea Pers was evaluated using a filter-paper disk method. The tested strains were cultivated on PDA plates at 28 °C. Compounds 1–9 were tested at a concentration of 5 mg/mL in MeOH using amphotericin B (5 mg/mL in DMSO) as the positive control. The tested compound solutions (5 μL) were transferred onto filtering paper disks (Φ = 5 mm) placed in the center of assay plates. After 48 h incubation at 28 °C, the diameters of inhibition zones were measured to evaluate the activity, a bigger size of the diameter indicated a stronger inhibition. All experiments were repeated in triplicate.
4. Conclusion
Three new compounds, 6-O-α-d-ribosylasperentin (1), 6-O-α-d-ribosyl-8-O-methylasperentin (2) and 5-hydroxyl-6-O-methylasperentin (3), along with six known asperentin derivatives (4–9), isolated and identified from the fermentation products of a halotolerant fungus Aspergillums sp. strain F00785 from the inner tissue of the marine alga. 1 and 2, were found to be the first members with d-ribofuranose via α-glycosidic linkage. To the best of our knowledge, the α-d-ribofuranosyl moiety is unusual in metabolites from fungi, with only four natural products containing α-d-ribofuranosyl moiety reported up to now [19,21,23,24]; their functions in microorganisms need to be further investigated. Among the nine compounds, compound 4 exhibited strong inhibitory activity against three crop pathogens. Preliminary study of the structure activity relationships (SARs) of these nine compounds indicated that free hydroxyls at C-6 and C-8 are critical to the antifungal activity, while the hydroxyl substitution at the 4′ or 5′ position of the pyranoid ring is not favorable. In summary, discovery of these new secondary metabolites suggest that chemical investigations of marine microorganisms in relatively uninvestigated environments may provide significant natural chemical diversity for drug discovery.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (81422045 and U1405223), the Science and Technology Plan Project of Xiamen (3502Z20133010), and the Fundamental Research Funds for the Central Universities of China (2011121030, and 2013121032) to X. Deng, and the Science and Technology Plan Project of Xiamen (3502Z20123010), the Fundamental Research Funds for the Central Universities of China (2010121092) to Q.-Y. Xu. We thank ChemAxon for giving academic license to access free software products.
Supplementary Files
Author Contributions
Q. Tang carried out most of the experiments and wrote the manuscript. X.-Y. Li performed the bioassay. K.Guo contributed to the structure determination. Z.-H. Zheng identified the fungus. X.-Y. Zheng and X.-J. Kong contributed to the X-ray crystal structure study. Q.-Y. Xu and X. Deng conceived and designed the experiments and wrote the manuscript. All authors read and revised the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Pettit R.K. Culturability and secondary metabolite diversity of extreme microbes: Expanding contribution of deep sea and deep-sea vent microbes to natural product discovery. Mar. Biotechnol. 2011;13:1–11. doi: 10.1007/s10126-010-9294-y. [DOI] [PubMed] [Google Scholar]
- 2.Fenical W. Chemical studies of marine bacteria: Developing a new resource. Chem. Rev. 1993;93:1673–1683. doi: 10.1021/cr00021a001. [DOI] [Google Scholar]
- 3.Schumacher M., Kelkel M., Dicato M., Diederich M. Gold from the sea: marine compounds as inhibitors of the hallmarks of cancer. Biotechnol. Adv. 2011;29:531–547. doi: 10.1016/j.biotechadv.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Molinski T.F., Dalisay D.S., Lievens S.L., Saludes J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009;8:69–85. doi: 10.1038/nrd2487. [DOI] [PubMed] [Google Scholar]
- 5.Berdy J. Bioactive microbial metabolites. J. Antibiot. 2005;58:1–26. doi: 10.1038/ja.2005.1. [DOI] [PubMed] [Google Scholar]
- 6.Martins A., Vieira H., Gaspar H., Santos S. Marketed marine natural products in the pharmaceutical and cosmeceutical industries: Tips for success. Mar. Drugs. 2014;12:1066–1101. doi: 10.3390/md12021066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D’Incalci M., Galmarini C.M. A review of trabectedin (ET-743): A unique mechanism of action. Mol. Cancer Ther. 2010;9:2157–2163. doi: 10.1158/1535-7163.MCT-10-0263. [DOI] [PubMed] [Google Scholar]
- 8.Monk B.J., Dalton H., Benjamin I., Tanovic A. Trabectedin as a new chemotherapy option in the treatment of relapsed platinum sensitive ovarian cancer. Curr. Pharm. Des. 2012;18:3754–3769. doi: 10.2174/138161212802002814. [DOI] [PubMed] [Google Scholar]
- 9.Bauer A., Bronstrup M. Industrial natural product chemistry for drug discovery and development. Nat. Prod. Rep. 2014;31:35–60. doi: 10.1039/c3np70058e. [DOI] [PubMed] [Google Scholar]
- 10.Mayer A.M., Glaser K.B., Cuevas C., Jacobs R.S., Kem W., Little R.D., McIntosh J.M., Newman D.J., Potts B.C., Shuster D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010;31:255–265. doi: 10.1016/j.tips.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Mayer A.M. Marine Pharmaceuticals: The Clinical Pipeline. [(accessed on 16 October 2014)]. Available online: http://marinepharmacology.midwestern.edu/clinPipeline.htm.
- 12.Aguilar A., Ingemansson T., Magnien E. Extremophile microorganisms as cell factories: Support from the European Union. Extremophiles. 1998;2:367–373. doi: 10.1007/s007920050080. [DOI] [PubMed] [Google Scholar]
- 13.Kamekura M. Diversity of extremely halophilic bacteria. Extremophiles. 1998;2:289–295. doi: 10.1007/s007920050071. [DOI] [PubMed] [Google Scholar]
- 14.Koch A.L. Genetic Response of Microbes to Extreme Challenges. J. Theor. Biol. 1993;160:1–21. doi: 10.1006/jtbi.1993.1001. [DOI] [PubMed] [Google Scholar]
- 15.Mejanelle L., Lopez J.F., Gunde-Cimerman N., Grimalt J.O. Ergosterol biosynthesis in novel melanized fungi from hypersaline environments. J. Lipid Res. 2001;42:352–358. [PubMed] [Google Scholar]
- 16.Springer J.P., Cutler H.G., Crumley F.G., Cox R.H., Davis E.E., Thean J.E. Plant-growth regulatory effects and stereochemistry of cladosporin. J. Agric. Food Chem. 1981;29:853–855. doi: 10.1021/jf00106a044. [DOI] [Google Scholar]
- 17.Grove J.F. New metabolic products of Aspergillus flavus. I. Asperentin, its methyl ethers, and 5′-hydroxyasperentin. J. Chem. Soc. Perkin Trans. 1. 1972;19:2400–2406. doi: 10.1039/p19720002400. [DOI] [PubMed] [Google Scholar]
- 18.Scott P.M., Van Walbeek W., MacLean W.M. Cladosporin, a new antifungal metabolite from Cladosporium cladosporioides. J. Antibiot. 1971;24:747–755. doi: 10.7164/antibiotics.24.747. [DOI] [PubMed] [Google Scholar]
- 19.Li Y., Li X., Lee U., Kang J.S., Choi H.D., Son B.W. A new radical scavenging anthracene glycoside, asperflavinribofuranoside, and polyketides from a marine isolate of the fungus Microsporum. Chem. Pharm. Bull. 2006;54:882–883. doi: 10.1248/cpb.54.882. [DOI] [PubMed] [Google Scholar]
- 20.Zinner H. Die Acetate der d-Ribose. Chem. Ber. 1953;86:817–824. doi: 10.1002/cber.19530860630. [DOI] [Google Scholar]
- 21.Pretsch E.B.P., Affolter C. Structure Determination of Organic Compounds, Tables of Spectral Data. 3rd ed. Springer-Verlag Berlin; Heidelberg, Germany: 2000. p. 152. [Google Scholar]
- 22.Grove J.F. New metabolic products of Aspergillus flavus. IV. 4′-Hydroxyasperentin and 5′-hydroxyasperentin 8-methyl ether. J. Chem. Soc. Perkin. 1. 1973;22:2704–2706. doi: 10.1039/p19730002704. [DOI] [PubMed] [Google Scholar]
- 23.Liu R., Zhou Z.Y., Jiang M.Y., Wang F., Liu J.K. A new isoprenyl phenyl ether riboside from the culture of basidiomycete Laccaria amethystea. J. Asian Nat. Prod. Res. 2010;12:723–726. doi: 10.1080/10286020.2010.499855. [DOI] [PubMed] [Google Scholar]
- 24.Li D.L., Li X.M., Wang B.G. Natural anthraquinone derivatives from a marine mangrove plant-derived endophytic fungus Eurotium rubrum: Structural elucidation and DPPH radical scavenging activity. J. Microbiol. Biotechnol. 2009;19:675–680. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.