

Abstract
L6 rat myoblasts undergo differentiation and myotube formation when cultured in medium containing a low-concentration of serum, but the underlying mechanism is not well understood. The role of atrogin-1, an E3 ligase with well-characterized roles in muscle atrophy, has not been defined in muscle differentiation. Myocardin is a coactivator of serum response factor (SRF), which together promotes smooth muscle differentiation. Myocardin is transiently expressed in skeletal muscle progenitor cells with inhibitory effects on the expression of myogenin and muscle differentiation. It remains unknown whether myocardin, which undergoes ubiquitination degradation, plays a role in L6 cell differentiation. The current study aimed to investigate the potential roles of myocardin and atrogin-1 in differentiation of L6 cells. As reported by many others, shifting to medium containing 2% serum induced myotube formation of L6 cells. Differentiation was accompanied by up-regulation of atrogin-1 and down-regulation of myocardin, suggesting that both may be involved in muscle differentiation. As expected, over-expression of atrogin-1 stimulated the expression of troponin T and myogenin and differentiation of the L6 myoblasts. Co-expression of myocardin with atrogin-1 inhibited atrogin-1-induced myogenin expression. Over-expression of atrogin-1 decreased myocardin protein level, albeit without affecting its mRNA level. Small-interfering RNA-mediated knockdown of atrogin-1 increased myocardin protein. Consistently, ectopic expression of myocardin inhibited myogenic differentiation. Unexpectedly, myocardin decreased the expression of atrogin-1 without involving Foxo1. Taken together, our results have demonstrated that atrogin-1 plays a positive role in skeletal muscle differentiation through down-regulation of myocardin.
Undifferentiated mononucleate myoblasts proliferate through repeated cycles of cell division, but they can undergo differentiation during tissue culture under various conditions. Cells first cease dividing, followed by cell fusion to form multi-nucleate myotubes with synthesis of various contractile proteins specific to muscle tissue, such as α-actin, tropomyosin and myosin. It is well known that manipulation of medium conditions promotes myotube formation in various myoblasts, such as rat myogenic L6 myoblasts (Yaffe, 1968; Pinset and Whalen, 1984). Medium containing 2% fetal bovine serum (FBS) has been widely used for differentiation of L6 myoblasts (Sorci et al., 2003; Wedhas et al., 2005). Myf5, MyoD, myogenin and MRF4 are myogenic basic helix-loop-helix transcription factors responsible for activation, proliferation, differentiation and fusion of the skeletal muscle of muscle satellite cells, the quiescent mononucleated myogenic cells (Kuang et al., 2008; Brack and Rando, 2012). However, the exact mechanism underlying low serum-induction of muscle differentiation remains to be fully elucidated.
Treatment of cultured L6 myotubes with dexamethasone or corticosterone represents a well-established in vitro muscle atrophy model in which mRNA level of atrogin-1, a muscle-specific ubiquitin ligase encoded by the F-box protein 32 (FBXO32) gene, is upregulated (Menconi et al., 2008). Atrogin-1 is highly induced during muscle atrophy (Gomes et al., 2001). Some evidence suggests that atrogin-1 can induce muscle atrophy through the induction of ubiquitination-proteasomal degradation of MyoD (Lagirand-Cantaloube et al., 2009) and eIF3-f (Lagirand-Cantaloube et al., 2008), but other studies suggest that the expression of atrogin-1 is not correlated with rates of protein breakdown in muscles and myotubes (Murton et al., 2008; Attaix and Baracos, 2010). Transcription of atrogin-1 is reported to be controlled by the Foxo transcription factor family. Recently, the atrogin-1 gene has been found to be transcriptionally activated by myogenin, an early differentiation marker of skeletal muscle (Hasty et al., 1993), during atrophy of denervated skeletal muscle (Moresi et al., 2010; Macpherson et al., 2011), suggesting a potential link between atrogin-1 and muscle differentiation. However, it remains to be investigated whether atrogin-1 plays a role in myoblast differentiation.
Skeletal muscle differentiation is inhibited by myocardin, a co-transcriptional activator of serum response factor (SRF), which together stimulate expression of contractile genes of smooth and cardiac muscle (Wang et al., 2001; Chen et al., 2011). During mouse development, myocardin is transiently expressed in embryonic skeletal muscle (Long et al., 2007). Over-expression of myocardin inhibits muscle differentiation by functioning as a transcriptional suppressor of myogenin (Long et al., 2007). Notably, over-expression of myocardin inhibits myogenin mRNA expression, but increases the expression of smooth muscle calponin and smooth muscle myosin heavy chain in C2C12 and BC3H1 myogenic cell lines (Long et al., 2007).
Taken together, the roles of myocardin together with atrogin-1 in low-serum-induced differentiation of L6 cells have not been investigated. Our current studies revealed that during myotube differentiation of rat L6 myoblasts, gene expression of atrogin-1 was up-regulated and myocardin down-regulated. Further studies demonstrated that myocardin and atrogin-1 had opposite roles with a potential interrelationship in muscle differentiation.
Materials and Methods
Reagents and antibodies
Dulbecco’s Modified Eagle Medium (DMEM) and fetal bovine serum (FBS) were purchased from Invitrogen (Burlington, Canada). Antibodies for myocardin (M-16), atrogin-1 (MAFbx, H-300), myogenin (D-10), Foxo1 (H-130), Foxo3a (H-144) and Foxo4 (N-19) were purchased from Santa Cruz (Santa Cruz, CA). Myocardin antibody for immunostaining was purchased from abcam (Abcam Inc, Toronto, Canada). Troponin T antibody was purchased from Sigma–Aldrich (Oakville, Canada).
Cell culture, plasmids, siRNA transfection and adenovirus transduction
Rat L6 myoblast cells (CRL-1458, ATCC) were cultured in DMEM supplemented with 10% FBS (growth medium, GM). Cells from passage 2–4 were used for the experiments. To induce differentiation, subconfluent L6 cells were cultured with media containing 2% FBS (differentiation medium, DM) as previously described (Sorci et al., 2003; Wedhas et al., 2005). Differentiation was indicated by detection of troponin T/myogenin expression and/or myotube formation (Sarker and Lee, 2004). L6 cells in growth medium were transfected with 100 nM control siRNA or siRNA against atrogin-1 (Thermo Fisher Scientific, Lafayette, CO) via Lipofectamine 2000 (Invitrogen) for 6 h, or transduced with adenoviral vectors at indicated doses in the figure legends for 24 h. Adenoviral vector encoding myocardin was a kind gift from Dr. Joseph M. Miano (University of Rochester, NY). Adenoviral vectors encoding GFP or atrogin-1 were purchased from Vector Biolabs, PA. The efficiency of adenoviral vector transduction was approximately 90% as visualized by immunofluorescence analysis for GFP. Subsequently, cells were incubated in DM for 1, 3, 5, or 7 d, followed by extraction of protein and RNA for Western blot and reverse transcription quantitative polymerase chain reaction (RT-qPCR), respectively.
Reverse transcription and qPCR
Total RNA was extracted from cultured cells, followed by cDNA synthesis using the iScript cDNA Synthesis Kit (Bio-Rad, Mississauga, Canada). qPCR was performed using predesigned primers (Qiagen, Mississauga, Canada) and gene expression was normalized using the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) housekeeping gene, as described in our previous study (Chen et al., 2011).
Immunoprecipitation (IP) and Western blot
Protein for IP was extracted with RIPA buffer (Sigma, Oakville, Canada). For direct Western blot, protein was extracted with non-reducing sample buffer [50 mM Tris–HCl, pH 6.8, 2% (w/v) SDS, 10% (v/v) glycerol]. Cell lysates were subjected to IP with Foxo1 antibody or normal IgG, followed by Western blot analysis with myocardin antibody, as previously described (Yin et al., 2011).
Immunofluorescence analysis
Indirect immunofluorescence analysis was conducted as previously described (Yin et al., 2011). Cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100, followed by blocking with 5% skim milk and incubation with primary antibodies against atrogin-1, myocardin, troponin T, myogenin or individual Foxo isoforms. Their subcellular localization was visualized using Alexa-Flour 488- or Rhodamine Red-X-conjugated secondary antibody. The nuclei were counterstained with 4′6-diamidino-2-phenylindole (DAPI). The images were acquired using a digital, charge-coupled device camera through an epifluorescence microscope.
Chromatin immunoprecipitation (ChIP)
ChIP was performed using the SimpleChIP Enzymatic Chromatin IP kit (Cell Signaling, Danvers, MA) according to the manufacturer’s protocol. After DNA-protein cross-linking with formaldehyde, chromatin DNA was extracted and digested with sonification and micrococcal nuclease. For each group, digested chromatin DNA from one 10 cm culture dish was subjected to IP overnight with 2 μg Foxo1 antibody or non-immune rabbit IgG. Immunoprecipitated DNA, as well as input genomic fragments containing Foxo-binding sites from the rat Fbxo32 gene promoter were quantified using qPCR analysis, as previously described (Yin et al., 2011). The occupancy of Foxo1 on the Fbxo32 gene promoter was expressed as the percent recovery of input DNA from IP with Foxo1 antibody.
Statistical analysis
Data are expressed as mean ± SEM. The number of replicates (n) represents the number of independent assays. Differences were evaluated using the Student’s t-test or one-way ANOVA with Instat3.0 (Graphpad Software, La Jolla, CA).
Results
L6 myoblast differentiation is accompanied by up-regulation of atrogin-1 and down-regulation of myocardin
Using cultured rat L6 myoblasts, we first examined the expression of atrogin-1 and myocardin during muscle differentiation. L6 myoblast differentiation, as indicated by myotube formation, was induced by DM (differentiation medium) containing 2% serum for 3 d (Sorci et al., 2003; Wedhas et al., 2005). More significant myotube formation was observed after 5 d (Fig. 1A). Our RT-qPCR results showed that, after 2-d differentiation, preceding the appearance of myotubes, atrogin-1 mRNA was already significantly up-regulated (Fig. 1B). In contrast, myocardin mRNA expression was significantly down-regulated after 2-d differentiation, and further decreased until 4 d (Fig. 1C). Additionally, a significant decrease in myocardin protein content was detected using Western blot analysis (Fig. 1D,E). Moreover, the expression of myogenin gene was up-regulated after 1- or 2-d differentiation (Fig. 1F).
Fig. 1.

Differentiation of L6 myoblasts is accompanied by up-regulation of atrogin-1 and down-regulation of myocardin A: Representative phase-contrast micrographs showing myotube formation as indicated by arrows. Day 0 (D0, left), L6 myoblasts were cultured in growth medium containing 10% FBS. D3 (middle) and D5 (right), cells were incubated in differentiation medium containing 2% FBS for 3 and 5 d, respectively. B: RT-qPCR results showing mRNA levels of atrogin-1 during L6 myoblast differentiation. C,D: RT-qPCR results and representative Western blots showing mRNA (C) and protein (D) levels of myocardin (Myocd), respectively, during L6 myoblast differentiation. E: Cumulative Western blot results. *P <0.05, **P <0.01, n = 3.
Atrogin-1 induces L6 myoblast differentiation
To test whether atrogin-1 plays a positive role in skeletal muscle differentiation, we over-expressed atrogin-1 in L6 myoblasts through adenoviral transduction, and then determined the expression of troponin T, a skeletal muscle differentiation marker (Zeschnigk et al., 1995). As shown in Figure 2A, our Western blot results showed that over-expression of atrogin-1 induced the ectopic expression of troponin T at day 5 or 7 even in growth medium. In control cells without atrogin-1 over-expression, no troponin was detected at day 5 or 7 under the same culture conditions (Fig. 2A). Furthermore, the immunostaining results showed that cells over-expressing atrogin-1 were positively stained for troponin T at day 5 in growth medium and much more significantly at day 7 (Fig. 2B). However, there was no troponin T expression detected in control cells even at day 7 (Fig. 2B).
Fig. 2.

Atrogin-1 induces L6 myoblast differentiation L6 myoblasts were transduced with adenovirus to over-express atrogin-1 (Ad-atrogin-1, 100 MOI) for 1 d, followed by incubation in growth medium for up to 7 d (A–C). A: Western blots results showing the expression of atrogin-1 and troponin T. C: D0 control without atrogin-1 transduction. D3, 5, and 7: L6 myoblasts cultured in growth medium for 3, 5 and 7 d, respectively. B, C: The same treatments were applied to cells cultured on coverslips, followed by fixation and immunostaining atrogin-1 (green), troponin T (B, red) and myogenin (C, red). Control 0, 5 and 7 in B and C indicate control cells cultured under same conditions at different time points. The nuclei were counterstained with DAPI (blue). A5 and A7 represent cells expressing atrogin-1 for 5 and 7 d, respectively. D: Immunostaining showing nuclear localization of myogenin in L6 myoblasts in response to differentiation medium for 3 d (D3). E, F, G: L6 myoblasts were transduced with adenovirus to over-express atrogin-1 (Ad-atrogin-1), myocardin (Ad-Myocd) or GFP (Ad-GFP, 100 MOI for control group in RT-qPCR, 50 MOI to supplement Ad-atrogin-1 only or Ad-Myocd only group) for 1 d, followed by incubation with DM for 1 d and subsequent extraction of RNA for RT-qPCR. E: RT-qPCR analysis confirming myocardin expression in Ad-myocardin-transduced cells. F, G: RT-qPCR results showing mRNA levels of myogenin and atrogin-1, respectively, in response to over-expression of atrogin-1 and myocardin as indicated. *P <0.05 when compared to Ad-GFP-transduced cells, #P <0.05 when compared to Ad-atrogin-1–transduced cells, n = 3. H, I: L6 myoblasts were cultured in growth medium without (Control) and with viral expression of atrogin-1 (Ad-atrogin-1) for 10 d, followed by examination of myotube formationunder a phase contrast microscope (H) and immunostaining for α-tubulin (I). H: Representative micrographs showing myotube formation as indicated by arrow heads in cells expressing atrogin-1 for 10 din growth medium. I: α-tubulin staining (red) of cells with (ad-atrogin-1) and without (Control) atrogin-1 expression for 10 d in growth medium with the nuclei counterstained with DAPI (blue). Arrow indicates myotube formation in cells expressing atrogin-1 for 10 d in growth medium.
In addition, we found that cells expressing atrogin-1 were positively stained for myogenin, a major inducer of skeletal muscle differentiation (Hasty et al., 1993), at day 5 or 7 in growth medium (Fig. 2C). As expected, there was no significant staining for myogenin in control cells without atrogin-1 over-expression (Fig. 2C). Interestingly, atrogin-1-induced expression of myogenin was detected predominantly in the cytoplasm of L6 myoblasts in growth medium, but differentiation medium-induced expression of myogenin was mainly in their nucleus (Fig. 2D). RT-qPCR analysis further confirmed over-expression of myocardin in L6 cells in response to adenoviral transduction of myocardin (Fig. 2E). Our RT-qPCR results showed that, compared with expression of GFP, adenoviral over-expression of atrogin-1 significantly increased the expression of myogenin in L6 myoblasts after 1d differentiation (Fig. 2F). Importantly, co-expression of myocardin blocked myogenin expression induced by atrogin-1 (Fig. 2F). Notably, myocardin expression did not have any effect on the mRNA level of ectopically expressed atrogin-1(Fig. 2G), but decreased endogenous atrogin-1 mRNA level (Fig. 2G), suggesting a negative regulation of atrogin-1 expression by myocardin.
Finally, we determined whether expression of atrogin-1 is capable of inducing terminal differentiation in L6 myoblasts in the presence of growth medium (10% serum). As shown in Figure 2H, in growth medium, atrogin-1 expression through adenoviral transduction induced ectopic myotube formation, albeit with a smaller size as compared with those induced by differentiation medium. This finding was further confirmed by immunostaining for α-tubulin with the nuclei counterstained with DAPI (blue) (Fig. 2I). Taken together, our data suggest that atrogin-1 induces L6 myoblasts differentiation.
Atrogin-1 down-regulates the expression of myocardin protein, but not mRNA
Since increased atrogin-1 mRNA was accompanied by decreased myocardin mRNA and protein during L6 differentiation (Fig. 1), we examined whether over-expression of atrogin-1 directly affects the expression levels of myocardin mRNA and protein. Our results showed that adenoviral over-expression of atrogin-1 did not significantly down-regulate myocardin mRNA levels (Fig. 3A). However, myocardin protein levels were moderately reduced by atrogin-1 expression, as detected in Western blot analysis (Fig. 3B). Conversely, siRNA-mediated knockdown of atrogin-1(Fig. 3C) resulted in a statistically significant increase in myocardin protein levels, as assessed by Western blot analysis (Fig. 3D).
Fig. 3.

Atrogin-1 down-regulates myocardin protein, but not mRNA, expression L6 myoblasts were transduced with adenovirus to express atrogin-1 (Ad-atrogin-1, 50 MOI), or GFP (Ad-GFP, 50 MOI) or transfected with siRNA against atrogin-1 or control siRNA (100 nM) for 1 d. Cells were then cultured with DM for 1 d, followed by extraction of RNA and protein for RT-qPCR and Western blot analysis, respectively. A: RT-qPCR results showing mRNA levels of myocardin with and without adenoviral expression of atrogin-1. B: Western blots showing myocardin protein levels in L6 myoblasts in response to adenoviral expression of atrogin-1. C: RT-qPCR results showing siRNA knockdown of atrogin-1 mRNA. D: Western blot analysis showing myocardin protein levels in L6 myoblasts in response to treatment with atrogin-1 siRNA. *P <0.05, n = 3. E: L6 myoblasts were cultured in growth medium for 48 h, followed by transduction without (left, Control) and with atrogin-1 (right, Ad-atrogin-1, 100 MOI). Cells were maintained in growth medium for 0, 3, 5, and 7 d, followed by immunostaining for atrogin-1 (green) and myocardin (red) with the nuclei counterstained with DAPI.
Therefore, we performed indirect immunofluorescence analysis to determine whether atrogin-1 negatively regulates myocardin in the same cell. To do so, we over-expressed atrogin-1 in L6 myoblasts cultured in growth medium through adenoviral transduction, followed by detection of myocardin in atrogin-1-expressing cells. As shown in Figure 3E, there was a decrease in myocardin expression in atrogin-1-expressing cells at day 5 and no signal was detected at day 7, compared with control cells at the same time points.
Myocardin inhibits myogenic differentiation and atrogin-1 gene expression in L6 myoblasts
Since myocardin expression is down-regulated during L6 differentiation (Fig. 1), we examined whether ectopic expression of myocardin inhibits L6 differentiation. As anticipated, adenoviral expression of myocardin drastically suppressed the mRNA expression of myogenin at 3-d differentiation (Fig. 4A). Given that only a marginal inhibitory effect of myocardin on myogenin mRNA levels was observed after 1-d differentiation (Fig. 2F), our data suggested that unscheduled myocardin expression is detrimental to myogenic differentiation of L6 myoblasts at a later stage, which is consistent with previous reports using mouse BC3H1 and C2C12 myoblasts (Long et al., 2007).
Fig. 4.
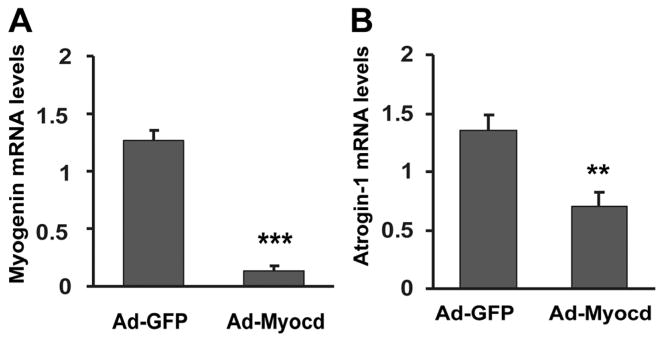
Myocardin inhibits atrogin-1 expression L6 myoblasts were transduced with adenovirus to express atrogin-1 (Ad-atrogin-1, 50 MOI), or GFP (Ad-GFP, 50 MOI). RT-qPCR results show mRNA levels of myogenin at 3-d differentiation (A) and atrogin-1 (B). **P <0.01, ***P <0.001, n = 3.
Intriguingly, RT-qPCR analysis also showed that adenoviral over-expression of myocardin significantly down-regulated endogenous atrogin-1 mRNA expression in differentiating L6 myoblasts compared with empty vector control (Fig. 4B). This suggested that myocardin could reciprocally suppress endogenous expression of atrogin-1, while atrogin-1 reduces myocardin protein abundance without affecting its mRNA level (Fig. 3).
Myocardin effect on atrogin-1 gene expression is independent of Foxo protein binding to its promoter
We wished to further investigate the mechanism underlying myocardin inhibition of atrogin-1 gene expression. Since members of the Foxo family are involved in transactivation of the atrogin-1 gene in skeletal muscle atrophy (Sandri et al., 2004), we examined whether myocardin inhibits atrogin-1 gene expression by interrupting the transcriptional functions of the Foxo family. First, we performed indirect immunofluorescence analysis to determine the nuclear localization of three mammalian Foxo family members: Foxo1, Foxo3a and Foxo4. Our results revealed the nuclear localization of Foxo1, but not Foxo3a and Foxo4, in differentiating L6 myoblasts (Fig. 5A). However, ChIP-qPCR assays showed that overexpression of myocardin did not significantly change the enrichment of Foxo1 on the atrogin-1 promoter (Fig. 5B). IP and Western blot analyses revealed no detectable interaction between myocardin and Foxo1 in differentiating L6 myoblasts (Fig. 5C), although myocardin is reported to bind to Foxo4 in smooth muscle cells (Liu et al., 2005). These results suggested that members of the Foxo family are unlikely to be involved in the inhibitory effects of myocardin on atrogin-1 expression.
Fig. 5.

Myocardin does not diminish atrogin-1 gene promoter binding of Foxo protein A: Immunofluorescence analysis showing subcellular localization of Foxo1, Foxo3a, and Foxo4 at D2 of L6 myoblast differentiation. Red fluorescent signals indicate the presence of Foxo protein. The nuclei of L6 cells were counterstained with DAPI. B: ChIP-qPCR results showing the occupancy of Foxo1 on the promoter of the Fbxo32 (atrogin-1) gene in the presence or absence of adenoviral expression of myocardin as described in the Methods. C: Representative IP-Western blots showing interaction between Foxo1 and myocardin. IP, immunoprecipitation; IB, immunoblotting. L6 myoblasts were transduced with adenovirus to over-express myocardin for 1 d, and then incubated in growth medium for 24 h before ChIP and IP assays.
Discussion
The current study has shown that culture with differentiation medium resulted in myotube formation and muscle differentiation of L6 myoblasts. During cell differentiation, atrogin-1 and myocardin were up-regulated and down-regulated, respectively. Over-expression studies with atrogin-1 and/or myocardin in L6 cells further established a stimulatory effect of atrogin-1 and an inhibitory effect of myocardin on muscle differentiation and also provided evidence for myocardin as a potential substrate of atrogin-1.
Our conclusion that atrogin-1 plays a critical role in muscle differentiation was based on two main findings. First, the increase of atrogin-1 expression level during the time course of L6 myoblast differentiation could suggest that up-regulation of atrogin-1 may be involved in muscle differentiation. Over-expression of atrogin-1 in L6 cells resulted in expression of troponin T and myogenin and myotube formation, even in growth medium, clearly establishing a role of atrogin-1 in late events of muscle differentiation. It is interesting to note that differentiation medium induced the expression of myogenin localized in the nuclei, but myogenin induced by atrogin-1 was also found in the cytoplasm. It was reported that nuclear translocation of myogenin serves as an early marker of myogenic differentiation (De Arcangelis et al., 2003). Myogenin was previously found in the cytoplasm of pre-differentiated C2C12 myoblasts, which translocated to the nucleus in response to induction of differentiation (Ferri et al., 2009). Therefore, our data suggest that in the presence of growth medium atrogin-1 expression induced a delayed differentiation of L6 cells, which was also supported by our observation that the formation of myotubes in growth medium was observed 10 d after atrogin-1 expression. This may also explain why atrogin-1-induced myotube in growth medium was not as large or mature as those induced by differentiation medium. Our data also suggest that some factors present in growth medium exert an inhibitory effect on atrogin-1-induced L6 cell differentiation.
Additionally, myogenin has recently been identified as a transcriptional activator for the atrogin-1 gene during muscle denervation (Moresi et al., 2010). Our data present in the current study have therefore suggested a functional link between myogenin and atrogin-1 in the context of skeletal muscle differentiation. However, more studies are warranted to clearly establish the link between atrogin-1 and myogenin in skeletal muscle differentiation.
We found that atrogin-1 down-regulates myocardin, an identified suppressor of skeletal muscle differentiation. This mechanism is, at least partially, responsible for its positive effect on L6 myoblast differentiation. Because atrogin-1 is a ubiquitin ligase that mediates ubiquitination degradation of proteins in skeletal muscle, and myocardin is known to undergo ubiquitination degradation as reported by our group and others (Xie et al., 2009; Jiang et al., 2010), the finding of up-regulation of atrogin-1 accompanied by down-regulation of myocardin during differentiation strongly suggests that myocardin undergoes ubiquitination-proteasomal degradation due to increased E3 ligase activity of atrogin-1 (Gomes et al., 2001). This speculation was supported by three other findings: 1) atrogin-1 over-expression reduced the expression of myocardin protein without affecting its mRNA level, 2) atrogin-1 siRNA up-regulated myocardin protein in L6 cells, and 3) adenoviral atrogin-1 over-expression significantly down-regulated the expression of myocardin. In addition, overexpression of myocardin blocked induction of myogenin gene expression by atrogin-1, further suggesting that myocardin may serve as a functional substrate of atrogin-1 in L6 cell differentiation.
Although we have revealed a role of atrogin-1 in L6 cell differentiation, it remains unknown how atrogin-1 is up-regulated in L6 cells when cultured in differentiation medium. Atrogin-1 gene expression was reportedly regulated by the Foxo family (Sandri et al., 2004), but our results showing no change in atrogin-1 promoter occupancy by Foxo suggests that the inhibitory effect of myocardin on atrogin-1 gene expression is independent of Foxo transcription factors. Given that myogenin maintains expression of the atrogin-1 gene under some circumstances (Moresi et al., 2010), it is our speculation that initial induction of myogenin may serve as a mechanism to turn on atrogin-1 gene expression during L6 cell differentiation (Long et al., 2007).
We also observed that myocardin was down-regulated during L6 myoblast differentiation and ectopic expression of myocardin inhibited the induction of myogenin gene expression in response to differentiation medium, indicating an inhibitory role of myocardin in L6 cell differentiation. This conclusion was further supported by our observation that over-expression of myocardin completely blocked atrogin-1 induced myogenin expression in L6 cells. Supportively, a previous study has also shown that myocardin expression is down-regulated during skeletal muscle differentiation in development, and over-expression of myocardin inhibits myogenin expression in C2C12 and BC3H1 skeletal muscle cell lines (Long et al., 2007). Notably, myogenin induction by lowering serum concentration is a relatively rapid event, which usually occurs within 24–48 h (Arnold et al., 1992; Ferri et al., 2009). Whether myocardin down-regulation occurs, as a preceding or subsequent event of myogenin induction still remains unknown, which requires further experiments to elaborate temporal relationship of myocardin and myogenin during the very early stage of myoblast differentiation. Nevertheless, our study has established a negative regulatory role of myocardin during, at least some later stage of, skeletal muscle differentiation.
We also noted that changing to differentiation medium also reduced the expression of myocardin mRNA in L6 cells, which likely plays a role in skeletal muscle differentiation. Given that our data showed that atrogin-1 down-regulated myocardin protein without significant effects on myocardin mRNA expression, it is possible that some other mechanism(s) contribute to downregulation of myocardin mRNA in L6 cells during differentiation in response to differentiation medium. Reciprocally, increased atrogin-1 leads to further decreases in myocardin abundance, probably through ubiquitination degradation. Subsequent reduction of myocardin will also alleviate its inhibitory effects on the expression of the atrogin-1 gene and muscle differentiation. In this regard, we show that maintenance of myocardin levels via ectopic expression of myocardin significantly inhibited expression of both myogenin and atrogin-1 gene expression, terminating the positive feedback loop in muscle differentiation. Nevertheless, further investigation is warranted to test this crosstalk model.
Taken together, our results have revealed that the promoting role for atrogin-1 in skeletal muscle differentiation is through down-regulation of myocardin. Although it remains unknown how differentiation medium results in up-regulation of atrogin-1 and down-regulation of myocardin, our findings have suggested critical roles for atrogin-1 and myocardin as a potential regulatory mechanism in skeletal muscle differentiation.
Acknowledgments
Contract grant sponsor: Canadian Institutes of Health Research;
Contract grant number: CIHR MOP-119511.
Contract grant sponsor: Health Solutions AIHS.
This research was supported by the Canadian Institutes of Health Research (CIHR MOP-119511 to X.-L.Z.). X.-L. Zheng is the recipient of a Senior Scholar Award of Alberta Innovates—Health Solutions (AIHS).
Literature Cited
- Arnold HH, Braun T, Bober E, Buchberger A, Winter B, Salminen A. Regulation of myogenin expression in normal and transformed myogenic cell lines. Symp Soc Exp Biol. 1992;46:37–51. [PubMed] [Google Scholar]
- Attaix D, Baracos VE. MAFbx/Atrogin-1 expression is a poor index of muscle proteolysis. Curr Opin Clin Nutr Metab Care. 2010;13:223–224. doi: 10.1097/MCO.0b013e328338b9a6. [DOI] [PubMed] [Google Scholar]
- Brack AS, Rando TA. Tissue-specific stem cells: Lessons from the skeletal muscle satellite cell. Cell Stem Cell. 2012;10:504–514. doi: 10.1016/j.stem.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Yin H, Jiang Y, Radhakrishnan SK, Huang ZP, Li J, Shi Z, Kilsdonk EP, Gui Y, Wang DZ, Zheng XL. Induction of microRNA-1 by myocardin in smooth muscle cells inhibits cell proliferation. Arterioscler Thromb Vasc Biol. 2011;31:368–375. doi: 10.1161/ATVBAHA.110.218149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Arcangelis V, Coletti D, Conti M, Lagarde M, Molinaro M, Adamo S, Nemoz G, Naro F. IGF-I-induced differentiation of L6 myogenic cells requires the activity of cAMP-phosphodiesterase. Mol Biol Cell. 2003;14:1392–1404. doi: 10.1091/mbc.E02-03-0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri P, Barbieri E, Burattini S, Guescini M, D’Emilio A, Biagiotti L, Del Grande P, De Luca A, Stocchi V, Falcieri E. Expression and subcellular localization of myogenic regulatory factors during the differentiation of skeletal muscle C2C12 myoblasts. J Cell Biochem. 2009;108:1302–1317. doi: 10.1002/jcb.22360. [DOI] [PubMed] [Google Scholar]
- Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci USA. 2001;98:14440–14445. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasty P, Bradley A, Morris JH, Edmondson DG, Venuti JM, Olson EN, Klein WH. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature. 1993;364:501–506. doi: 10.1038/364501a0. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Yin H, Zheng XL. MicroRNA-1 inhibits myocardin-induced contractility of human vascular smooth muscle cells. J Cell Physiol. 2010;225:506–511. doi: 10.1002/jcp.22230. [DOI] [PubMed] [Google Scholar]
- Kuang S, Gillespie MA, Rudnicki MA. Niche regulation of muscle satellite cell self-renewal and differentiation. Cell Stem Cell. 2008;2:22–31. doi: 10.1016/j.stem.2007.12.012. [DOI] [PubMed] [Google Scholar]
- Lagirand-Cantaloube J, Cornille K, Csibi A, Batonnet-Pichon S, Leibovitch MP, Leibovitch SA. Inhibition of atrogin-1/MAFbx mediated MyoD proteolysis prevents skeletal muscle atrophy in vivo. PLoS ONE. 2009;4:e4973. doi: 10.1371/journal.pone.0004973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagirand-Cantaloube J, Offner N, Csibi A, Leibovitch MP, Batonnet-Pichon S, Tintignac LA, Segura CT, Leibovitch SA. The initiation factor eIF3-f is a major target for atrogin1/ MAFbx function in skeletal muscle atrophy. EMBO J. 2008;27:1266–1276. doi: 10.1038/emboj.2008.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZP, Wang Z, Yanagisawa H, Olson EN. Phenotypic modulation of smooth muscle cells through interaction of Foxo4 and myocardin. Dev Cell. 2005;9:261–270. doi: 10.1016/j.devcel.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Long X, Creemers EE, Wang DZ, Olson EN, Miano JM. Myocardin is a bifunctional switch for smooth versus skeletal muscle differentiation. Proc Natl Acad Sci USA. 2007;104:16570–16575. doi: 10.1073/pnas.0708253104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson PC, Wang X, Goldman D. Myogenin regulates denervation-dependent muscle atrophy in mouse soleus muscle. J Cell Biochem. 2011;112:2149–2159. doi: 10.1002/jcb.23136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menconi M, Gonnella P, Petkova V, Lecker S, Hasselgren P-O. Dexamethasone and corticosterone induce similar, but not identical, muscle wasting responses in cultured L6 and C2C12 myotubes. J Cell Biochem. 2008;105:353–364. doi: 10.1002/jcb.21833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moresi V, Williams AH, Meadows E, Flynn JM, Potthoff MJ, McAnally J, Shelton JM, Backs J, Klein WH, Richardson JA, Bassel-Duby R, Olson EN. Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell. 2010;143:35–45. doi: 10.1016/j.cell.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murton AJ, Constantin D, Greenhaff PL. The involvement of the ubiquitin proteasome system in human skeletal muscle remodelling and atrophy. Biochim Biophys Acta. 2008;1782:730–743. doi: 10.1016/j.bbadis.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Pinset C, Whalen RG. Manipulation of medium conditions and differentiation in the rat myogenic cell line L6. Dev Biol. 1984;102:269–277. doi: 10.1016/0012-1606(84)90192-1. [DOI] [PubMed] [Google Scholar]
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarker KP, Lee KY. L6 myoblast differentiation is modulated by Cdk5 via the PI3K-AKT-p70S6K signaling pathway. Oncogene. 2004;23:6064–6070. doi: 10.1038/sj.onc.1207819. [DOI] [PubMed] [Google Scholar]
- Sorci G, Riuzzi F, Agneletti AL, Marchetti C, Donato R. S100B inhibits myogenic differentiation and myotube formation in a RAGE-independent manner. Mol Cell Biol. 2003;23:4870–4881. doi: 10.1128/MCB.23.14.4870-4881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, Krieg PA, Olson EN. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell. 2001;105:851–862. doi: 10.1016/s0092-8674(01)00404-4. [DOI] [PubMed] [Google Scholar]
- Wedhas N, Klamut HJ, Dogra C, Srivastava AK, Mohan S, Kumar A. Inhibition of mechanosensitive cation channels inhibits myogenic differentiation by suppressing the expression of myogenic regulatory factors and caspase-3 activity. FASEB J. 2005;19:1986–1997. doi: 10.1096/fj.05-4198com. [DOI] [PubMed] [Google Scholar]
- Xie P, Fan Y, Zhang H, Zhang Y, She M, Gu D, Patterson C, Li H. CHIP represses myocardin-induced smooth muscle cell differentiation via ubiquitin-mediated proteasomal degradation. Mol Cell Biol. 2009;29:2398–2408. doi: 10.1128/MCB.01737-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe D. Retention of differentiation potentialities during prolonged cultivation of myogenic cells. Proc Natl Acad Sci USA. 1968;61:477–483. doi: 10.1073/pnas.61.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Jiang Y, Li H, Li J, Gui Y, Zheng XL. Proteasomal degradation of myocardin is required for its transcriptional activity in vascular smooth muscle cells. J Cell Physiol. 2011;226:1897–1906. doi: 10.1002/jcp.22519. [DOI] [PubMed] [Google Scholar]
- Zeschnigk M, Kozian D, Kuch C, Schmoll M, Starzinski-Powitz A. Involvement of M-cadherin in terminal differentiation of skeletal muscle cells. J Cell Sci. 1995;108:2973–2981. doi: 10.1242/jcs.108.9.2973. [DOI] [PubMed] [Google Scholar]