

Abstract
Human immunodeficiency virus type 1 (HIV-1) subtype C viruses are associated with nearly half of worldwide HIV-1 infections and are most predominant in India and the southern and eastern parts of Africa. Earlier reports from India identified the preponderance of subtype C and a small proportion of subtype A viruses. Subsequent reports identifying multiple subtypes suggest new introductions and/or their detection due to extended screening. The southern parts of India constitute emerging areas of the epidemic, but it is not known whether HIV-1 infection in these areas is associated with subtype C viruses or is due to the potential new introduction of non-subtype C viruses. Here, we describe the development of a specific and sensitive PCR-based strategy to identify subtype C-viruses (C-PCR). The strategy is based on amplifying a region encompassing a long terminal repeat and gag in the first round, followed by two sets of nested primers; one amplifies multiple subtypes, while the other is specific to subtype C. The common HIV and subtype C-specific fragments are distinguishable by length differences in agarose gels and by the difference in the numbers of NF-κB sites encoded in the subtype C-specific fragment. We implemented this method to screen 256 HIV-1-infected individuals from 35 towns and cities in four states in the south and a city in the east. With the exception of single samples of subtypes A and B and a B/C recombinant, we found all to be infected with subtype C viruses, and the subtype assignments were confirmed in a subset by using heteroduplex mobility assays and phylogenetic analysis of sequences. We propose the use of C-PCR to facilitate rapid molecular epidemiologic characterization to aid vaccine and therapeutic strategies.
The development of a vaccine against human immunodeficiency virus type 1 (HIV-1) has been affected by the ability of HIV to acquire high levels of genetic heterogeneity, which is also expected to pose problems in expanding therapeutic intervention to resource-poor countries. HIV-1 is divided into distinct groups and subtypes or clades based on genetic differences (22) that are unevenly distributed in different parts of the world. Group M HIV-1, which is most prevalent, has been divided into at least 10 different subtypes and a number of intersubtype circulating recombinant forms. Among the various subtypes, subtype C is predominant and is associated with >50% of all HIV-1 infections (10, 12, 38) and with the establishment and rapid growth of epidemics in India, China, Brazil, and the southern and eastern parts of Africa. Although the preponderance of HIV infections in India appear to be associated with subtype C viruses, several studies have reported the prevalences of subtypes A, B, D, AE, and Thai B (2, 13, 19, 53), as well as the recombinant forms (27), at rates varying from 0 to 100% (5, 8, 15, 28, 45, 49). It is not clear if the extensive proliferation of subtype C viruses might be attributed to demographic or genetic factors unique to these populations or to the presence of intrinsic viral biologic properties that offer potential advantages (11, 40, 41). For example, subtypes C and A have been documented since early stages of the epidemic in India, but the reason why subtype C viruses have come to predominate is not clear.
Most studies in India have involved a small number of samples and/or were restricted to city or local levels and hence do not offer reliable estimates of subtype prevalence. Furthermore, these studies have also left out the newly emerging high-prevalence areas. For example, characterization of HIV subtypes from the southern states has not been reported, even though a high population prevalence of HIV infection has been predicted on the basis of >1% prevalence among antenatal clinic subjects and a high prevalence among high-risk groups (http://www.naco.nic.in). Recent reports of the potential spread of subtype B infections (15) illustrate a need to distinguish whether this is an artifact of the examination of an isolated cluster or the nucleus of a change in the epidemic.
Among the strategies used for subtyping viruses, sequencing viral genomes followed by phylogenetic analyses is the de facto standard but is an expensive and labor-intensive option. Heteroduplex mobility assays (HMA) (6) are the most often used cost-effective alternative and have excellent concordance with phylogeny-based subtyping (1). In spite of HMA being a technically simpler technique, processing a large number of samples requires running parallel electrophoresis gels with multiple subtypes for each sample and is labor-intensive. Therefore, approaches and methods that allow rapid answers to straightforward questions, such as scoring a given sample for subtype assignment, will facilitate processing large numbers of samples and provide greater coverage of the infected population. Subtype sequence-specific PCR is one such method that targets sequences differentially conserved among subtypes. Analogous to sequence-specific (or allele-specific) PCR that has been used extensively in genomic studies (9, 34, 35), this method exploits genetic differences at the level of primer sequences to differentially amplify fragments specific to a given subtype and has been used with other subtypes of HIV-1 (4, 39). Here, we document a PCR strategy (C-PCR) that exploits differences in the long terminal repeat (LTR) region between subtype C and non-subtype C sequences to generate common HIV-1 and subtype C-specific fragments with mobilities distinguishable in an agarose gel. We have optimized the method and implemented it to screen 256 HIV-infected individuals from 35 cities and towns in five states in the southern and eastern parts of India. We found a preponderance of these individuals to be infected with subtype C viruses.
MATERIALS AND METHODS
Clinical samples and DNA extraction.
Blood samples from individuals who were identified as HIV seropositive by multiple enzyme-linked immunosorbent assays and/or Western blots following high-risk exposure and/or with symptoms characteristic of HIV infection were collected in EDTA vacutainers (Becton Dickinson, San Diego, Calif.). The study subjects were voluntary participants under the care of government hospitals, private clinics, and referral centers dedicated to the service of HIV-AIDS in the southern Indian states of Karnataka, Tamil Nadu, Andhra Pradesh, and Kerala. Subjects from the eastern state of West Bengal were part of a high-risk HIV study group at the School of Tropical Medicine in Kolkata, India (14). Blood samples were collected from the patients after informed consent was obtained and under the approval of institutional biosafety and ethics committees at participating institutions. Out of the 256 seropositive samples collected for this study, clinical information was available for 250. Twenty subjects were 15 years old or younger, and 82 were females. The mean ages for the males and the females were 35.2 (range, 17 to 60) and 28.6 (range, 16 to 51) years, respectively. The subjects represented a heterogeneous community of social and demographic groups. The blood samples were transported at room temperature to the Virology Laboratory at the Jawaharlal Nehru Centre for Advanced Scientific Research for processing within 4 days of collection. Genomic DNA was extracted from 0.2 ml of whole blood by using a commercial DNA extraction kit (NucleoSpin Blood; Macherey-Nagel Gmbh & Co. KG, Düren, Germany), eluted in a 100-μl volume, and stored at 4°C until it was used. Reference plasmids containing DNAs from various subtypes were obtained from the National Institutes of Health (NIH) AIDS Repository Program. Carryover contamination of PCR products was prevented by adherence to strict procedural and physical safeguards that included reagent preparation and PCR setup, amplification, and post-PCR processing of samples in separate rooms (24).
Primer design.
We targeted a genomic region extending from the LTR into gag, containing sequences highly conserved among multiple subtypes, as well as stretches of sequences differentially conserved between subtype C and other subtypes. We designed three sets of primers (Table 1). One set of outer primers was designed to amplify a 975-bp LTR-gag fragment from multiple subtypes. A second set of internal primers was designed to amplify a 138-bp LTR fragment specific to subtype C sequences while being refractory to amplification from non-subtype C sequences. A third set of highly conserved internal primers was designed to amplify a 232-bp LTR leader-gag region fragment from all subtypes (Fig. 1).
TABLE 1.
Sequences for primers used in C-PCR
| Nature of PCR | Primer name | HXB2 genome coordinates | Sequencea |
|---|---|---|---|
| First round | N419F | 130-157 | 5′-GAT GGT GCT TCA AGC TAG TRC CAG TTG A |
| N424R | 1075-1104 | 5′-CTC TAT YTT RTC TAA RGC TTC YTT GGT GTC | |
| Common-HIV | N420F | 683-709 | 5′-CTC TCG ACG CAG GAC TCG GCT TGC TGA |
| N423R | 890-915 | 5′-TTC YAG CTC CCT GCT TGC CCA TAC TA | |
| Subtype C specific | N415F | 256-284 | 5′-AGT GGA AGT TTG ACA GTC AMC TAG CAC RC |
| N417R | 367-393 | 5′-CGC CCA GAC CAC WCC TCC TGR AMC GC |
International Union of Pure and Applied Chemistry codes were used for positions with redundant nucleotides. The sequences for all the reverse primers are presented as the inverse of antisense.
FIG.1.

C-PCR strategy, genomic position, and variability in sequences corresponding to subtype C-specific primers. (Top) LTR/gag region corresponding to primers used in this study and their relative positions (not drawn to scale). The upstream subtype C-specific primer, N415F, was anchored on the upstream stimulatory factor site in the LTR, while the downstream primer was anchored on the NF-κB site. (Bottom) Levels of variability in sequences corresponding to these primers among various subtypes. For each subtype, using the number of sequences indicated, the proportion of different nucleotides at each position was calculated and plotted as a percentage. Subtype C sequences from India (CIN) are shown separately. The numbers below the subtypes indicate the average uncorrected nucleotide distances between the primer and the corresponding subtype. The asterisks indicate the four positions that accounted for much of the variability within subtype C sequences. International Union of Pure and Applied Chemistry codes were used for positions with redundant nucleotides.
Subtype C-specific and common HIV primers were designed after a thorough review of all the sequences available in the database. Variability across the genome was evaluated, and regions containing conserved as well as subtype-specific differences were explored in the design of common HIV and subtype C-specific primers. Figure 1 illustrates the variability over the region of the subtype C-specific primers N415F and N417R. For this analysis, sequences for the corresponding regions were collected from the database for the full subtype B genome (n = 103), as well as all the available sequences for subtypes A, AE, C, D, and G. The sequences were aligned using Clustal X (52) and were manually edited. The frequencies of individual nucleotides along the primer sequence were determined for each subtype and plotted as percentages. Uncorrected average nucleotide distances between primers and sequences corresponding to each of the subtypes were computed using MEGA (23). N415F was divergent by averages of 4.3% from Indian subtype C sequences and 9.5% from the rest of the subtype C sequences. Variations at four positions accounted for many of these differences, and a few sequences in the database appeared to be recombinants with other subtypes in the region. Nevertheless, these differences did not appear to influence the sensitivity or specificity of amplification. Primer N417R was more conserved within subtype C, being <2% divergent. Compared to other subtypes, N415F was ∼20% or more divergent, while N417R was divergent in excess of 14%.
As is evident in Fig. 1, N415F and N417R were specifically designed to contain mismatches at the 3′ end to support specific amplification of subtype C sequences while being refractory to amplification from non-subtype C sequences. N415F was anchored on the 3-base sequence in the upstream stimulatory factor motif within the LTR-U3 region, while N417R was anchored on the NF-κB motif that is proximal to the Sp1 sites. As expected from the high levels of heterogeneity observed in HIV-1, we could not find sequences that encoded the exact complement of subtype C-specific primers in the 3′ end. However, this does not appear to be a hurdle, since none of the subtype c samples tested thus far have failed to amplify and the few that did not amplify were found to be non-subtype C. This is the first subtype-specific PCR report involving a comprehensive characterization of region-encoding primer sequences. The lack of similar characterizations may account for the less than widespread use of previously published primers.
PCR and cloning.
PCR amplifications were performed using 500 ng of genomic DNA in a 25-μl volume containing 3 mM MgCl2, 200 μM (each) deoxynucleoside triphosphates, 25 pmol of each primer, and 0.625 U of Taq DNA polymerase (Amersham, Piscataway, N.J.). The first-round PCR conditions were as follows: 94°C for 1 min, 60°C for 1 min, and 72°C for 1 min for a total of 25 cycles on a thermocycler (Minicycler; M.J. Research). Two microliters of first-round PCR products was transferred to a second-round PCR that contained the four internal primers. The PCR conditions were as follows: 94°C for 1 min, 65°C for 40 s, and 72°C for 40 s for 35 cycles. After the second round, the PCR products were resolved on a 1.5% agarose gel or a 3% NuSieve agarose gel (FMC Bioproducts, Rockland, Maine), and the ethidium bromide-stained DNA bands were captured using a gel documentation system (Alpha Innotech, San Leandro, Calif.). The quality control measures included the negative controls containing template DNAs from uninfected individuals and blind analyses of samples from different known subtypes.
Annealing temperatures and Mg concentrations were optimized for sensitivity and specificity, individually and in multiplex PCR amplifications using DNA templates from two unrelated molecular clones of subtype C (pINDIE [30] and pMJ4 [33]) and one of subtype B (pYU-2 [25]). Defined concentrations of plasmid DNA were serially diluted in the presence of 500 ng of salmon sperm DNA to derive precise template copy numbers ranging from <1 to 10,000 copies in each reaction. We found the subtype C-specific and common HIV primers to consistently amplify 1 to 10 copies of the template DNA in uniplex PCR experiments (Fig. 2). Conditions optimized in uniplex PCR amplifications were found to work in multiplex PCR with no decrease in sensitivity and/or specificity. These findings suggest consistent and reliable detection of small numbers of copies of HIV using the primer pairs and amplification conditions outlined here.
FIG. 2.

Specificity and sensitivity of C-PCR. (A) The Specificity of C-PCR was assessed using molecular clones derived from different subtypes of HIV-1 in the following order (from left): A (p92UG037.1), A (p90CF402.1.8), B (pBH10), B (pYU2), C2 (pMJ4), C3 (pINDIE), C4 (p92BR025.8), D (p94UG114.1), D (p94UG114.1.6), D (p84ZR085.1), E (p90CF402.1), F (p93BR020.1), G (p92NG003.1), G (p92NG083.2), and H (p90CF056.1). Lane M, DNA size standard; −, control lane. The extra NF-κB site present in the subtype C4 molecular clone can be seen to lead to increased size of the subtype C-specific fragment. (B) Sensitivity of C-PCR. The subtype C molecular clone pINDIE was used at the copy numbers shown above the lanes. Optimization of uniplex PCR conditions and their subsequent use in multiplex PCR with no loss of sensitivity are seen. −, control lane.
The env C2-V5 region was amplified, cloned, and sequenced from a subset of 45 samples using a nested-PCR amplification strategy as described previously (48). In addition, sequences spanning env V3 sequences were obtained from eight additional subjects and used for subtyping but were not included in the phylograms due to their dissimilar lengths and ambiguous base calls. The LTR region for sequencing studies was amplified using a nested-PCR amplification with primers N299F (1-TGGAWGGGYTAATTTACTCCMARAAA) and N306R (763-CTCTCTCCTTCTAGCCTCCGCTAGTCA) to amplify a 789-bp fragment in the first round and primers N300F (106-GGGTCAGATATCCACTGACCTTTGGAT) and N304R (722-CAGTCGCCGCCCCTCGCCTCTT) in the second nested round to amplify a 638-bp fragment. The numbers in the primer sequences represent positions corresponding to HXB2 (accession no. K03455). The PCR conditions used for LTR amplification were the same as those for the first round of C-PCR except that the annealing temperature in the first-round PCR for LTR was 50°C. The PCR products were purified using a Spinprep PCR clean-up kit (Novagen, Madison, Wis.) and directly sequenced using internal primers. env PCR fragments were cloned into a TA cloning vector (Invitrogen, Carlsbad, Calif.) and sequenced using dye terminator chemistry. Chromatograms from ABI377 and Beckman CEQ8000 sequencers were edited using Sequencher (GeneCodes, Ann Arbor, Mich.).
HMA and sequence analysis.
HMA was performed according to methods outlined by Delwart et al. (6), using the HMA-subtyping kit supplied by the NIH AIDS Reference and Reagent Program. An ∼700-bp fragment of the env gene, spanning the C2 to V5 domains, was amplified using the primers ES7 and ES8. Heteroduplexes were formed by mixing equal amounts of amplified DNA (∼500 ng of each) in a volume of 35 μl and boiling the mixture in the presence of 100 mM NaCl and 2 mM EDTA for 5 min, followed by a quick chill and incubation on ice for 90 min. The heteroduplexes were separated on 5% polyacrylamide gels, stained with ethidium bromide, and scored for subtype assignment. Forty-eight of the 256 clinical samples were tested for concordance among HMA, C-PCR, and phylogenetic analysis.
Sequences corresponding to the LTR and env C2V5 regions used for phylogenetic reconstructions were collected from multiple subtypes from the Los Alamos database (22), aligned at the codon level, and gap stripped. The rates of nucleotide substitution and gamma distribution parameters were estimated using Paup* (51) as described earlier (47). Subtype assignments and depiction of phylogenetic relationships were accomplished using the codeml (with discrete NS site model) and baseml programs for the env and LTR sequences, respectively, in PAML (56).
Nucleotide sequence accession numbers.
The GenBank accession numbers for the sequences in this study are AY567495 to AY567539 and AY567474 to AY567486.
RESULTS
Standardization of C-PCR.
We optimized C-PCR conditions to detect 1 to 10 copies of plasmid templates containing full genomes of single and/or multiple subtypes in individual and multiplex PCR. As illustrated in Fig. 2, the 232-bp Leader/gag common HIV region was amplified from all the subtypes tested thus far, while the 138-bp fragment specific to subtype C was restricted to subtype C (Fig. 2A). All of the C2, C3, and C4 subtype standard sequences supported C-PCR amplification to similar levels. The presence of an additional NF-κB site in clone C4 was reflected in the increased size of the subtype C-specific fragment. In addition to these prototype clones, C-PCR accurately subtyped DNAs extracted from primary cultures of subtype C2, C3, A1, and B2 viruses (provided by the National AIDS Research Institute, Pune, India). Consistent amplification of common HIV and subtype C-specific fragments using template copies ranging from 10,000 to <1 in uniplex and multiplex PCRs is illustrated in Fig. 2B. The overall subtype specificity and the ability of each subtype C variant to support PCR amplification suggested that these primers are highly specific to subtype C viruses and that they amplify fragments of predicted sizes from different representative subtype C viruses.
Subtyping clinical samples using C-PCR.
Subtype-specific C-PCR was implemented to screen DNAs isolated from 256 samples obtained from 35 towns or cities spread over four states in the south and one city in the east of India. These samples were derived from individuals previously diagnosed as having been HIV infected using serologic assays. Among the five states, Karnataka, Andhra Pradesh, and Tamil Nadu have been considered high-prevalence states based on >1% prevalence among women attending antenatal clinics and a high prevalence among sexually transmitted disease patients. Kerala is considered a low-prevalence state based on low prevalence among sexually transmitted disease patients and <0.1% prevalence among antenatal-clinic patients. Subtype C viruses have been identified as the major subtype circulating in West Bengal State in earlier studies (28, 29, 47), and these samples were included to allow comparison with previous findings.
Each of the clinical samples tested in this study generated fragments of the predicted sizes suggestive of the presence of three or four NF-κB sites (Fig. 3A). The heteroduplex mobility patterns of selected representative samples are presented below the corresponding C-PCR lanes. With the exception of one sample from Bangalore, all of the samples identified as subtype C in C-PCR were also determined to be subtype C by HMA (Fig. 3B). All 177 samples from Karnataka screened by C-PCR and 32 env sequences obtained showed the presence of subtype C. One sample was discordant between C-PCR and HMA analyses, suggesting potential infection by a recombinant virus (Fig. 3D). Analysis of nucleotide sequences over the LTR and env regions of this sample confirmed the presence of a B/C recombinant. This is the first report identifying a subtype B/C recombinant virus in India. Each of the 13 samples from Kerala and 11 from Tamil Nadu State was also identified as subtype C by C-PCR. Subtype designation of seven of these samples by using HMA found all of them to be concordant with C-PCR results. Of the 15 samples from Andhra Pradesh that were analyzed, 14 were found to be subtype C by C-PCR and one sample failed to amplify a subtype C-specific fragment and was subsequently identified as subtype B by HMA (Fig. 3C). Similarly, one sample out of the 40 from West Bengal State failed to amplify a subtype C-specific fragment and was identified as subtype A by analysis of the env sequence (results not shown). Overall, C-PCR screening of 256 samples from different states indicated an absence of subtype C sequences in only two samples, both of which were validated using HMA and sequencing.
FIG. 3.

Subtyping clinical samples with C-PCR, HMA, and sequencing. (A) Illustration of typical gel profiles of subtype C sequences obtained using C-PCR. Each lane represents a different clinical sample. −, control reaction mixture that contained DNA from an individual not infected with HIV-1. (B) HMA analysis of a subset of samples identified as subtype C by C-PCR. These representative samples demonstrate the typical HMA profiles of subtype C viruses. The labeling above the lanes indicates the subtype standards used. Although additional subtype standards were used in the analysis, only profiles with subtype A and C standards are shown. −, control lanes that did not include subtype standards. (C) A non-C virus identified as subtype B by HMA. C-PCR failed to amplify the LTR fragment from this sample. (D) A single recombinant virus identified in this study was subtype C in the LTR and subtype B in env. (E) Insertion of additional sequences resembling NF-κB motif is common in subtype C strains and results in bands with lower mobility than expected (Fig. 1A). The insertion of a 15-bp sequence between two authentic NF-κB motifs of one such clinical sample has been confirmed by sequencing the LTR. An NF-κB-like motif is highlighted.
The LTR fragments of a number of samples migrated more slowly than expected, suggesting the insertion of additional sequences. DNA sequencing of the LTR region identified the insertion of NF-κB-like sequences in these cases (Fig. 3E). Similar to that observed for a subtype C4 clone (Fig. 2A) and clinical samples (Fig. 3A), of the 254 subtype C-specific fragments, 35 contained larger fragments, suggesting the presence of four NF-κB sites. These 35 samples were distributed over 17 towns among the states studied here (Table 2).
TABLE 2.
Subtype assignments of HIV-1 using three methods on samples obtained from Indian states
| State | Town/city | Distance from state's urban center (km) | Populataion (103) | No. subtype C (non-subtype C or recombinant) by:
|
|||
|---|---|---|---|---|---|---|---|
| C-PCR
|
HMA | Phylogenetic | |||||
| Subtype score | Extra NF-κB | ||||||
| Karnataka | Total | 177 (0) | 24 | 34 (1a) | 32 (1a) | ||
| Bangalore | 0 | 4,461 | 78 | 10 | 16 (1a) | 16 (1a) | |
| Bellary | 301 | 329 | 2 | 0 | 2 | 2 | |
| Bhadravati | 227 | 167 | 4 | 0 | 1 | 0 | |
| Chikamagalore | 251 | 105 | 4 | 2 | 1 | 1 | |
| Coorg | 263 | 33 | 4 | 1 | 0 | 1 | |
| Davanagere | 264 | 378 | 20 | 2 | 3 | 1 | |
| Gulbarga | 663 | 445 | 11 | 0 | 1 | 2 | |
| Hassan | 187 | 122 | 5 | 1 | 1 | 1 | |
| Haveri | 333 | 58 | 6 | 1 | 1 | 2 | |
| Hospet | 289 | 170 | 4 | 1 | 1 | 0 | |
| Kadur | 240 | 32 | 2 | 0 | 0 | 0 | |
| Kanakapura | 78 | 49 | 3 | 1 | 1 | 1 | |
| Kolar | 70 | 118 | 11 | 3 | 3 | 1 | |
| Mandya | 97 | 136 | 4 | 0 | 1 | 0 | |
| Mysore | 139 | 771 | 12 | 2 | 1 | 3 | |
| Raichur | 508 | 214 | 2 | 0 | 0 | 0 | |
| Tumkur | 70 | 258 | 5 | 0 | 1 | 1 | |
| Andhra Pradesh | Total | 14 (1b) | 4 | 5 (1b) | 13 | ||
| Hyderabad | 0 | 3,586 | 4 | 1 | 2 | 3 | |
| Ananthapur | 306 | 229 | 3 (1b) | 2 | 2 (1b) | 3 | |
| Chittoor | 480 | 159 | 1 | 0 | 0 | 1 | |
| Hindupur | 413 | 130 | 1 | 0 | 0 | 1 | |
| Mallapuram | 80 | 1 | 0 | 0 | 1 | ||
| Rayadurg | 327 | 56 | 1 | 0 | 0 | 1 | |
| R.R Dist | 60 | 2,528 | 2 | 1 | 1 | 2 | |
| Warangal | 179 | 550 | 1 | 0 | 0 | 1 | |
| Tamil Nadu | Total | 11 (0) | 1 | 4 | 6 | ||
| Chennai | 0 | 4,382 | 1 | 0 | 0 | 0 | |
| Dharmapuri | 250 | 67 | 3 | 1 | 2 | 1 | |
| Hosur | 266 | 88 | 2 | 0 | 1 | 1 | |
| Kanniyakumari | 659 | 20 | 3 | 0 | 0 | 3 | |
| Krishnagiri | 262 | 68 | 2 | 0 | 1 | 1 | |
| Kerala | Total | 13 (0) | 2 | 3 | 13 | ||
| Thiruvananthapuram | 0 | 775 | 4 | 0 | 1 | 4 | |
| Kayankulam | 123 | 68 | 1 | 1 | 0 | 1 | |
| Kovalam | 15 | 18 | 8 | 1 | 2 | 6 | |
| Vasak | 50 | 4 | 2 | 0 | 0 | 2 | |
| West Bengal | Total | 39 (1c) | 3 | 0 | 0 | ||
| Kolkata | 0 | 4,580 | 39 (1c) | 3 | 0 | 0 | |
| Overall total | 254 (2) | 34 | 46 (2) | 64 (1a) | |||
C(LTR)/B(env) recombinant.
Subtype B.
Subtype A.
A subset of 48 samples from 22 towns and cities was chosen by randomly selecting samples from each of the states (35, 6, 4, and 3 samples from Karnataka, Andhra Pradesh, Tamil Nadu, and Kerala, respectively) for subtyping by HMA. To confirm the subtype assignment using phylogenetic approaches, additional subsets of 45 and 13 samples were chosen to derive env and LTR sequence data, respectively. As illustrated in Table 2, subtyping by HMA and sequencing was representative because it included a number of different towns. The heteroduplex mobility patterns of representative samples are illustrated below the corresponding subtype C-PCR lanes in Fig. 3B. With the exception of one subtype B/C recombinant shown, env HMA correctly subtyped all samples that were classified as subtype C by C-PCR. Two samples in this study, one from the state of Andhra Pradesh (Fig. 3C) and the other from West Bengal (not shown), failed to amplify the subtype C-specific fragment while yielding correctly sized common HIV-1 fragments. The sample from Andhra Pradesh was found to belong to subtype B by analysis of HMA and LTR sequences, while the other sample from West Bengal was found to be subtype A by HMA and analysis of the env sequence.
We analyzed env C2V5 and LTR sequences from subsets of 45 and 13 samples, respectively (Table 2 and Fig. 4). In phylogenetic reconstructions, env sequences sampled from each of the 45 individuals drawn from the four southern states clustered unambiguously with other subtype C sequences from India and other parts of the world. Similarly, all 13 LTR sequences sampled in this study also clustered with other subtype C sequences. These analyses showed that subtyping viruses based on subtype-specific PCR of the LTR region is consistent with the subtype assignments for the env genes and LTRs in these samples. As illustrated in Fig. 5, the study subjects were from different areas in the southern part of the country. The places where non-subtype C viruses were identified are indicated.
FIG. 4.

Phylograms depicting the relationship between env and LTR sequences obtained in this study (solid circles) and sequences obtained from the HIV database. Single non-subtype C env sequences obtained from the database are identified by the final digits of their accession numbers. Subtype C sequences from eight other countries were included in the analysis and are identified by open triangles along with their two-letter country codes and the accession numbers. The open circles identify subtype C sequences from India available in the database. The phylograms were obtained from gap-stripped alignments and edited. The log likelihood (lnL) scores for the phylograms and the scale bars corresponding to substitutions per codon are indicated.
FIG. 5.
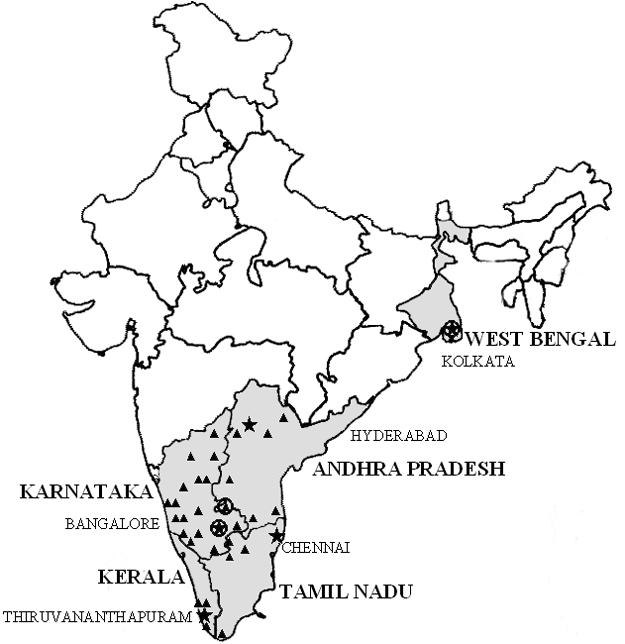
Geographic spread in the assessment of subtype prevalence in India. The four states in the south and the one state in the east of India from which the clinical samples for this study were collected are shaded. The urban centers of the states are indicated by stars, and the towns and cities are indicated by triangles. Subtype C viruses were found in all of these places, and the three places where non-subtype C viruses or recombinants were detected are circled.
DISCUSSION
We have documented the development of a sensitive and specific PCR-based strategy to identify the presence of subtype C viruses. Using this C-PCR, we document a predominance of subtype C viruses in samples derived from different parts of four southern states and from an eastern city in India. We confirmed C-PCR findings, using HMA and sequence analysis in a subset of samples, as well as the consistency of previous reports of subtype prevalence in Kolkata. This is the first report of molecular epidemiologic characterization of viruses from the southern part of India. Additionally, to date this is the largest study of molecular characterization of the HIV-1 subtype distribution in India. While early studies suggested a preponderance of subtype C with a small number of subtype A viruses in India, recent studies have indicated a potentially increasing prevalence of non-subtype C viruses (2, 13, 15, 19, 29, 45, 53). Based on our studies, we conclude that newly emerging areas of epidemic HIV disease in the south continue to be associated with a preponderance of subtype C viruses. However, it is necessary to examine more samples drawn from wider geographic areas to further validate our findings. In addition, we propose designing primers specific to other subtypes in the use of a subtype identification strategy similar to C-PCR to confirm the absence of non-subtype C viruses.
Even though we designed both of our primers to contain subtype-specific mismatches, it is possible to discriminate subtypes by using a single subtype-specific primer. With the exception of subtype A viruses, which show high variability in the 3′ end of N415F (Fig. 1), the region we have targeted appears to be highly suited for designing primers corresponding to each of the other subtypes. However, we focused this study solely on identifying the presence of subtype C sequences because no information on molecular epidemiology is available from the southern parts of India. These regions represent major epicenters of viral infection, and in view of recent reports documenting the presence of non-subtype C viruses, the need to identify subtype profiles in the emerging epidemics in the southern parts of India is of utmost importance and urgency.
In contrast to most earlier studies that have characterized HIV from major urban epicenters, this study was specifically targeted to explore the natures of viruses circulating in rural parts of India that are geographically distant from major epicenters in each state. While demographic and other information from nonurban areas of the country is slowly becoming available, the proper design of vaccine and antiretroviral therapy programs also requires molecular characterization. Even though we have examined a small and disproportionate number of samples, the study design incorporating a wide geographic area makes up for this deficiency. Regardless of the small number of non-subtype C viruses identified, this study indicates a strong association between subtype C viruses and the epidemic in the southern parts of India. Documentation of the presence of intersubtype recombinants in this study and in others makes it necessary to design strategies specifically targeted to identify them. This can be accomplished by implementing a strategy analogous to C-PCR in a more distant region of the genome. Previous studies have documented recombinants using a similar strategy but involving testing for discordance between gag and env subtype assignments by using HMA (3, 16).
The region of the viral genome targeted for the development of C-PCR has a unique significance among subtype C viruses, which was the basis for our choice. Primer N417R was specifically designed to exploit the features conserved within and flanking the NF-κB motif. While most subtype C viruses contain three NF-κB sites in their LTRs (17, 20, 21, 32), a number of them have been reported to possess an additional fourth motif that resembles an NF-κB site (18, 31, 46). Although the functional significance of these additional NF-κB sites is not clear, their presence in the subtype C LTR is believed to enhance the transactivation property of the viral promoter (31, 43, 44). The link between variable numbers of NF-κB sites and potential biological significance, and the conservation of sequences in the targeted region, supports a wider use of C-PCR to identify subtype C viruses in other parts of the world. In addition to identifying the presence of subtype C viruses, C-PCR can also help to identify and score the number of NF-κB sites.
With the C-PCR strategy, it is important to note that the enhanced sensitivity of nested PCR is invariably accompanied by higher susceptibility to carryover contamination. To make sure that such contamination did not influence any of the results outlined here, we implemented strict procedural controls that included template preparation, PCR setup, and gel electrophoresis in physically separated laboratories. In addition, each PCR experiment included replicates containing DNAs from individuals who were not infected with HIV. Therefore, it is necessary to take similar uncompromising precautions to prevent artifactual interpretations. The possibility of contamination, however, is not a unique problem for C-PCR but also applies to HMA analysis that is also designed with a nested-PCR format.
C-PCR is highly specific, sensitive, and discriminatory for subtype-C strains. Unlike HMA, C-PCR is amenable to automation, and detection is simpler and economical. C-PCR, therefore, could be of use for large-scale molecular epidemiology studies. Especially in a country like India, where subtype-C viruses cause a large proportion of infections, C-PCR offers the advantage of rapid and less expensive screening at the primary level. Importantly, C-PCR in combination with HMA could detect recombinant viruses, as the two techniques target different regions of the virus (Fig. 3D). C-PCR, by design, is a multiplexing of two individual regions of the subtype-C virus, thus offering the advantage of reducing false-negative results due to genetic diversity. The Leader/gag region is one of the most conserved regions of the virus. C-PCR, therefore, is highly sensitive to detect all infections by HIV-1 regardless of the subtype. In our experience with hundreds of primary clinical samples, amplification of the LTR/Leader/gag region failed on very rare occasions. This is in contrast to frequent failures encountered in the amplification of the env region for HMA. LTR/Leader/gag amplification occurred even in a few cases where we could not amplify the env region for HMA analysis. A large number of samples could not be genotyped using HMA due to ambiguous band patterns (18, 45). In one study, 17 out of 52 samples were untypeable by HMA (28). We also encountered several samples that could not be characterized by HMA; however, C-PCR identified all of these samples without ambiguity.
Identifying subtype prevalence, especially in regions where multiple subtypes circulate, is necessary for a number of reasons. Subtype characterization impacts strongly on our understanding of the epidemic and approaches to tackle it. No clear evidence linking genetic subtypes with differences in disease progression is available, but the link cannot be ruled out. Subtype differences are also expected to impact strongly on the efficacy of a vaccine and are most relevant to resource-poor countries. The presence of differences in the pattern of cytotoxic-T-lymphocyte epitope recognition (36, 37) suggests that the high level of sequence differences among subtypes is an important variable in the design of a vaccine. Pending the availability of a potent vaccine, emphasis is being placed on making antiretroviral therapies available in resource-poor settings (42). However, the impact of subtype differences on the outcome following therapy is not clear, since nearly all the available antiretroviral agents were designed in a subtype B setting. Even though most of these agents appear to be effective in suppressing non-subtype B viruses to similar extents, significant differences in enzyme kinetics and mutations associated with reduced sensitivity have been documented (50). For example, subtype C and A polymerases appear to exhibit reduced affinities to a panel of antiretroviral agents (54) and to amplify the effect of mutations associated with reduced susceptibility (55). In addition, Diallo et al. and Loemba et al. have documented distinct differences between subtypes in the rates at which mutations associated with reduced sensitivity to antiretroviral agents evolve and the differences in mutations within subtypes C and B that are associated with reduced sensitivity to antiretroviral agents (7, 26). These studies suggest that identifying subtypes at individual and population levels is an important step in monitoring and managing the epidemic.
In summary, we have optimized the conditions for specific amplification of subtype C sequences using C-PCR and implemented it in the analysis of 256 samples derived from a wide geographic area in the southern part of India. With the exception of single samples containing subtypes A and B and a B/C recombinant, the rest contained subtype C viruses. This is the first report of subtype prevalence in the emerging areas of high prevalence in the southern part of India. This study addresses the need for identifying the proportion of infections caused by different subtypes in the design and implementation of vaccine and antiretroviral therapy programs.
Acknowledgments
U.R. acknowledges institutional financial support from JNCASR and the AIDS International Training and Research Program (NIH D43-TW01403) of the Albert Einstein College of Medicine. N.B.S. and P.K.D. are recipients of CSIR fellowships from the Government of India. R.S. was supported by NIH grant AI 41870 and by the Center for Genomic Sciences, Allegheny Singer Research Institute.
The HMA kit and other reagents were received from the NIH AIDS Research and Reference Reagent Program and the Centralised Facility for AIDS Reagents, National Institute for Biological Standards and Control, UNAIDS. Help from the following individuals in collecting samples from HIV-seropositive individuals is gratefully acknowledged: A. O. Saroja, Consultant Neurologist, KLES Hospital, Belgaum, Karnataka, India; M. N. Balamurugan, Consultant Neurologist, Salem, Tamil Nadu, India; E. Srikanth Reddy, Consultant Neurologist, Vijayawada, Andhra Pradesh, India; James Joseph, Seva Free Clinic, Bangalore, India; and Phalguni Gupta, Department of Infectious Diseases and Microbiology, University of Pittsburgh, Pittsburgh, Pa. R.S. acknowledges G. Ehrlich and C. Post for support and encouragement. The Human Brain Tissue Repository for Neurobiological Studies at NIMHANS, Bangalore, India, is acknowledged for providing samples from the Body Fluid Bank.
REFERENCES
- 1.Bongertz, V., D. C. Bou-Habib, L. F. Brigido, M. Caseiro, P. J. Chequer, J. C. Couto-Fernandez, P. C. Ferreira, B. Galvao-Castro, D. Greco, M. L. Guimaraes, M. I. Linhares de Carvalho, M. G. Morgado, C. A. Oliveira, S. Osmanov, C. A. Ramos, M. Rossini, E. Sabino, A. Tanuri, M. Ueda, et al. 2000. HIV-1 diversity in Brazil: genetic, biologic, and immunologic characterization of HIV-1 strains in three potential HIV vaccine evaluation sites. J. Acquir. Immun. Defic. Syndr. 23:184-193. [DOI] [PubMed] [Google Scholar]
- 2.Cassol, S., B. G. Weniger, P. G. Babu, M. O. Salminen, X. Zheng, M. T. Htoon, A. Delaney, M. O'Shaughnessy, and C. Y. Ou. 1996. Detection of HIV type 1 env subtypes A, B, C, and E in Asia using dried blood spots: a new surveillance tool for molecular epidemiology. AIDS Res. Hum. Retrovir. 12:1435-1441. [DOI] [PubMed] [Google Scholar]
- 3.Cham, F., L. Heyndrickx, W. Janssens, K. Vereecken, K. De Houwer, S. Coppens, G. Van der Auwera, H. Whittle, and G. van Der Groen. 2000. Development of a one-tube multiplex reverse transcriptase-polymerase chain reaction assay for the simultaneous amplification of HIV type 1 group M gag and env heteroduplex mobility assay fragments. AIDS Res. Hum. Retrovir. 16:1503-1505. [DOI] [PubMed] [Google Scholar]
- 4.Chen, M. Y., W. K. Wang, M. C. Lee, S. J. Twu, S. I. Wu, and C. N. Lee. 2002. Rapid detection of human immunodeficiency virus type 1 subtype e infection by PCR. J. Clin. Microbiol. 40:3805-3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choudhury, S., M. A. Montano, C. Womack, J. T. Blackard, J. K. Maniar, D. G. Saple, S. Tripathy, S. Sahni, S. Shah, G. P. Babu, and M. Essex. 2000. Increased promoter diversity reveals a complex phylogeny of human immunodeficiency virus type 1 subtype C in India. J. Hum. Virol. 3:35-43. [PubMed] [Google Scholar]
- 6.Delwart, E. L., E. G. Shpaer, J. Louwagie, F. E. McCutchan, M. Grez, H. Rubsamen-Waigmann, and J. I. Mullins. 1993. Genetic relationships determined by a DNA heteroduplex mobility assay: analysis of HIV-1 env genes. Science 262:1257-1261. [DOI] [PubMed] [Google Scholar]
- 7.Diallo, K., B. Brenner, M. Oliveira, D. Moisi, M. Detorio, M. Gotte, and M. A. Wainberg. 2003. The M184V substitution in human immunodeficiency virus type 1 reverse transcriptase delays the development of resistance to amprenavir and efavirenz in subtype B and C clinical isolates. Antimicrob. Agents Chemother. 47:2376-2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dietrich, U., M. Grez, H. von Briesen, B. Panhans, M. Geissendorfer, H. Kuhnel, J. Maniar, G. Mahambre, W. B. Becker, and M. L. Becker. 1993. HIV-1 strains from India are highly divergent from prototypic African and US/European strains, but are linked to a South African isolate. AIDS 7:23-27. [DOI] [PubMed] [Google Scholar]
- 9.Donohoe, G. G., M. Laaksonen, K. Pulkki, T. Ronnemaa, and V. Kairisto. 2000. Rapid single-tube screening of the C282Y hemochromatosis mutation by real-time multiplex allele-specific PCR without fluorescent probes. Clin. Chem. 46:1540-1547. [PubMed] [Google Scholar]
- 10.Esparza, J., and N. Bhamarapravati. 2000. Accelerating the development and future availability of HIV-1 vaccines: why, when, where, and how? Lancet 355:2061-2066. [DOI] [PubMed] [Google Scholar]
- 11.Essex, M. 1998. State of the HIV pandemic. J. Hum. Virol. 1:427-429. [PubMed] [Google Scholar]
- 12.Essex, M. 1999. Human immunodeficiency viruses in the developing world. Adv. Virus Res. 53:71-88. [DOI] [PubMed] [Google Scholar]
- 13.Gadkari, D. A., D. Moore, H. W. Sheppard, S. S. Kulkarni, S. M. Mehendale, and R. C. Bollinger. 1998. Transmission of genetically diverse strains of HIV-1 in Pune, India. Indian J. Med. Res. 107:1-9. [PubMed] [Google Scholar]
- 14.Gupta, P., L. Kingsley, H. W. Sheppard, L. H. Harrison, R. Chatterjee, A. Ghosh, P. Roy, and D. K. Neogi. 2003. High incidence and prevalence of HIV-1 infection in high risk population in Calcutta, India. Int. J. STD AIDS 14:463-468. [DOI] [PubMed] [Google Scholar]
- 15.Halani, N., B. Wang, Y. C. Ge, H. Gharpure, S. Hira, and N. K. Saksena. 2001. Changing epidemiology of HIV type 1 infections in India: evidence of subtype B introduction in Bombay from a common source. AIDS Res. Hum. Retrovir. 17:637-642. [DOI] [PubMed] [Google Scholar]
- 16.Heyndrickx, L., W. Janssens, L. Zekeng, R. Musonda, S. Anagonou, G. Van Der Auwera, S. Coppens, K. Vereecken, K. De Witte, R. Van Rampelbergh, M. Kahindo, L. Morison, F. E. McCutchan, J. K. Carr, J. Albert, M. Essex, J. Goudsmit, B. Asjo, M. Salminen, and G. van Der. 2000. Simplified strategy for detection of recombinant human immunodeficiency virus type 1 group M isolates by gag/env heteroduplex mobility assay. J. Virol. 74:363-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hunt, G., and C. T. Tiemessen. 2000. Occurrence of additional NF-κB-binding motifs in the long terminal repeat region of South African HIV type 1 subtype C isolates. AIDS Res. Hum. Retrovir. 16:305-306. [DOI] [PubMed] [Google Scholar]
- 18.Hunt, G. M., D. Johnson, and C. T. Tiemesse. 2001. Characterisation of the long terminal repeat regions of South African human immunodeficiency virus type 1 isolates. Virus Genes 23:27-34. [DOI] [PubMed] [Google Scholar]
- 19.Jameel, S., M. Zafrulla, M. Ahmad, G. P. Kapoor, and S. Sehgal. 1995. A genetic analysis of HIV-1 from Punjab, India reveals the presence of multiple variants. AIDS 9:685-690. [DOI] [PubMed] [Google Scholar]
- 20.Jeeninga, R. E., M. Hoogenkamp, M. Armand-Ugon, M. de Baar, K. Verhoef, and B. Berkhout. 2000. Functional differences between the long terminal repeat transcriptional promoters of human immunodeficiency virus type 1 subtypes A through G. J. Virol. 74:3740-3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johansson, B., K. Sherefa, and A. Sonnerborg. 1995. Multiple enhancer motifs in HIV type 1 strains from Ethiopia. AIDS Res. Hum. Retrovir. 11:761-764. [DOI] [PubMed] [Google Scholar]
- 22.Kuiken, C. L., B. Foley, B. Hahn, B. Korber, F. McCutchan, P. A. Marx, J. W. Mellors, J. I. Mullins, J. Sodroski, and S. Wolinsky. 2002. Human retroviruses and AIDS: a compilation and analysis of nucleic acid and amino acid sequences. Los Alamos National Laboratory, Los Alamos, N. Mex.
- 23.Kumar, S., K. Tamura, I. B. Jakobsen, and M. Nei. 2001. MEGA2: molecular evolutionary genetics analysis software. Bioinformatics 17:1244-1245. [DOI] [PubMed] [Google Scholar]
- 24.Kwok, S., and R. Higuchi. 1989. Avoiding false positives with PCR. Nature 339:237-238. [DOI] [PubMed] [Google Scholar]
- 25.Li, Y., J. C. Kappes, J. A. Conway, R. W. Price, G. M. Shaw, and B. H. Hahn. 1991. Molecular characterization of human immunodeficiency virus type 1 cloned directly from uncultured human brain tissue: identification of replication-competent and -defective viral genomes. J. Virol. 65:3973-3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loemba, H., B. Brenner, M. A. Parniak, S. Ma'ayan, B. Spira, D. Moisi, M. Oliveira, M. Detorio, and M. A. Wainberg. 2002. Genetic divergence of human immunodeficiency virus type 1 Ethiopian clade C reverse transcriptase (RT) and rapid development of resistance against nonnucleoside inhibitors of RT. Antimicrob. Agents Chemother. 46:2087-2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lole, K. S., R. C. Bollinger, R. S. Paranjape, D. Gadkari, S. S. Kulkarni, N. G. Novak, R. Ingersoll, H. W. Sheppard, and S. C. Ray. 1999. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73:152-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mandal, D., S. Jana, S. K. Bhattacharya, and S. Chakrabarti. 2002. HIV type 1 subtypes circulating in eastern and northeastern regions of India. AIDS Res. Hum. Retrovir. 18:1219-1227. [DOI] [PubMed] [Google Scholar]
- 29.Mandal, D., S. Jana, S. Panda, S. Bhattacharya, T. C. Ghosh, S. K. Bhattacharya, and S. Chakrabarti. 2000. Distribution of HIV-1 subtypes in female sex workers of Calcutta, India. Indian J. Med. Res. 112:165-172. [PubMed] [Google Scholar]
- 30.Mochizuki, N., N. Otsuka, K. Matsuo, T. Shiino, A. Kojima, T. Kurata, K. Sakai, N. Yamamoto, S. Isomura, T. N. Dhole, Y. Takebe, M. Matsuda, and M. Tatsumi. 1999. An infectious DNA clone of HIV type 1 subtype C. AIDS Res. Hum. Retrovir. 15:1321-1324. [DOI] [PubMed] [Google Scholar]
- 31.Montano, M. A., V. A. Novitsky, J. T. Blackard, N. L. Cho, D. A. Katzenstein, and M. Essex. 1997. Divergent transcriptional regulation among expanding human immunodeficiency virus type 1 subtypes. J. Virol. 71:8657-8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naghavi, M. H., M. O. Salminen, A. Sonnerborg, and A. Vahlne. 1999. DNA sequence of the long terminal repeat of human immunodeficiency virus type 1 subtype A through G. AIDS Res. Hum. Retrovir. 15:485-488. [DOI] [PubMed] [Google Scholar]
- 33.Ndung'u, T., B. Renjifo, and M. Essex. 2001. Construction and analysis of an infectious human immunodeficiency virus type 1 subtype C molecular clone. J. Virol. 75:4964-4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newton, C. R., A. Graham, L. E. Heptinstall, S. J. Powell, C. Summers, N. Kalsheker, J. C. Smith, and A. F. Markham. 1989. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res. 17:2503-2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nollau, P., C. Wagener, et al. 1997. Methods for detection of point mutations: performance and quality assessment. Clin. Chem. 43:1114-1128. [PubMed] [Google Scholar]
- 36.Novitsky, V., H. Cao, N. Rybak, P. Gilbert, M. F. McLane, S. Gaolekwe, T. Peter, I. Thior, T. Ndung'u, R. Marlink, T. H. Lee, and M. Essex. 2002. Magnitude and frequency of cytotoxic T-lymphocyte responses: identification of immunodominant regions of human immunodeficiency virus type 1 subtype C. J. Virol. 76:10155-10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Novitsky, V., N. Rybak, M. F. McLane, P. Gilbert, P. Chigwedere, I. Klein, S. Gaolekwe, S. Y. Chang, T. Peter, I. Thior, T. Ndung'u, F. Vannberg, B. T. Foley, R. Marlink, T. H. Lee, and M. Essex. 2001. Identification of human immunodeficiency virus type 1 subtype C Gag-, Tat-, Rev-, and Nef-specific ELISPOT-based cytotoxic T-lymphocyte responses for AIDS vaccine design. J. Virol. 75:9210-9228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osmanov, S., C. Pattou, N. Walker, B. Schwardlander, and J. Esparza. 2002. Estimated global distribution and regional spread of HIV-1 genetic subtypes in the year 2000. J. Acquir. Immun. Defic. Syndr. 29:184-190. [DOI] [PubMed] [Google Scholar]
- 39.Peeters, M., F. Liegeois, F. Bibollet-Ruche, D. Patrel, N. Vidal, E. Esu-Williams, S. Mboup, N. E. Mpoudi, B. Koumare, N. Nzila, J. L. Perret, and E. Delaporte. 1998. Subtype-specific polymerase chain reaction for the identification of HIV-1 genetic subtypes circulating in Africa. AIDS 12:671-673. [PubMed] [Google Scholar]
- 40.Peeters, M., and P. M. Sharp. 2000. Genetic diversity of HIV-1: the moving target. AIDS 14:S129-S140. [PubMed] [Google Scholar]
- 41.Ranga, U. 2002. Human immunodeficiency virus-1 subtypes: could genetic diversity translate to differential pathogenesis? J. Indian Inst. Sci. 82:73-91. [Google Scholar]
- 42.Reynolds, S. J., J. G. Bartlett, T. C. Quinn, C. Beyrer, and R. C. Bollinger. 2003. Antiretroviral therapy where resources are limited. N. Engl. J. Med. 348:1806-1809. [DOI] [PubMed] [Google Scholar]
- 43.Rodenburg, C. M., Y. Li, S. A. Trask, Y. Chen, J. Decker, D. L. Robertson, M. L. Kalish, G. M. Shaw, S. Allen, B. H. Hahn, and F. Gao. 2001. Near full-length clones and reference sequences for subtype C isolates of HIV type 1 from three different continents. AIDS Res. Hum. Retrovir. 17:161-168. [DOI] [PubMed] [Google Scholar]
- 44.Roof, P., M. Ricci, P. Genin, M. A. Montano, M. Essex, M. A. Wainberg, A. Gatignol, and J. Hiscott. 2002. Differential regulation of HIV-1 clade-specific B, C, and E long terminal repeats by NF-κB and the Tat transactivator. Virology 296:77-83. [DOI] [PubMed] [Google Scholar]
- 45.Sahni, A. K., V. V. Prasad, and P. Seth. 2002. Genomic diversity of human immunodeficiency virus type-1 in India. Int. J. STD AIDS 13:115-118. [DOI] [PubMed] [Google Scholar]
- 46.Scriba, T. J., F. K. Treurnicht, M. Zeier, S. Engelbrecht, and E. J. van Rensburg. 2001. Characterization and phylogenetic analysis of South African HIV-1 subtype C accessory genes. AIDS Res. Hum. Retrovir. 17:775-781. [DOI] [PubMed] [Google Scholar]
- 47.Shankarappa, R., R. Chatterjee, G. H. Learn, D. Neogi, M. Ding, P. Roy, A. Ghosh, L. Kingsley, L. Harrison, J. I. Mullins, and P. Gupta. 2001. Human immunodeficiency virus type 1 env sequences from Calcutta in eastern India: identification of features that distinguish subtype C sequences in India from other subtype C sequences. J. Virol. 75:10479-10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shankarappa, R., J. B. Margolick, S. J. Gange, A. G. Rodrigo, D. Upchurch, H. Farzadegan, P. Gupta, C. R. Rinaldo, G. H. Learn, X. He, X. L. Huang, and J. I. Mullins. 1999. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol. 73:10489-10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Soto-Ramirez, L. E., S. Tripathy, B. Renjifo, and M. Essex. 1996. HIV-1 pol sequences from India fit distinct subtype pattern. J. Acquir. Immun. Defic. Syndr. Hum. Retrovirol. 13:299-307. [DOI] [PubMed] [Google Scholar]
- 50.Spira, S., M. A. Wainberg, H. Loemba, D. Turner, and B. G. Brenner. 2003. Impact of clade diversity on HIV-1 virulence, antiretroviral drug sensitivity and drug resistance. J. Antimicrob. Chemother. 51:229-240. [DOI] [PubMed] [Google Scholar]
- 51.Swofford, D. L. PAUP* 4.0: phylogenetic analysis using parsimony (* and other methods). 2002. Sinauer Associates, Inc., Sunderland, Mass.
- 52.Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G. Higgins. 1997. The CLUSTAL_X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsuchie, H., J. K. Maniar, N. Yoshihara, M. Imai, T. Kurimura, and T. Kitamura. 1993. Sequence analysis of V3 loop region of HIV-1 strains prevalent in India. Jpn. J. Med. Sci. Biol. 46:95-100. [DOI] [PubMed] [Google Scholar]
- 54.Velazquez-Campoy, A., M. J. Todd, S. Vega, and E. Freire. 2001. Catalytic efficiency and vitality of HIV-1 proteases from African viral subtypes. Proc. Natl. Acad. Sci. USA 98:6062-6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Velazquez-Campoy, A., S. Vega, and E. Freire. 2002. Amplification of the effects of drug resistance mutations by background polymorphisms in HIV-1 protease from African subtypes. Biochemistry 41:8613-8619. [DOI] [PubMed] [Google Scholar]
- 56.Yang, Z. 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13:555-556. [DOI] [PubMed] [Google Scholar]