

Abstract
The current paper describes the synthesis and biological evaluation of dihydrophthalazine-appended 2,4-diaminopyrimidine (DAP) inhibitors (1) oxidized at the methylene bridge linking the DAP ring to the central aromatic ring and (2) modified at the central ring ether groups. Structures 4a-b incorporating an oxidized methylene bridge showed a decrease in activity, while slightly larger alkyl groups (CH2CH3 versus CH3) on the central ring oxygen atoms (R2 and R3) had a minimal impact on the inhibition. Comparison of the potency data for previously reported RAB1 and BN-53 with the most potent of the new derivatives (19b and 20a-b) showed similar values for inhibition of cellular growth and direct enzymatic inhibition (MICs 0.5-2 μg/mL). Compounds 29-34 with larger ester and ether groups containing substituted aromatic rings at R3 exhibited slightly reduced activity (MICs 2-16 μg/mL). One explanation for this attenuated activity could be encroachment of the extended R3 into the neighboring NADPH co-factor. These results indicate that modest additions to the central ring oxygen atoms are well tolerated, while larger modifications have the potential to act as dual-site inhibitors of dihydrofolate reductase (DHFR).
Keywords: Bacillus anthracis inhibition, Dihydrofolate reductase, Antifolates, Antibiotics, Antimicrobial agents
1. Introduction
Bacillus anthracis, a Gram-positive bacterium, is the etiological agent responsible for the acute infectious disease anthrax.1 The Center for Disease Control and Prevention (CDC) classifies B. anthracis as a Category A potential high-priority bioterror threat agent, and it is well documented that certain strains of these bacteria have been modified to produce weapons of mass destruction to humans and animals.2 It is also well known that these engineered strains have innate resistance to current commercial drugs.3-6 Thus, there is a compelling and imminent need to develop new therapeutic agents to treat these resistant bacteria.
Previous studies from our research group have identified dihydrophthalazine-appended 2,4-diaminopyrimidine (DAP) derivatives as inhibitors of B. anthracis.7-9 These DAP inhibitors target the key enzyme dihydrofolate reductase (DHFR) in the folate pathway, which is intended to respond to intrinsic anti-folate antibiotic resistance found in this organism.10 Apart from our research group, there are a number of programs throughout the world developing antifolates. Targets similar to those reported herein have been studied as broad-spectrum antibiotics by Basilea Pharmaceutica.11-14 Other examples include Iclaprim by Acino Pharma,15 AR-709 by Evolva,16 and 7-aryl-2,4- diaminoquinazolines Trius Therapeutics.17 In the current study, potential DAP inhibitors, incorporating modifications from our potent lead structures RAB1 and BN-53 (Figure 1), were prepared and evaluated for their biological activity against B. anthracis.
Figure 1.
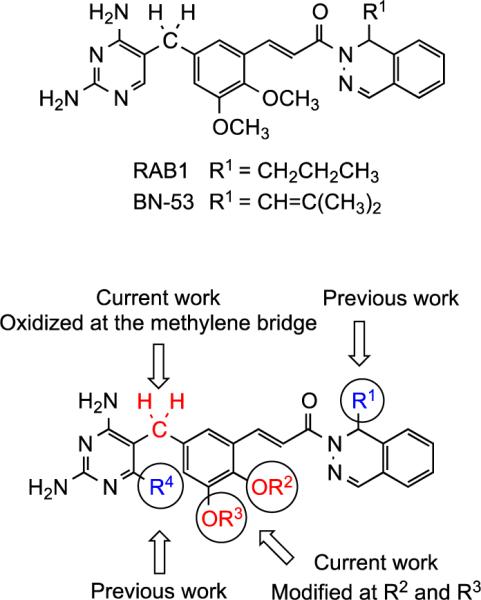
Structural modifications of the DAP-based inhibitor and lead compounds
The basic structural motif of the target is composed of three ring systems: a 2,4-diaminopyrimidine ring linked via a methylene bridge to a central 3,4-dimethoxybenzene ring, which is, in turn, tethered through an acryloyl chain to a substituted dihydrophthalazine (Figure 1). Based on previous work by Basilea Pharmaceutica11-14 and Barrow et al.,4 a number of DAP inhibitors were designed, synthesized, and evaluated for their biological activities. The initial modifications were made at R1 on the dihydrophthalazine ring. A series of substrates were prepared with different R1 substituents,7, 9 and from this set, RAB1 (minimum inhibitory concentration, MIC 1-4 μg/mL) and BN-53 (MIC 0.5 μg/mL) were identified as potential drugs.7, 10 The second alterations were carried out at R4 on the DAP ring, but increased activity was not observed.8 In a further quest for improved potency over RAB1 and BN-53, the current study investigated analogs resulting from oxidation of the methylene bridge linking the DAP fragment to the central ring, as well as changes at R2 and R3, and assessed the biological activities of the resulting compounds.
2. Results and Discussion
2.1 Chemistry
The molecular targets in Scheme 1 incorporate a ketone function in the methylene bridge between the DAP moiety and the central dimethoxybenzene ring, but retain the propyl and isobutenyl groups at R1 on the dihydrophthalazine. The preparation of these structures was a two-step process involving benzylic oxidation of 1 to ketone 2, followed a by Heck coupling to the acrylamide derivative 3 to yield 4. Benzylic oxidation of 1 was performed in 78% yield using potassium dichromate in acetic acid at reflux. This was followed by a palladium(II) acetate-mediated Heck reaction of the resulting ketone 2 with (±)-1-(1-alkyl-2(1H)-phthalazinyl)-2-propen-1-ones 3a and 3b in DMF at 140 °C to give the target molecules 4a (79%) and 4b (75%), respectively.7, 8, 18, 19
Scheme 1.
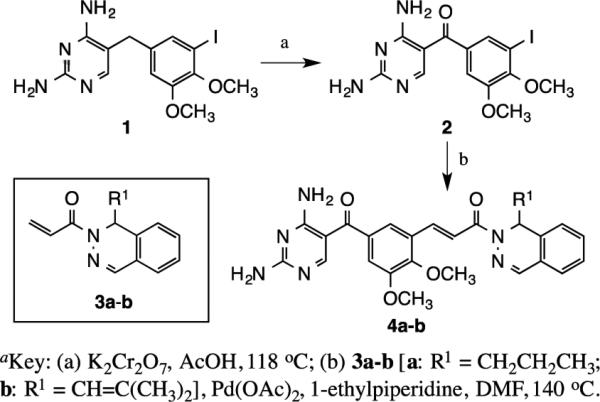
Synthesis of structures incorporating an oxidized methylene bridge.a
The synthesis of drug candidates, modified at the central ring ether groups (R2 and R3), while retaining the propyl and isobutenyl substitution on the dihydrophthalazine, are shown in Scheme 2. Benzaldehydes 6-9, derived from 5-iodovanillin, were prepared using methodology previously described.20 Condensation of 6-9 with 3-morpholinopropionitrile (10) using sodium methoxide in dry DMSO gave adducts 11-14, which were treated sequentially in dry EtOH with aniline hydrochloride and guanidine hydrochloride, both in the presence of sodium methoxide, to afford the substituted 2,4-diamino-5-benzyl-1,3-pyrimidines 15-18 in 72-80% yields. Heck coupling of 15-18 with 3a and 3b then generated the target compounds 19a-b, 20a-b, 21a and 22a in 70-78% yields.
Scheme 2.
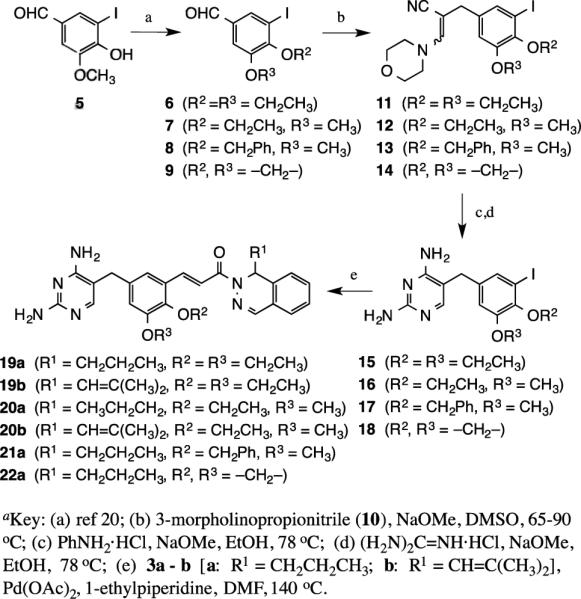
Synthesis of structures modified at R2 and R3
Phenol 27, which would allow variation of groups at R3 on the central ring of RAB1, was synthesized (Scheme 3) from the 3-methoxymethyl (MOM)-substituted benzaldehyde 23, prepared by modification of our previously reported procedure.20 Sodium methoxide promoted condensation of 10 with 23 produced 24, which was cyclocondensed with aniline hydrochloride and guanidine hydrochloride to give the substituted 2,4-diamino-5-benzyl-1,3-pyrimidine 25 in 83% yield. Heck coupling of 25 with 3a gave the MOM-protected catechol 26, which was deprotected using methanolic hydrogen chloride to give phenol 27 (58%). Finally, treatment of 27 with DBU at room temperature in dichloromethane, followed by addition of propyl iodide, benzoyl chloride, 4-methoxybenzoyl chloride, 4-nitrobenzoyl chloride, 4-(trifluoromethoxy)benzoyl chloride, 4- fluorobenzoyl chloride, or 4-(trifluoromethyl)benzoyl chloride, afforded targets 28-34 in 62-92% yields.
Scheme 3.
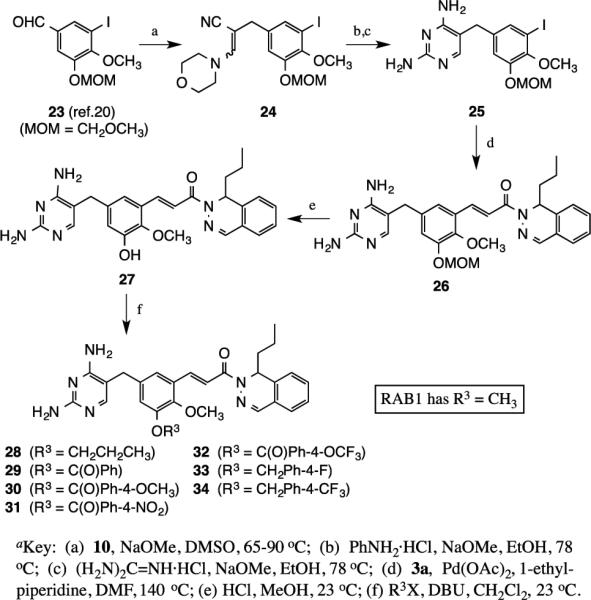
Synthesis of analogues of RAB1 modified at R3a
2.2 Biology
The biological activities of our new dihydrophthalazine-appended DAP derivatives are summarized in Table 1. Compounds incorporating a ketone function between the DAP ring and the central dimethoxybenzene ring, as found in 4a (R1 = propyl, R2 = R3 = CH3) and 4b (R1 = isobutenyl, R2 = R3 = CH3), exhibited low activity. The impact on direct inhibition of the DHFR enzyme did display a preference for the isobutenyl moiety at R1, but this change did not rescue the lack of inhibition of bacterial cell growth.
Table 1.
Biological activity of new DAP inhibitors relative to earlier compounds
| Compound | MIC (μg/mL) B. anthracis Sterne | Ki (nM) ± SEM B anthracis DHFR | Ki (nM) ± SEM B anthracis NADPH first |
|---|---|---|---|
| TMPa | > 2048 | 77233 | |
| RAB1b | 1-4 | 9.4 ± 0.2 | |
| BN-53b | 0.5 | 8.3 ± 0.2 | |
| 4a | > 128 | 83.4 ± 2.3 | |
| 4b | > 128 | 15.1 ± 0.6 | |
| 19a | 4 | 20.2 ± 0.2 | |
| 19b | 1-2 | 9.3 ± 0.2 | |
| 20a | 2 | 9.9 ± 0.2 | |
| 20b | 0.5 | 8.8 ± 0.4 | |
| 21a | 4 | 38.8 ± 0.6 | 14.6 ± 0.4 |
| 22a | 32 | 1300 ± 11 | 1090 ± 167 |
| 28 | 4 | 13.8 ± 0.2 | 14.6 ± 0.4 |
| 29 | 4-16 | 2200 ± 20 | 720 ± 20 |
| 30 | 8-16 | > 2300 | 2200 ± 30 |
| 31 | 8 | 310 ± 3 | 260 ± 6 |
| 32 | 8 | > 2100 | 220 ± 6 |
| 33 | 8-16 | > 2400 | > 2400 |
| 34 | 8-16 | > 2200 | 700 ± 10 |
Alterations of the central-ring ether groups to incorporate slightly larger moieties at either R2 alone, or in addition to R3, had little impact on the potency. For example, when compared with unmodified RAB1 (R1 = propyl, R2 = R3 = CH3), 19a (R1 = propyl, R2 = R3 = CH2CH3) and 20a (R1 = propyl, R2 = CH2CH3, R3 = CH3) showed similar activity. Substitution of larger groups, such as a benzyl at R2 in 21a (R1 = propyl, R2 = benzyl, R3 = CH3) or a propyl at R3 in 28 (R1 = propyl, R2 = CH3, R3 = propyl), exerted a small, but measurable, affect on the growth of the bacterial cells and on the purified enzyme. In contrast, compounds having a smaller methylene moiety bridging R2 and R3, as in 22a (R1 = propyl, R2 = R3 = –CH2–), showed reduced activity. Comparison of the activity data for unmodified BN-53 (R1 = isobutenyl, R2 = R3 = CH3) with that for 19b (R1 = isobutenyl, R2 = R3 = CH2CH3) and 20b (R1 = isobutenyl, R2 = CH2CH3, R3 = CH3) revealed no measurable change in either inhibition of cellular growth or direct enzymatic inhibition. When contextualized within the binding pocket of the DHFR enzyme from B. anthracis, small extensions (replacing CH3 with CH2CH3) at R2 and R3 would be readily accommodated without unfavorable steric interaction with any protein atoms. Finally, the comparable reduction in activity for the much larger benzyl at R2 in 21a versus the smaller propyl at R3 in 28 suggests that the R3 position has a greater impact on potency.
Further modification in the central ring installed larger groups at the R3 position to give compounds 29-34 (R1 = propyl, R2 = CH3, R3 = variable). These compounds exhibited lower efficacy, and this was revealed more dramatically in the enzyme inhibition assay. In reactions with purified DHFR protein, four of the six R3 derivatives were unable to achieve at least 50% inhibition at the limit of compound solubility if the compound was added after the NADPH. Only two derivatives, 29 and 31, effectively inhibited the enzyme with this order of addition. Structure 29 contained the minimum addition of a benzoyl group at R3, although the Ki was barely measurable. When compounds were added prior to the NADPH co-factor, the inhibition improved remarkably such that all but one compound had measurable Ki values. Compound 31 (R3 = 4-nitrobenzoyl), the only polarized structure tested, stood out as remarkably better than the others in this series. Nevertheless, it was not as efficacious as RAB1 or BN-53, and the MIC value did not indicate the same remarkable gain in potency that the Ki value revealed.
The compounds containing the larger extensions from R3 present an interesting picture when viewed in the context of the DHFR substrate site. These inhibitors are known to dock with the DAP moiety, which closely mimics the natural folate substrate.10, 22 Based on our structural data to date, it is likely that each compound within the inhibitor series binds with a relatively conserved orientation.10 We hypothesize that these larger extensions from R3 are approaching the neighboring NADPH co-factor site. This hypothesis is supported, in part, by experiments of enzyme inhibition in which the compounds were instead added prior to the NADPH co-factor. In this situation, the measurable Ki values decreased and three additional compounds showed inhibition. It is of note that compound 28 was relatively unchanged by the order of addition experiment, as it was predicted to not encroach on the co-factor site. If our hypothesis of dual-site binding is correct, the Ki values would no longer be reflective of the enzymatic inhibition as it would now be a double-competitive reaction with both the folate substrate and with the co-factor. This may contribute to the immeasurable Ki values while retaining inhibitory activity at the whole cell level.
3. Conclusion
The current investigation describes the synthesis and biological evaluation of dihydrophthalazine-appended DAP inhibitors oxidized at the methylene bridge linking the DAP ring to the structure and modified at the ether groups of the central aromatic ring. The indication from activity studies of 4a and 4b is a requirement for flexibility in the methylene linkage between the DAP group and the central dialkoxy-substituted ring. Alteration of this tetrahedral geometry to a trigonal planar arrangement, as found in the ketone-derivatized structures, abolished all cellular growth inhibition (Table 1). Alterations at R2 and R3 are well tolerated when the added group is small and conservative, such as the addition of ethyl groups in compounds 19a-b and 20a-b. This is also true when a larger and hydrophobic benzyl moiety is added at R2, as in 21a, or propyl at R3, as in 22a (Figure 2). This is particularly striking when viewed versus the ketone modifications in compounds 4a and 4b, and strongly suggests locations in the inhibitory compounds that can accept substitutions while maintaining potency. However, larger additions at R3 (viz. 29-34) are less well tolerated at the level of enzymatic inhibition, while those modifications have a more tempered effect on the inhibition of whole cell growth. Further experimentation is warranted to completely explore the reason for the reduced potency and possible mechanisms for dual-site inhibition. Our working hypothesis is that this effect results from extension of the compound beyond the targeted folate substrate pocket. If this occurs, it is sub-optimal due to the strained conformation the compound would have to adopt to fill this pocket. It is reasonable that this happens in only a fraction of the interactions, and in the remaining binding region, this added compound bulk would promote unfavorable interplay with the solvent surrounding the substrate-binding site. While we cannot rule out cross-reaction with bacterial targets aside from DHFR in this setting, the concept of creating a dual-pocket inhibitor is a promising area in antifolate development.23-25 Future design of DHFR inhibitors involving this important class of 2,4-diaminopyrimidines should incorporate more restrained linkages and/or more spatially targeted modifications to assess this possibility.
Figure 2.
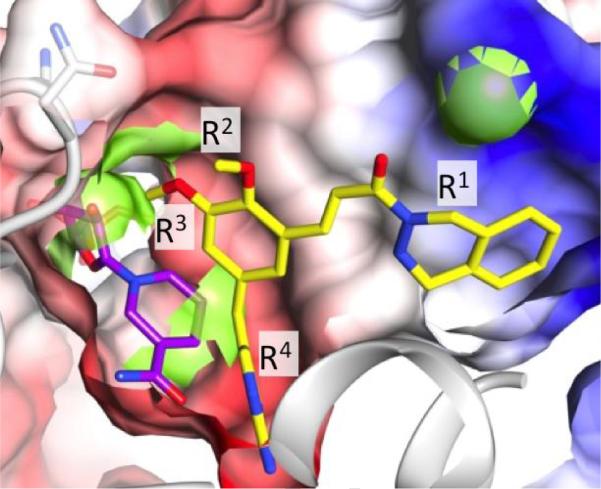
Extension from the R3 position of the DAP inhibitor is predicted to clash with co-factor binding. The DHFR binding site accommodates an NADPH co-factor, shown with purple carbon atoms, as well as a folate substrate or inhibitor (the dihydrophthalazine-appended DAP inhibitor is shown with yellow carbon atoms). The protein is displayed as a Coulombic-shaded van der Waals surface (red is acidic, blue is basic) and with a grey backbone ribbon. Modifications of the inhibitor scaffold are labeled as discussed in the text. Clashes between the protein and modified inhibitors were calculated and are visualized by green surface shading. Of note are green shadings proximal to the R3 modification site, the methylene bridge and the R1 site. The green sphere is Arg-53 of the DHFR enzyme, which can interact with R1 of the inhibitor.
4. Experimental section
Commercial anhydrous N,N-dimethylformamide (DMF) and dimethyl sulfoxide (DMSO) were stored under dry nitrogen and transferred by syringe into reactions when needed. Tetrahydrofuran (THF) was dried over potassium hydroxide pellets and distilled from lithium aluminum hydride prior to use. All other commercial reagents were used as received.
Unless otherwise specified, all reactions were run under dry nitrogen in oven-dried glassware. Reactions were monitored by thin layer chromatography on silica gel GF plates (Analtech, No. 21521). Preparative separations were performed by column chromatography on silica gel (Davisil®, grade 62, 60-200 mesh) mixed with UV-active phosphor (Sorbent Technologies, No. UV-05); band elution was monitored using a hand held UV lamp. Saturated NaHCO3, NaCl and NH4Cl used in work-up procedures refer to aqueous solutions. Melting points were uncorrected. FT-IR spectra were run as thin films on sodium chloride disks. 1H- and 13C-NMR spectra were measured on a Varian GEMINI 300 instrument at 300 MHz and 75 MHz, respectively, and referenced to internal tetramethylsilane. Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, GA 30071.
4.1 Synthesis of structures incorporating an oxidized methylene bridge (Scheme 1)
4.1.1 (2,4-Diaminopyrimidin-5-yl)(3-iodo-4,5-dimethoxyphenyl)methanone (2)
To a stirred solution of 1 (10.0 g, 25.9 mmol) in AcOH (80 mL) was added K2Cr2O7 (30.5 g, 0.104 mol, 4 equiv), and the solution was placed in a preheated oil bath at 120 °C and refluxed for a period of 6 h. The reaction mixture was cooled to 0 °C and poured into ice cold water with stirring. The mixture was then extracted with EtOAc (3 × 200 mL). The organic extracts were washed with saturated NaHCO3 (3 × 100 mL), water, saturated NaCl, dried (MgSO4), filtered, and concentrated under vacuum to give a thick brown liquid. The compound was purified using a silica gel column and eluted with CH2Cl2:MeOH:Et3N (10:4:1). Evaporation of the solvent yielded product 2 (8.08 g, 78%) as an off-white solid, mp 206-208 °C. IR: 3448, 3382, 3326, 1598 cm−1; 1H-NMR (DMSO-d6): δ 8.28 (br s, 1H), 8.14 (s, 1H), 7.54 (br s, 1H), 7.45 (d, J = 1.6 Hz, 1H), 7.20 (d, J = 1.6 Hz, 1H), 7.13 (br s, 2H), 3.85 (s, 3H), 3.78 (s, 3H); 13C-NMR (DMSO-d6): δ 191.5, 164.3, 163.8, 163.6, 152.0, 150.1, 137.2, 129.8, 113.4, 103.3, 92.4, 60.0, 56.1.
4.1.2. (±)-(E)-3-[5-(2,4-Diaminopyrimidine-5-carbonyl)-2,3-dimethoxyphenyl]-1-(1-propyl-2(1H)-phthalazinyl)-2-propen-1-one (4a)
To a stirred solution of 2 (1.00 g, 2.50 mmol) in dry DMF (7 mL) was added a solution of (±)-3a (657 mg, 2.88 mmol, 1.15 equiv)7 in DMF (1 mL), followed by N-ethylpiperidine (340 mg, 0.41 mL, 3.00 mmol 1.2 equiv) and Pd(OAc)2 (20 mg, 0.089 mmol). The reaction mixture was heated at 140 °C for 20 h and then gradually cooled to 0 °C. Isolation of the product was achieved by pouring the cooled reaction mixture directly onto a 50-cm × 2.5-cm silica gel flash chromatography column packed in CH2Cl2. Impurities were eluted using CH2Cl2, and the final product was collected using CH2Cl2:MeOH:Et3N (95:5:1). Evaporation of the solvent gave a pale yellow solid, which was further purified using a 15-cm × 2-cm silica gel column, packed with CH2Cl2:MeOH:Et3N (95:5:1). This second chromatography removed colored impurities as well as minor contaminants. Evaporation of the solvent gave 4a as an off-white solid. The compound was further purified by recrystallization from MeOH to give a white solid (990 mg, 79%), mp 212-214 °C. IR: 3377, 3143, 1660, 1593 cm−1; 1HNMR (DMSO-d6): δ 8.33 (br s, 2H), 8.18 (s, 1H), 7.93 (s, 1H), 7.89 (d, J = 16.5 Hz, 1H), 7.68 (d, J = 16.5 Hz, 1H), 7.58-7.37 (complex m, 5H), 7.24 (s, 1H), 7.12 (br s, 2H), 5.84 (t, J = 6.6 Hz, 1H), 3.88 (s, 3H), 3.87 (s, 3H), 1.54 (m, 2H), 1.17 (sextet, J = 7.1 Hz, 2H), 0.82 (t, J = 7.1 Hz, 3H); 13C-NMR (DMSO-d6): δ 192.8, 165.4, 164.4, 163.9, 163.6, 152.5, 149.3, 143.0, 135.9, 135.5, 133.6, 131.7, 128.3, 128.0, 126.5, 126.1, 123.6, 119.2, 119.0, 11.7, 103.5, 60.9, 56.1, 50.4, 36.9, 17.8, 13.7. Anal. Calcd for C27H28N6O4: C, 64.79; H, 5.64; N, 16.79. Found: C, 64.63; H, 5.75; N, 16.51.
4.1.3. (±)-(E)-3-[5-(2,4-Diaminopyrimidine-5-carbonyl)-2,3-dimethoxyphenyl]-1-(1-isobutenyl-2(1H)-phthalazinyl)-2-propen-1-one (4b)
This compound was prepared as above using 2 (1.00 g, 2.50 mmol), (±)-3b (691 mg, 2.88 mmol, 1.15 equiv),7 N-ethylpiperidine (340 mg, 0.41 mL, 3.00 mmol 1.2 equiv), and Pd(OAc)2 (20 mg, 0.089 mmol) in dry DMF (8 mL) under N2 to give 4b (960 mg, 75%) as an off-white solid, mp 138-140 °C. IR: 3320, 3179, 1655, 1594, cm−1; 1H-NMR (DMSO-d6 : δ 8.65 (br s, 1H), 8.39 (br s, 1H), 8.11 (d, J = 16.5 Hz, 1H), 7.67 (d, J = 16.5 Hz, 1H), 7.62 (s, 1H), 7.49 (d, J = 1.6 Hz, 1H), 7.43 (td, J = 7.1, 1.6 Hz, 1H), 7.33 (t, J = 7.1 Hz, 1H), 7. 27 (d, J = 6.6 Hz, 1H), 7.16 (d, J = 6.6 Hz, 1H), 7.15 (d, J = 1.6 Hz, 1H), 6.58 (d, J = 9.9 Hz, 1H), 5.67 (br s, 1H), 5.47 (br s, 2H), 5.30 (d, J = 9.9 Hz, 1H), 3.93 (s, 3H), 3.91 (s, 3H), 2.05 (s, 3H), 1.65 (s, 3H); 13C-NMR (DMSO-d6): δ 194.3, 166.2, 164.6, 164.4, 163.4, 153.1, 150.7, 141.5, 136.3, 134.6, 134.5, 134.2, 131.9, 129.5, 127.9, 126.2, 125.8, 123.3, 122.1, 120.1, 119.7, 113.2, 105.2, 61.5, 56.0, 50.0, 25.7, 18.6. Anal. Calcd for C28H28N6O4·0.4 H2O: C, 64.70; H, 5.58; N, 16.17. Found: C, 64.67; H, 5.63; N, 15.83.
4.2. Synthesis of structures modified at R2 and R3 (Scheme 2)
4.2.1. Benzaldehydes 6-9 and 23
These compounds were prepared on a 54-mmol scale via a two-step literature procedure20 in the following yields: 6 (80%), 7 (92%), 8 (92%), 9 (78%) and 23 (88%). The spectral data matched those reported.20
4.2.2. 3-Morpholinopropionitrile (10)
This compound was prepared on a 0.28-mol scale according to a literature procedure.26 The crude product was vacuum distilled to give 10 (37.2 g, 95%) as a colorless liquid, bp 88-90 °C (0.5 mm Hg) [lit.26 bp 149 °C (20 mm Hg)].
4.2.3. 5-(3,4-Diethoxy-5-iodobenzyl)pyrimidine-2,4-diamine (15)
To a stirred solution of NaOMe (1.69 g, 31.3 mmol, 1.0 equiv) in DMSO (25 mL) was added dropwise 3-morpholinopropionitrile 10 (5.25 g, 37.5 mmol, 1.2 equiv) at 65 °C. The reaction mixture was heated to 90 °C, followed by the addition of a warm solution of 6 (10.0 g, 31.3 mmol) in DMSO (15 mL) over 20 min. Stirring was continued at 90 °C for 45 min. The crude reaction mixture was then cooled in an ice bath, poured into ice-cold water, and extracted with CH2Cl2 (3 × 100 mL). The combined organic extracts were washed with saturated NaCl (1 × 100 mL), dried (MgSO4), filtered, and concentrated under vacuum to give crude 11 as dark red oil.
The crude product 11 was re-dissolved in dry ethanol (150 mL), aniline hydrochloride (4.46 g, 34.4 mol, 1.1 equiv) was added, and the reaction mixture was heated under reflux for 1 h. Guanidine hydrochloride (7.16 g, 75.0 mmol, 2.4 equiv), followed by NaOMe (6.75 g, 125 mmol, 4 equiv), was then added to the reaction mixture under hot conditions, and refluxing was continued for 4 h. The reaction mixture was concentrated to 1/4 of the volume under vacuum, and ice-cold water was added, resulting in the formation of a pale yellow solid. The solid was collected, washed with ice-cold ethanol, water, and finally with Et2O to give an off-white solid. The product was recrystallized using ethanol:water (4:1) to give 15 (10.4 g, 80%) as a white solid, mp 173-175 °C. IR: 3472, 3327, 3176 cm−1; 1H-NMR (DMSO-d6): δ 7.57 (s, 1H), 7.15 (s, 1H), 6.95 (s, 1H), 6.19 (br s, 2H), 5.82 (br s, 2H), 4.01 (q, J = 7.1 Hz, 2H), 3.93 (q, J = 7.1 Hz, 2H), 3.53 (s, 2H), 1.32 (coincident t, J = 7.1 Hz, 6H); 13C-NMR (DMSO-d6): δ 162.4, 162.2, 156.0, 151.1, 145.8, 138.5, 129.1, 114.6, 105.3, 93.2, 68.2, 63.9, 31.7, 15.7, 14.6.
4.2.4. 5-(4-Ethoxy-3-iodo-5-methoxybenzyl)pyrimidine-2,4-diamine (16)
This compound was prepared as described above for compound 15 using 7 (10.0 g, 32.7 mmol), 10 (5.49 g, 39.2 mmol, 1.2 equiv), and NaOMe (1.77 g, 32.7 mmol, 1.0 equiv) in DMSO (40 mL) to give crude 12 as a dark red oil. This oil in ethanol (150 mL) was further treated with PhNH2·HCl (5.08 g, 39.2 mmol, 1.2 equiv), guanidine·HCl (7.50 g, 78.5 mmol, 2.4 equiv), and NaOMe (7.06 g, 130.7 mmol, 4.0 equiv) to give 16 (10.2 g, 78%) as a white solid, mp 170-172 °C. IR: 3477, 3323, 3174 cm−1; 1H-NMR (DMSO-d6): δ 7.57 (s, 1H); 7.14 (d, J = 1.8 Hz, 1H), 6.97 (d, J = 1.8 Hz, 1H), 6.16 (br s, 2H), 5.77 (br s, 2H), 3.90 (q, J = 7.0 Hz, 2H), 3.76 (s, 3H), 3.53 (s, 2H), 1.31 (t, J = 7.0 Hz, 3H); 13C-NMR (DMSO-d6): δ 162.4, 162.1, 156.1, 152.0, 145.5, 138.7, 129.1, 113.7, 105.3, 93.2, 68.2, 55.8, 31.7, 15.6.
4.2.5. 5-[4-(Benzyloxy)-3-iodo-5-methoxybenzyl]pyrimidine-2,4-diamine (17)
This compound was prepared as described above for compound 15 using 8 (10.0 g, 27.2 mmol), 10 (4.57 g, 32.6 mmol, 1.2 equiv), and NaOMe (1.47 g, 27.2 mmol, 1.0 equiv) in DMSO (40 mL) to give crude 13 as a dark brown oil. This oil in ethanol (150 mL) was further treated with PhNH2·HCl (4.22 g, 32.6 mmol, 1.2 equiv), guanidine·HCl (6.23 g, 65.2 mmol, 2.4 equiv), and NaOMe (5.87 g, 109 mol, 4.0 equiv) to afford the product. The work-up procedure was altered for this compound. After completion, the reaction mixture was concentrated to dryness under vacuum, 100 mL of ice-cold water was added, and the compound was extracted with EtOAc (3 × 150 mL). The combined organic extracts were washed with saturated NaCl (1 × 100 mL), dried (MgSO4), filtered, and concentrated under vacuum. The crude mixture was then purified by column chromatography using a 30-cm × 2.5-cm silica gel column eluted with CH2Cl2:MeOH:Et3N (98:2:1) to afford 17 (9.03 g, 72%) as a yellow solid, mp 158-160 °C. IR: 3330, 3178 cm−1; 1H-NMR (DMSO-d6): δ 7.59 (s, 1H), 7.53 (m, 2H), 7.38 (m, 3H), 7.19 (s, 1H), 7.03 (s, 1H), 6.21 (br s, 2H), 5.82 (br s, 2H), 4.89 (s, 2H), 3.82 (s, 3H), 3.57 (s, 2H); 13C-NMR (DMSO-d6): δ 162.4, 162.2, 156.0, 152.1, 145.1, 139.0, 137.1, 129.3, 128.2 (2C), 128.0, 113.8, 105.3, 92.9, 73.7, 55.9, 31.8.
4.2.6. 5-(3-Iodo-4,5-methylenedioxybenzyl)pyrimidine-2,4-diamine (18)
This compound was prepared as described above for compound 15 using 9 (9.03 g, 32.7 mmol), 10 (5.49 g, 39.2 mmol, 1.2 equiv), and NaOMe (1.77 g, 32.7 mmol, 1.0 equiv) in DMSO (40 mL) to give crude 14 as a dark red oil. This oil in ethanol (150 mL) was further treated with PhNH2·HCl (5.08 g, 39.2 mmol, 1.2 equiv), guanidine·HCl (7.49 g, 78.5 mmol, 2.4 equiv), and NaOMe (7.06 g, 130.7 mmol, 4.0 equiv) to give 18 (9.07 g, 75%) as a white solid, mp 218-220 °C. IR: 3440, 3322, 3214 cm−1; 1H-NMR (CDCl3): d 7.54 (s, 1H), 7.02 (s, 1H), 6.78 (s, 1H), 6.14 (br s, 2H), 6.03 (s, 2H), 5.76 (br s, 2H), 3.49 (s, 2H); 13C-NMR (CDCl3): d 162.2, 162.1, 155.9, 147.3, 146.2, 136.5, 128.9, 108.9, 105.6, 100.5, 71.3, 31.7.
4.2.7. (±)-(E)-3-{5-[(2,4-Diamino-5-pyrimidinyl)methyl]-2,3-diethoxyphenyl}-1-(1-propyl-2(1H)-phthalazinyl)-2-propen-1-one (19a)
This compound was prepared as described above for 4a using 15 (1.00 g, 2.42 mmol), (±)-3a (634 mg, 2.78 mmol, 1.15 equiv), N-ethylpiperidine (328 mg, 0.40 mL, 2.90 mmol 1.2 equiv), and Pd(OAc)2 (20 mg, 0.089 mmol) in dry DMF (8 mL) under N2 to give 19a (968 mg, 78%) as an off-white solid, mp 118-120 ° C. IR: 3334, 1652, 1630 cm−1; 1H-NMR (DMSO-d6): δ 7.93 (s, 1H), 7.89 (d, J = 15.9 Hz, 1H), 7.64 (d, J = 15.9 Hz, 1H), 7.57 (s, 1H), 7.52 (m, 2H), 7.45 (d, J = 6.8 Hz, 1H), 7.39 (t, J = 7.1 Hz, 1H), 7.23 (s, 1H), 6.97 (s, 1H), 6.53 (br s, 2H), 6.06 (br s, 2H), 5.84 (t, J = 6.6 Hz, 1H), 4.01 (overlapping q, J = 7.1 Hz, 4H), 3.59 (s, 2H), 1.54 (m, 2H), 1.34 (t, J = 6.6 Hz, 3H), 1.30 (t, J = 7.1 Hz, 3H), 1.17 (sextet, J = 7.1 Hz, 2H), 0.82 (t, J = 7.1 Hz, 3H); 13C-NMR (DMSO-d6): δ 165.6, 162.5, 161.0, 153.4, 151.8, 145.3, 142.8, 137.2, 135.9, 133.6, 131.7, 128.3, 128.2, 126.5, 126.1, 123.7, 118.7, 117.7, 115.6, 106.3, 68.7, 63.7, 50.4, 36.8, 32.3, 17.8, 15.5, 14.7, 13.7. Anal. Calcd for C29H34N6O3·2.5 H2O: C, 62.24; H, 7.06; N, 15.02. Found: C, 62.46; H, 7.26; N, 15.05.
4.2.8. (±)-(E)-3-{5-[(2,4-Diamino-5-pyrimidinyl)methyl]-2,3-diethoxyphenyl}-1-(1-isobutenyl-2(1H)-phthalazinyl)-2-propen-1-one (19b)
This compound was prepared as described above for 4a using 15 (1.00 g, 2.42 mmol), (±)-3b (667 mg, 2.78 mmol, 1.15 equiv), N-ethylpiperidine (328 mg, 0.40 mL, 2.90 mmol 1.2 equiv), and Pd(OAc)2 (20 mg, 0.089 mmol) in dry DMF (8 mL) under N2 to give 19b (915 mg, 72%) as an off-white solid, mp 251-252 °C. IR: 3325, 3147, 1661, 1590 cm−1; 1H-NMR (DMSO-d6): δ 7.91 (s, 1H), 7.87 (d, J = 15.9 Hz, 1H), 7.63 (d, J = 15.9 Hz, 1H), 7.54 (s, 1H), 7.51 (m, 2H), 7.42 (m, 3H), 7.31 (d, J = 7.1 Hz, 1H), 7.26 (s, 1H), 7.00 (s, 1H), 6.89 (br s, 2H), 6.50 (d, J = 9.9 Hz, 1H), 5.24 (d, J = 9.9 Hz, 1H), 4.02 (m, 4H), 3.63 (s, 2H), 1.96 (s, 3H), 1.60 (s, 3H), 1.35 (t, J = 7.1 Hz, 3H), 1.31 (t, J = 7.1, 3H); 13C-NMR (DMSO-d6): δ 165.3, 163.3, 156.9, 151.9, 145.5, 142.0, 137.2, 134.4, 133.8, 133.5, 132.2, 128.3, 128.2, 126.2 (2C), 123.1, 122.1, 119.0, 117.9, 115.8, 107.8, 68.7, 63.9, 49.2, 31.8, 25.3, 18.4, 15.5, 14.7 (one aromatic C unresolved). Anal. Calcd for C30H34N6O3·5.7 H2O: C, 57.26; H, 7.27; N, 13.35. Found: C, 57.12; H, 7.00; N, 13.27.
4.2.9. (±)-(E)-3-{5-[(2,4-Diamino-5-pyrimidinyl)methyl]-2-ethoxy-3-methoxyphenyl}-1-(1-propyl-2(1H)-phthalazinyl)-2-propen-1-one (20a)
This compound was prepared as described above for 4a using 16 (1.00 g, 2.50 mmol), (±)-3a (656 mg, 2.88 mmol, 1.15 equiv), N-ethylpiperidine (339 mg, 0.41 mL, 3.00 mmol 1.2 equiv), and Pd(OAc)2 (20 mg, 0.089 mmol) in dry DMF (8 mL) under N2 to give 20a (925 mg, 74%) as an off-white solid, mp 145-147 °C. IR: 3334, 3196, 1650, 1603 cm−1; 1H-NMR (DMSO-d6): δ 7.94 (s, 1H), 7.89 (d, J = 15.9 Hz, 1H), 7.67 (d, J = 15.9 Hz, 1H), 7.56 (s, 1H), 7.53 (m, 2H), 7.46 (d, J = 6.8 Hz, 1H), 7.40 (t, J = 7.1 Hz, 1H), 7.27 (s, 1H), 7.07 (br s, 2H), 7.02 (s, 1H), 6.57 (br s, 2H), 5.84 (t, J = 6.6 Hz, 1H), 3.97 (q, J = 7.1 Hz, 2H), 3.79 (s, 3H), 3.63 (s, 2H), 1.54 (m, 2H), 1.30 (t, J = 7.1 Hz, 3H), 1.17 (sextet, J = 7.1 Hz, 2H), 0.82 (t, J = 7.1 Hz, 3H); 13C-NMR (DMSO-d6): δ 165.6, 163.0, 158.4, 152.7, 148.4, 145.2, 142.7, 137.1, 135.0, 133.6, 131.9, 131.7, 128.2, 126.5, 126.1, 123.6, 118.9, 117.9, 114.8, 107.2, 68.7, 55.8, 50.3, 36.8, 32.0, 17.8, 15.4, 13.6. Anal. Calcd for C28H32N6O3·4.5 H2O: C, 57.82; H, 7.10; N, 14.45. Found: C, 57.89; H, 6.94; N, 14.49.
4.2.10. (±)-(E)-3-{5-[(2,4-Diamino-5-pyrimidinyl)methyl]-2-ethoxy-3-methoxyphenyl}-1-(1-isobutenyl-2(1H)-phthalazinyl)-2-propen-1-one (20b)
This compound was prepared as described above for 4a using 16 (1.00 g, 2.50 mmol), (±)-3b (690 mg, 2.88 mmol, 1.15 equiv), N-ethylpiperidine (339 mg, 0.41 mL, 3.00 mmol 1.2 equiv), and Pd(OAc)2 (20 mg, 0.089 mmol) in dry DMF (8 mL) under N2 to give 20b (896 mg, 70%) as an off-white solid, mp 140-142 °C. IR: 3354, 3146, 1662, 1591 cm−1; 1H-NMR (DMSO-d6): δ 7.93 (s, 1H), 7.89 (d, J = 15.9 Hz, 1H), 7.64 (d, J = 15.9 Hz, 1H), 7.60 (s, 1H), 7.52 (m, 2H), 7.43 (t, J = 7.1 Hz, 1H), 7.31 (d, J = 7.1 Hz, 1H), 7.25 (s, 1H), 7.01 (s, 1H), 6.66 (br s, 2H), 6.51 (d, J = 9.9 Hz, 1H), 6.19 (br s, 2H), 5.25 (d, J = 9.9 Hz, 1H), 3.97 (q, J = 7.1 Hz, 2H), 3.80 (s, 3H), 3.63 (s, 2H), 1.97 (s, 3H), 1.61 (s, 3H), 1.31 (t, J = 7.1 Hz, 3H); 13C-NMR (DMSO-d6): δ 165.3, 162.6, 160.3, 152.6, 152.0, 145.1, 142.0, 137.2, 135.7, 133.8, 133.5, 132.2, 128.2 (2C), 126.2, 126.1, 123.1, 122.2, 118.8, 117.9, 114.7, 106.5, 68.7, 55.8, 49.2, 32.2, 25.3, 18.4, 15.4. Anal. Calcd for C29H32N6O3·2.4 H2O: C, 62.65; H, 6.38; N, 15.10. Found: C, 62.52; H, 6.10; N, 14.75.
4.2.11. (±)-(E)-3-{2-(Benzyloxy)-5-[(2,4-diamino-5-pyrimidinyl)methyl]-3-methoxyphenyl}-1-(1-propyl-2(1H)-phthalazinyl)-2-propen-1-one (21a)
This compound was prepared as described above for 4a using 17 (1.00 g, 2.16 mmol), (±)-3a (568 mg, 2.49 mmol, 1.15 equiv), N-ethylpiperidine (294 mg, 0.36 mL, 2.60 mmol 1.2 equiv), and Pd(OAc)2 (20 mg, 0.089 mmol) in dry DMF (8 mL) under N2 to give 21a (850 mg, 70%) as an off-white solid, mp 123-125 °C. IR: 3474, 3342, 3181, 1654, 1605 cm−1; 1H-NMR (DMSO-d6): δ 8.06 (d, J = 15.9 Hz, 1H), 7.80 (s, 1H), 7.64 (d, J = 15.9 Hz, 1H), 7.62 (s, 1H), 7.46 (m, 3H), 7.40-7.25 (complex m, 5H), 7.18 (d, J = 7.1 Hz, 1H), 7.11 (s, 1H), 6.67 (s, 1H), 5.90 (t, J = 6.6 Hz, 1H), 5.00 (s, 2H), 4.77 (br s, 2H), 4.60 (br s, 2H), 3.81 (s, 3H), 3.68 (s, 2H), 1.65 (m, 2H), 1.27 (m, 2H), 0.86 (t, J = 7.1 Hz, 3H); 13C-NMR (DMSO-d6): δ 166.5, 162.6, 162.2, 156.7, 153.7, 145.9, 142.2, 137.4, 137.0, 134.2, 134.1, 131.4 130.2, 128.6, 128.3, 128.00, 127.95, 126.5, 125.6, 124.0, 118.9, 118.8, 112.9, 106.4, 75.4, 55.9, 51.3, 37.3, 34.4, 18.3, 13.8. Anal. Calcd for C33H34N6O3·0.3 H2O: C, 69.77; H, 6.14; N, 14.79. Found: C, 69.70; H, 6.14; N, 14.44.
4.2.12. (±)-(E)-3-{5-[(2,4-Diaminopyrimidin-5-yl)methyl]-2,3-methylenedioxyphenyl}-1-(1-propylphthalazin-2(1H)-yl)-2-propen-1-one (22a)
This compound was prepared as described above for 4a using 18 (1.00 g, 2.70 mmol), (±)-3a (709 mg, 3.11 mmol, 1.15 equiv), N-ethylpiperidine (366 mg, 0.44 mL, 3.24 mmol 1.2 equiv), and Pd(OAc)2 (20 mg, 0.089 mmol) in dry DMF (8 mL) under N2 to give 22a (901 mg, 71%) as an off-white solid, mp 123-125 °C. IR: 3424, 3312, 3100, 1635, 1599 cm−1; 1H-NMR (DMSO-d6): d 7.96 (s, 1H), 7.74 (d, J = 16.0 Hz, 1H), 7.55 (s, 1H), 7.54-7.41 (complex, 3H), 7.50 (d, J = 16.0 Hz, 1H), 7.38 (d, J = 7.4 Hz, 1H), 7.06 (s, 1H), 6.82 (s, 1H), 6.13 (2s, 4H), 5.85 (t, J = 6.7 Hz, 1H), 5.73 (br s, 2H), 3.53 (s, 2H), 1.52 (m, 2H), 1.17 (sextet, J = 7.4 Hz, 2H), 0.81 (t, J = 7.4 Hz, 3H); 13C-NMR (DMSO-d6): d 165.5, 162.3, 162.1, 155.7, 147.8, 144.4, 143.0, 136.8, 134.5, 133.6, 131.7, 128.3, 126.5, 126.1, 123.6, 122.0, 119.2, 116.7, 110.0, 105.8, 101.7, 50.4, 36.8, 32.2, 17.8, 13.7. Anal. Calcd for C26H26N6O3·0.6 EtOH: C, 65.58; H, 5.99; N, 16.87. Found: C, 65.40; H, 5.85; N, 16.76.
4.3. Synthesis of compounds modified at R3 (Scheme 3)
4.3.1. 5-[3-Iodo-4-methoxy-5-(methoxymethoxy)benzyl]-pyrimidine-2,4-diamine (25)
This compound was prepared as described above for compound 15 using 2320 (10.0 g, 24.0 mmol), 10 (4.03 g, 28.8 mmol, 1.2 equiv) and NaOMe (1.30 g, 24.0 mmol, 1.0 equiv) in DMSO (40 mL) to give intermediate 24 (9.24 g, 90%) as a dark brown oil. This oil in ethanol (150 mL) was treated with PhNH2·HCl (3.10 g, 24.0 mmol, 1.0 equiv), guanidine·HCl (5.50 g, 57.6 mmol, 2.4 equiv), and NaOMe (5.18 g, 96.0 mmol, 4.0 equiv) to afford a dark yellow solution. The work-up procedure was altered for this compound. The crude reaction mixture was concentrated to dryness under vacuum, 100 mL of ice-cold water was added, and the compound was extracted with EtOAc (3 × 150 mL). The combined organic extracts were washed with saturated NaCl (1 × 100 mL), dried (MgSO4), filtered, and concentrated under vacuum. The crude mixture was then purified by column chromatography using 30-cm × 2.5-cm silica gel column eluted with CH2Cl2:MeOH:Et3N (98:2:1) to afford 25 (8.29 g, 83%) as a white solid, mp 123-125 °C. IR: 3461, 3153, 1625 cm−1; 1H-NMR (DMSO-d6): δ 7.54 (s, 1H), 7.22 (d, J = 1.6 Hz, 1H), 7.05 (d, J = 1.6 Hz, 1H), 6.14 (br s, 2H), 5.76 (br s, 2H), 5.9 (s, 2H), 3.70 (s, 3H), 3.51 (s, 2H), 3.40 (s, 3H); 13C-NMR (DMSO-d6): δ 162.4, 162.2, 156.1, 149.3, 147.3, 138.9, 130.7, 117.2, 105.1, 94.6, 92.6, 60.0, 56.0, 31.6.
4.3.2. (±)-(E)-3-{5-[(2,4-Diamino-5-pyrimidinyl)methyl]-2-methoxy-3-(methoxymethoxy)-phenyl]-1-(1-propyl-2(1H)-phthalazinyl)-2-propen-1-one (26)
This compound was prepared as described above for 4a using 25 (1.00 g, 2.40 mmol), with (±)-3a (630 mg, 2.76 mmol, 1.15 equiv), N-ethylpiperidine (326 mg, 0.40 mL, 2.88 mmol 1.2 equiv), and Pd(OAc)2 (20 mg, 0.089 mmol) in dry DMF (8 mL) under N2 to give 26 (719 mg, 58%) as a brown solid. 1H-NMR revealed the product 26 contained compound 27 (hydroxy derivative) in the ratio of (8:2) as a mixture. The product was taken to the next step without further purification or analysis.
4.3.3. (±)-(E)-3-{5-[(2,4-Diamino-5-pyrimidinyl)methyl]-3-hydroxy-2-methoxyphenyl}-1-(1-propyl-2(1H)-phthalazinyl)-2-propen-1-one (27)
To a stirred solution of crude 26 (1.00 g, 1.94 mmol) in CH2Cl2 (35 mL) was added MeOH·HCl (35 mL), and the reaction mixture was stirred for 24 h at room temperature. Evaporation of the solvent gave a dark brown liquid. To remove all traces of MeOH·HCl, a 50-mL portion of Et2O:CH2Cl2 (1:1) was added, and the solvent was removed under vacuum. This process was repeated 5-6 times to produce an off-white solid. This solid was triturated with Et2O, and the product was collected to give 27 (0.86 g, 94%) as a white solid, mp 135-137 °C. IR: 3481, 3345, 3204, 1652, 1617 cm−1; 1HNMR (DMSO-d6): δ 9.50 (br s, 1H), 7.94 (s, 1H), 7.86 (d, J = 15.9 Hz, 1H), 7.63 (d, J = 15.9 Hz, 1H), 7.58-7.48 (complex m, 3H), 7.46 (d, J = 7.1 Hz, 1H), 7.39 (d, J = 7.1 Hz, 1H), 7.15 (s, 1H), 6.70 (s, 1H), 6.16 (br s, 2H), 5.85 (t, J = 6.6 Hz, 1H), 5.79 (br s, 2H), 3.73 (s, 3H), 3.54 (s, 2H), 1.54 (m, 2H), 1.17 (sextet, J = 7.1 Hz, 2H), 0.82 (t, J = 7.1 Hz, 3H); 13C-NMR (DMSO-d6): δ 165.6, 162.3, 162.2, 155.6, 150.2, 145.0, 142.7, 137.1, 136.3, 133.6, 131.7, 128.2, 127.8, 126.5, 126.0, 123.7, 118.1, 117.3, 105,5, 60.5, 50.3, 36.8, 32.0, 17.8, 14.0, 13.7. Anal. Calcd for C26H28N6O3·0.5 H2O: C, 64.85; H, 6.07; N, 17.45. Found: C, 64.93; H, 5.94; N, 17.15.
4.3.4 (±)-(E)-3-{5-[(2,4-Diamino-5-pyrimidinyl)methyl]-2-methoxy-3-propoxyphenyl}-1-(1-propyl-2(1H)-phthalazinyl)-2-propen-1-one (28)
To a stirred solution of 27 (150 mg, 0.318 mmol) in CH2Cl2 (8 mL) was added DBU (53 mg, 0.052 mL, 0.350 mmol, 1.1 equiv), and the solution was stirred for 30 min. To the reaction mixture was added dropwise propyl bromide (43 mg, 0.032 mL, 0.350 mmol, 1.1 equiv), and stirring was continued for 20 h. The reaction mixture was evaporated to dryness and purified on a 20-cm × 20-cm silica gel, preparative thin layer chromatography plate eluted with CH2Cl2:MeOH:Et3N (95:5:1). The product band was washed with CH2Cl2:MeOH: Et3N (95:5:1) to afford an off-white solid. This solid was triturated with Et2O to remove Et3N, and the compound was filtered and dried under vacuum to afford 28 (144 mg, 88%) as a white solid, mp 194-195 °C. IR: 3342, 3157, 1659, 1637 cm−1; 1H-NMR (DMSO-d6): δ 7.94 (s, 1H), 7.87 (d, J = 15.9 Hz, 1H), 7.64 (d, J = 15.9 Hz, 1H), 7.53 (m, 3H), 7.46 (d, J = 7.1 Hz, 1H), 7.40 (d, J = 7.1 Hz, 1H), 7.28 (s, 1H), 7.09 (br s, 2H), 7.01 (s, 1H), 6.59 (br s, 2H), 5.85 (t, J = 6.3 Hz, 1H), 3.94 (t, J = 6.0 Hz, 2H), 3.78 (s, 3H), 3.62 (s, 2H), 1.77 (sextet, J = 6.6 Hz, 2H), 1.54 (m, 2H), 1.17 (sextet, J = 7.1 Hz, 2H), 1.01 (t, J = 7.1 Hz, 3H), 0.82 (t, J = 7.1 Hz, 3H); 13C-NMR (DMSO-d6): δ 165.6, 163.0, 158.4, 151.9, 148.3, 146.3, 142.8, 136.7, 135.1, 133.6, 131.7, 128.3, 127.9, 126.5, 126.1, 123.6, 118.6, 117.9, 115.7, 107.3, 69.9, 60.7, 50.3, 36.8, 32.0, 22.1, 17.8, 13.7, 10.6. Anal. Calcd for C29H34N6O3·3.7 H2O: C, 59.92; H, 7.18; N, 14.46. Found: C, 59.83; H, 7.19; N, 14.39.
4.3.5. (±)-(E)-5-[(2,4-Diamino-5-pyrimidinyl)methyl]-2-methoxy-3-[3-oxo-3-(1-propyl-2(1H)-phthalazinyl)-1-propen-1-yl]phenyl benzoate (29)
This compound was prepared as described above for compound 28 using 27 (150 mg, 0.318 mmol), DBU (53 mg, 0.052 mL, 0.350 mmol, 1.1 equiv), and benzoyl chloride (49 mg, 0.041 mL, 0.350 mmol, 1.1 equiv) in CH2Cl2 (8 mL) to give 29 (169 mg, 92%) as an off-white solid, mp 125-127 °C. IR: 3335, 3194, 1739, 1654, 1604 cm−1; 1HNMR (DMSO-d6): δ 8.20 (d, J = 7.7 Hz, 2H), 8.01 (d, J = 15.9 Hz, 1H), 7.78 (s, 1H), 7.71 (d, J = 15.9 Hz, 1H), 7.66 (m, 2H), 7.52 (t, J = 7.7 Hz, 2H), 7.44 (obscured, 1H), 7.42 (s, 1H), 7.36 (t, J = 7.1 Hz, 1H), 7.29 (d, J = 7.1 Hz, 1H), 7.18 (d, J = 7.1 Hz, 1H), 6.99 (s, 1H), 5.90 (t, J = 6.6 Hz, 1H), 5.05 (br s, 2H), 4.76 (br s, 2H), 3.81 (s, 3H), 3.72 (s, 2H), 1.64 (m, 2H), 1.26 (m, 2H), 0.87 (t, J = 7.1 Hz, 3H); 13C-NMR (DMSO-d6): δ 166.3, 164.7, 163.6, 161.8, 155.9, 144.9, 144.6, 142.5, 136.4, 134.2, 134.1, 133.8, 131.4, 130.9, 130.3, 128.9, 128.7, 128.0, 126.5, 125.7, 125.0, 124.2, 123.9, 119.6, 105.8, 62.1, 51.4, 37.3, 33.6, 18.3, 13.8. Anal. Calcd for C33H32N6O4·5.3 H2O: C, 58.97; H, 6.39; N, 12.50. Found: C, 58.98; H, 6.52; N, 12.50.
4.3.6. (±)-(E)-5-[(2,4-Diamino-5-pyrimidinyl)methyl]-2-methoxy-3-[3-oxo-3-(1-propyl-2(1H)-phthalazinyl)-1-propen-1-yl]phenyl 4-methoxybenzoate (30)
This compound was prepared as described above for compound 28 using 27 (150 mg, 0.318 mmol), DBU (53 mg, 0.052 mL, 0.350 mmol, 1.1 equiv), and 4-methoxybenzoyl chloride (60 mg, 0.047 mL, 0.350 mmol, 1.1 equiv) in CH2 Cl2 (8 mL) to give 30 (168 mg, 87%) as an off-white solid, mp 124-126 °C. IR: 3474, 3344, 3188, 1732, 1605 cm−1; 1H-NMR (DMSO-d6): δ 8.14 (d, J = 8.8 Hz, 2H); 8.01 (d, J = 15.9 Hz, 1H); 7.81 (s, 1H), 7.70 (d, J= 15.9 Hz, 1H), 7.67 (s, 1H), 7.48-7.40 (complex m, 2H), 7.36 (t, J = 7.1 Hz, 1H), 7.30 (d, J = 7.1 Hz, 1H), 7.18 (d, J = 7.1 Hz, 1H), 6.99 (complex m, 3H), 5.90 (t, J = 6.6 Hz, 1H), 4.77 (br s, 2H), 4.65 (br s, 2H), 3.90 (s, 3H), 3.80 (s, 3H), 3.72 (s, 2H), 1.65 (m, 2H), 1.28 (m, 2H), 0.87 (t, J = 7.1 Hz, 3H); 13C-NMR (DMSO-d6): δ 166.4, 164.4, 164.1, 162.5, 162.3, 157.1, 150.0, 144.7, 142.5, 136.6, 134.3, 134.2, 132.0, 131.4, 130.8, 128.0, 126.5, 125.7, 124.9, 124.4, 124.0, 121.2, 119.5, 113.9, 105.8, 62.0, 55.5, 51.4, 37.3, 33.7, 18.3, 13.8. Anal. Calcd for C34H34N6O5·H2O: C, 65.37; H, 5.81; N, 13.45. Found: C, 65.15; H, 5.65; N, 13.10.
4.3.7. (±)-(E)-5-[(2,4-Diamino-5-pyrimidinyl)methyl]-2-methoxy-3-[3-oxo-3-(1-propyl-2(1H)-phthalazinyl)-1-propen-1-yl]phenyl 4-nitrobenzoate (31)
This compound was prepared as described above for compound 28 using 27 (150 mg, 0.318 mmol), DBU (53 mg, 0.052 mL, 0.350 mmol, 1.1 equiv), and 4-nitrobenzoyl chloride (65 mg, 0.350 mmol, 1.1 equiv) in CH2Cl2 (8 mL) to give 31 (154 mg, 78%) as a yellow solid, mp 130-132 °C. IR: 3480, 3344, 3176, 1744, 1607 cm−1; 1H-NMR (DMSO-d6): δ 8.38 (s, 2H), 8.00 (d, J = 15.9 Hz, 1H), 7.81 (s, 1H), 7.72 (d, J = 15.9 Hz, 1H), 7.68 (s, 1H), 7.46 (complex m, 4H), 7.37 (t, J = 7.1 Hz, 1H), 7.30 (d, J = 6.6 Hz, 1H), 7.29 (d, J = 7.1 Hz, 1H), 6.98 (d, J = 1.6 Hz, 1H), 5.90 (t, J = 6.6 Hz, 1H), 4.78 (br s, 2H), 4.63 (br s, 2H), 3.80 (s, 3H), 3.74 (s, 2H), 1.65 (m, 2H), 1.28 (m, 2H), 0.87 (t, J = 7.1 Hz, 3H); 13C-NMR (DMSO-d6): δ 166.2, 162.9, 162.4, 162.3, 157.1, 151.0, 149.6, 144.3, 142.6, 136.1, 134.7, 134.3, 134.2, 131.5, 131.4, 131.1, 128.0, 126.5, 125.7, 125.6, 123.9, 123.8, 123.6, 120.0, 105.6, 62.3, 51.4, 37.3, 33.6, 18.3, 13.8. Anal. Calcd for C33H31N7O6·0.8 H2O: C, 62.31; H, 5.17; N, 15.41. Found: C, 62.28; H, 4.86; N, 15.53.
4.3.8. (±)-(E)-5-[(2,4-Diamino-5-pyrimidinyl)methyl]-2-methoxy-3-[3-oxo-3-(1-propyl-2(1H)-phthalazinyl)-1-propen-1-yl]phenyl 4-(trifluoromethoxy)benzoate (32)
This compound was prepared as described above for compound 28 using 27 (150 mg, 0.318 mmol), DBU (53 mg, 0.052 mL, 0.350 mmol, 1.1 equiv), and 4-(trifluoromethoxy)benzoyl chloride (79 mg, 0.055 mL, 0.350 mmol, 1.1 equiv) in CH2Cl2 (8 mL) to give 32 (172 mg, 82%) as an off-white solid, mp 118-120 °C. IR: 3339, 3182, 1743, 1656, 1606 cm −1; 1H-NMR (DMSO-d6): δ 8.25 (d, J = 8.8 Hz, 2H), 8.00 (d, J = 16.5 Hz, 1H), 7.81 (s, 1H), 7.70 (d, J = 16.5 Hz, 1H), 7.68 (s, 1H), 7.45 (m, 2H), 7.41-7.28 (complex m, 4H), 7.19 (d, J = 7.7 Hz, 1H), 6.97 (d, J = 1.6 Hz, 1H), 5.90 (t, J = 6.6, 1H), 4.79 (br s, 2H), 4.62 (br s, 2H), 3.80 (s, 3H), 3.73 (s, 2H), 1.65 (m, 2H), 1.28 (m, 2H), 0.87 (t, J = 7.1 Hz, 3H); 13C-NMR (DMSO-d6): δ 166.3, 163.5, 162.5, 162.2, 156.9, 153.2, 149.8, 144.5, 142.6, 136.3, 134.4, 134.2, 132.3, 131.5, 131.0, 128.0, 127.2, 126.5, 125.7, 125.3, 124.0, 123.9, 123.7 (q, J = 258.1 Hz), 120.5, 119.8, 105.7, 62.2, 51.4, 37.3, 33.6, 18.3, 13.8. Anal. Calcd for C34H31F3N6O5·5.5 H2O: C 53.75; H 5.57; N 11.06. Found: C, 53.96; H, 5.57; N, 11.04.
4.3.9. (±)-(E)-3-{5-[(2,4-Diamino-5-pyrimidinyl)methyl]-3-[(4-fluorobenzyl)oxy]-2-methoxyphenyl}-1-(1-propyl-2(1H)-phthalazinyl)-2-propen-1-one (33)
This compound was prepared as described above for compound 28 using 27 (150 mg, 0.318 mmol), DBU (53 mg, 0.052 mL, 0.350 mmol, 1.1 equiv), and 4-fluorobenzyl bromide (66 mg, 0.044 mL, 0.350 mmol, 1.1 equiv) in CH2Cl2 (8 mL) to give 33 (160 mg, 87%) as a yellow solid, mp 115-117 °C. IR: 3473, 3338, 3183, 1649, 1605 cm−1; 1H-NMR (CDCl3): δ 8.07 (d, J = 15.9 Hz, 1H), 7.78 (s, 1H), 7.67 (s, 1H), 7.66 (d, J = 15.9 Hz, 1H), 7.45 (t, J = 7.1 Hz, 1H), 7.40-7.26 (complex m, 4H), 7.19 (d, J = 7.1 Hz, 1H), 7.16 (s, 1H), 7.05 (t, J = 8.8 Hz, 2H), 6.66 (d, J = 1.1 Hz, 1H), 5.91 (t, J = 6.6 Hz, 1H), 5.00 (s, 2H), 4.78 (br s, 2H), 4.52 (br s, 2H), 3.87 (s, 3H), 3.65 (s, 2H), 1.65 (m, 2H), 1.28 (m, 2H), 0.87 (t, J = 7.1 Hz, 3H); 13C-NMR (CDCl3): δ 166.5, 162.55, 162.50 (d, J = 245.0 Hz), 162.2, 156.7, 152.3, 147.9, 142.4, 137.1, 134.3, 133.9, 132.3, 131.4, 130.0, 129.3 (J = 7.7 Hz), 128.0, 126.5, 125.7, 124.0, 119.4, 118.9, 115.5 (J = 21.3 Hz), 115.1, 106.3, 70.2, 61.4, 51.3, 37.3, 34.2, 18.3, 13.8. Anal. Calcd for C33H33FN6O3·0.5 H2O: C, 67.22; H, 5.81; N, 14.25. Found: C, 67.04; H, 5.99; N, 14.00.
4.3.10. (±)-(E)-3-{5-[(2,4-Diamino-5-pyrimidinyl)methyl]-2-methoxy-3-[(4-(trifluoromethyl)benzyl)oxy]phenyl}-1-(1-propyl-2(1H)-phthalazinyl)-2-propen-1-one (34)
This compound was prepared as described above for compound 28 using 27 (150 mg, 0.318 mmol), DBU (53 mg, 0.052 mL, 0.350 mmol, 1.1 equiv), and 4-(trifluoromethyl)benzyl bromide (84 mg, 0.054 mL, 0.350 mmol, 1.1 equiv) in CH2Cl2 (8 mL) to give 34 (124 mg, 62%) as a yellow solid, mp 145-147 °C. IR: 3328, 3175, 1659, 1620 cm−1; 1H-NMR (CDCl3): δ 8.01 (d, J = 15.9 Hz, 1H), 7,68 (d, J = 15.9 Hz, 1H), 7.66 (s, 1H), 7.60 (d, J = 8.0 Hz, 2H), 7.54 (d, J = 8.0 Hz, 2H), 7.42 (t, J = 7.1 Hz, 1H), 7.33 (m, 3H), 7.27 (s, 1H), 7.27 (d, J = 7.1 Hz, 1H), 7.14 (d, J = 7.1 Hz, 1H), 7.13 (s, 1H), 6.80 (s, 1H), 6.69 (br s, 4H), 5.86 (t, J = 6.6 Hz, 1H), 5.12 (s, 2H), 3.86 (s, 1H), 3.66 (s, 2H), 1.61 (m, 2H), 1.33 (m, 2H), 0.82 (t, J = 7.1 Hz, 3H); 13C-NMR (CDCl3): δ 166.6, 163.6, 157.1, 152.3, 147.9, 142.7, 140.6, 136.8, 134.1, 132.2, 131.5, 130.2, 130.0, 129.8, 128.1, 127.5, 126.5, 125.8, 125.3, 124.1 (d, J = 270.9 Hz), 123.9, 120.2, 119.4, 115.4, 108.3, 70.1, 61.4, 51.4, 37.3, 33.5, 18.3, 13.8. Anal. Calcd for C34H33F3N6O3·3.0 H2O: C, 59.64; H, 5.74; N, 12.27. Found: C, 59.70; H, 5.95; N, 12.37.
4.4 Biological measurements
The methods used to assess potency have been previously published.7, 8 Briefly, the minimum inhibitory concentration (MIC) was the lowest concentration of compound needed to block growth of a standardized culture of B. anthracis Sterne as measured by turbidity at 600 nm and by visual inspection.27 Enzymatic activity was reconstituted in vitro with saturating concentrations of co-factor and dihydrofolate reductase. The amount of tetrahydrofolate produced was monitored, and the concentration of compound that inhibited 50% of this reaction was then determined. This value, an IC50, was converted to an inhibition constant (Ki) by incorporating the strength of binding to dihydrofolate (Km) through the formalism of Cheng-Prusoff.28
Acknowledgments
We gratefully acknowledge support of this work by the National Institutes of Allergy and Infectious Diseases (R01-AI090685) of the NIH/NIAID and the Sitlington Chair in Infectious Diseases, both to WWB. NSF (BIR-9512269), the Oklahoma State Regents for Higher Education, the W. M. Keck Foundation, and Conoco, Inc, provided funding the 300 MHz and 400 MHz NMR spectrometers of the Oklahoma Statewide Shared NMR Facility. The authors also wish to thank the OSU College of Arts and Sciences for funds to upgrade our departmental FT-IR instruments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Beierlein JM, Anderson AC. Curr. Med. Chem. 2011;18:5083. doi: 10.2174/092986711797636036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Proliferation of weapons of mass destruction: Assessing the Risks. Available online atat: http://www.au.af.mil/au/awc/awcgate/ota/9341.pdf.
- 3.Bourne CR, Bunce RA, Bourne PC, Berlin KD, Barrow EW, Barrow WW. Antimicrob. Agents Chemother. 2009;53:3065. doi: 10.1128/AAC.01666-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrow EW, Dreier J, Reinelt S, Bourne PC, Barrow WW. Antimicrob. Agents Chemother. 2007;51:4447. doi: 10.1128/AAC.00628-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.D'Costa VM, King CE, Kalan L, Morar M, Sung WWL, Schwarz C, Froese D, Zazula G, Calmels F, Debruyne R, Golding GB, Poinar HN, Wright GD. Nature. 2011;477:457. doi: 10.1038/nature10388. [DOI] [PubMed] [Google Scholar]
- 6.Odendaal MW, Pieterson PM, Devos V. Onderstepoort J. Vet. Res. 1991;58:17. [PubMed] [Google Scholar]
- 7.Nammalwar B, Bunce RA, Berlin KD, Bourne CR, Bourne PC, Barrow EW, Barrow WW. Eur. J. Med. Chem. 2012;54:387. doi: 10.1016/j.ejmech.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nammalwar B, Bourne CR, Bunce RA, Wakeham N, Bourne PC, Ramnarayan K, Mylvaganam S, Berlin KD, Barrow EW, Barrow WW. ChemMedChem. 2012;7:1974. doi: 10.1002/cmdc.201200291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nammalwar B, Muddala NP, Bourne CR, Henry M, Bourne PC, Bunce RA, Barrow EW, Berlin KD, Barrow WW. Molecules. 2014;19:3231. doi: 10.3390/molecules19033231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bourne CR, Wakeham N, Nammalwar B, Tseitin V, Bourne PC, Barrow EW, Mylvaganam S, Ramnarayan K, Bunce RA, Berlin KD, Barrow WW. Biochim. Biophys. Acta-Prot. Proteom. 2013;1834:46. doi: 10.1016/j.bbapap.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caspers P, Bury L, Gaucher B, Heim J, Shapiro S, Siegrist S, Schmitt-Hoffmann A, Thenoz L, Urwyler H. Antimicrob. Agents Chemother. 2009;53:3620. doi: 10.1128/AAC.00377-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark C, Ednie LM, Lin G, Smith K, Kosowska-Shick K, McGhee P, Dewasse B, Beachel L, Caspers P, Gaucher B, Mert G, Shapiro S, Appelbaum PC. Antimicrob. Agents Chemother. 2009;53:1353. doi: 10.1128/AAC.01619-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bowker KE, Caspers P, Gaucher B, MacGowan AP. Antimicrob. Agents Chemother. 2009;53:4949. doi: 10.1128/AAC.00845-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guerry P, Hubschwerlen C, Jolidon S, Specklin J-L, Wyss P-C. 1998 World Patent WO9839328A1.; Chem. Abstr. 1998;129:230736. [Google Scholar]
- 15.Oefner C, Bandera M, Haldimann A, Laue H, Schulz H, Mukhija S, Parisi S, Weiss L, Lociuro S, Dale GE. J. Antimicrob. Chemother. 2009;63:687. doi: 10.1093/jac/dkp024. [DOI] [PubMed] [Google Scholar]
- 16.Smith K, Ednie LM, Appelbaum PC, Hawser S, Lociuro S. Antimicrob. Agents Chemother. 2008;52:2279. doi: 10.1128/AAC.01620-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li XM, Hilgers M, Cunningham M, Chen ZY, Trzoss M, Zhang JH, Kohnen L, Lam T, Creighton C, Kedar GC, Nelson K, Kwan B, Stidham M, Brown-Driver V, Shaw KJ, Finn J. Bioorg. Med. Chem. Lett. 2011;21:5171. doi: 10.1016/j.bmcl.2011.07.059. [DOI] [PubMed] [Google Scholar]
- 18.Nammalwar B, Bunce RA, Berlin KD, Bourne CR, Bourne PC, Barrow EW, Barrow WW. Org. Prep. Proced. Int. 2012;44:281. doi: 10.1080/00304948.2012.676823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nammalwar B, Bunce RA, Berlin KD, Bourne CR, Bourne PC, Barrow EW, Barrow WW. Org. Prep. Proced. Int. 2013;45:66. doi: 10.1080/00304948.2013.743755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nammalwar B, Bunce RA, Berlin KD, Bourne CR, Bourne PC, Barrow EW, Barrow WW. Org. Prep. Proced. Int. 2012;44:146. doi: 10.1080/00304948.2012.643700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barrow EW, Bourne PC, Barrow WW. Antimicrob. Agents Chemother. 2004;48:4643. doi: 10.1128/AAC.48.12.4643-4649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwalbe CH, Cody V. Crystallogr. Rev. 2006;12:267. [Google Scholar]
- 23.Yun M-K, Wu Y, Li Z, Zhao Y, Waddell MB, Ferreira AM, Lee RE, Bashford D, White SW. Science. 2012;335:1110. doi: 10.1126/science.1214641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi G, Shaw G, Li Y, Wu Y, Yan H, Ji X. Bioorg. Med. Chem. 2012;20:4303. doi: 10.1016/j.bmc.2012.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bennett BC, Wan Q, Ahmad MF, Langan P, Dealwis CG. J. Struct. Biol. 2009;166:162. doi: 10.1016/j.jsb.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whitmore FC, Mosher HS, Adams RR, Taylor RB, Chapin EC, Weisel C, Yanko W. J. Am. Chem. Soc. 1944;66:725. [Google Scholar]
- 27.Clinical Laboratory Standards Institute (CLSI) Methods for Dilution Antimicrobial Susceptibility Test for Bacteria That Grow Aerobically; Approved Standard–Eighth Edition. Wayne, PA, USA: 2009. CLSI document M7-A8. [Google Scholar]
- 28.Cheng Y, Prusoff WH. Biochem. Pharmacol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]