

ABSTRACT
Osteomyelitis is a difficult-to-eradicate bone infection typically caused by Staphylococcus aureus. In this study, we investigated the in vivo transcriptional adaptation of S. aureus during bone infection. To this end, we determined the transcriptome of S. aureus during the acute (day 7) and chronic (day 28) phases of experimental murine osteomyelitis using RNA sequencing (RNA-Seq). We identified a total of 180 genes significantly more highly expressed by S. aureus during acute or chronic in vivo infection than under in vitro growth conditions. These genes encoded proteins involved in gluconeogenesis, proteolysis of host proteins, iron acquisition, evasion of host immune defenses, and stress responses. At the regulatory level, sarA and -R and saeR and -S as well as the small RNA RsaC were predominantly expressed by S. aureus during in vivo infection. Only nine genes, including the genes encoding the arginine deiminase (ADI) pathway and those involved in the stringent response, were significantly more highly expressed by S. aureus during the chronic than the acute stage of infection. Analysis by quantitative reverse transcription-PCR (qRT-PCR) of a subset of these in vivo-expressed genes in clinical specimens yielded the same results as those observed in the murine system. Collectively, our results show that during acute osteomyelitis, S. aureus induced the transcription of genes that mediate metabolic adaptation, immune evasion, and replication. During the chronic phase, however, S. aureus switched its transcriptional response from a proliferative to a persistence mode, probably driven by the severe deficiency in nutrient supplies. Interfering with the survival strategies of S. aureus during chronic infection could lead to more effective treatments.
IMPORTANCE
The key to the survival success of pathogens during an infection is their capacity to rapidly adjust to the host environment and to evade the host defenses. Understanding how a pathogen redirects and fine-tunes its gene expression in response to the challenges of infection is central to the development of more efficient anti-infective therapies. Osteomyelitis is a debilitating infection of the bone predominantly caused by S. aureus. In this study, we evaluated the transcriptional response of S. aureus during bone infection. Our results indicate that S. aureus reprograms its genetic repertoire during the acute phase of infection to adapt to nutrient availability and to replicate within the host. During the chronic phase, S. aureus upregulates a survival genetic program activated in response to nutrient starvation. Thus, we have uncovered key survival pathways of S. aureus during acute and chronic osteomyelitis that can be used as therapeutic targets.
INTRODUCTION
Staphylococcus aureus is a major human pathogen and an important cause of death and morbidity worldwide. Of major concern are the continuous emergence and spread of antibiotic-resistant strains, such as methicillin-resistant S. aureus (MRSA), which have limited treatment options (1). Although S. aureus is part of the normal human microbiota as a commensal microorganism, it can also be the cause of a wide range of diseases if given the chance to access deeper tissues. S. aureus can establish infections in a range of different organs, including the lungs, kidneys, and heart, as well as in diverse host tissues, such as the skin, bones, and blood (2). The ability of S. aureus to cope with the biological pressure imposed by the immune response as well as by the shortage of nutrients within the different host microenvironments is central to its success as a pathogen. The rapid adaptation of S. aureus to changing environments is accompanied by reprogramming complex regulatory networks to activate the expression of genes essential for its survival in the new environment while repressing those that are unnecessary or potentially deleterious. Dissecting the transcriptional adaptation of S. aureus during in vivo infection is therefore central for understanding how this pathogen interacts with the host and causes disease.
The transcriptional adaptation of S. aureus during in vivo infection is relatively unexplored. Examination of S. aureus transcriptional response in a rabbit subcutaneous cage model (3), in a mouse model of S. aureus pneumonia (4), in human cutaneous abscesses (5), and in murine kidneys (5) using microarray technology has demonstrated that genes regulated in vivo differ greatly from those regulated in vitro. This underscores the need for in vivo model systems that mimic, as closely as possible, the pathophysiology of the human infection for understanding the transcriptional adaptation of S. aureus to the host during productive infection. We recently established a model of metastatic osteomyelitis initiated after intravenous inoculation of S. aureus whose pathological features closely resemble those of acute and chronic human disease (6). Osteomyelitis is an infection of the bone predominantly caused by S. aureus that can be associated with high levels of inflammation and bone tissue destruction (7). Despite long and high-dose treatment, the rate of clinical failure in osteomyelitis treatment is very high, and these infections frequently result in loss of function and extremity amputation (8). Our murine model of staphylococcal osteomyelitis represents a robust platform to investigate how S. aureus coordinates its gene expression during the acute and chronic phases of bone infection. This information could be used to develop novel anti-infective approaches aimed at interfering with S. aureus within-host adaptation, thereby enabling the immune system to eliminate the pathogen in a more natural way.
Most current research regarding staphylococcal pathogenesis has focused on the identification, functional properties, and regulation of individual virulence determinants. However, beyond the induction of genes encoding virulence determinants, the whole-genome expression profile of in vivo-infecting bacteria will comprise other genes that are likely to be essential for in vivo survival. The products encoded by these genes may not be harmful to the host but may help the microbe to subvert the host defenses, and thus they are not specifically ascribed a role as virulence determinants. Instead, these genes are likely to encode functions required for growth in vivo, although they may be dispensable for the bacterial growth under in vitro conditions. In the study presented here, we analyzed the genome-wide transcriptional expression of S. aureus isolated from the bones of experimentally infected mice during the acute and chronic phases of infection by deep RNA sequencing (RNA-Seq) using Illumina technology. The results in the murine model were validated in clinical specimens. The results of our study provide the first high-resolution mapping of the integral adaptive transcriptional response of S. aureus to the host during acute and chronic bone infection.
RESULTS
RNA-Seq analysis of S. aureus transcriptional adaptation during acute and chronic experimental osteomyelitis.
We used a previously described murine model of bone infection to determine the transcriptional profile of S. aureus during acute (AC; 7 days of infection) and chronic (CH; 28 days of infection) experimental osteomyelitis (6). In this model, S. aureus grown in vitro to exponential phase was injected intravenously into C57BL/6 mice and entered the bones through the bloodstream (6). Following an acute phase of progressive growth, the bacterial loads in the bones declined, and a surviving population remained relatively stable during the chronic phase of infection (6). Bacterial RNA was extracted from the bones of 8 to 10 mice at days 7 (AC) and 28 (CH) of infection. Integrated maps of genes and their level of expression in S. aureus during in vitro exponential (EX) growth in complex medium and AC and CH in vivo bone infection were then generated using Illumina deep-sequencing technology. Three independent biological replicates were performed for each set of samples. Sequence reads were subjected to a sequential filtering process and aligned to the reference genome of S. aureus strain 6850 (9). Although each of the samples produced between 53 and 105 million reads, the numbers of final bacterial mRNA reads differed considerably between conditions (see Table S1 in the supplemental material). The initial data matrix comprises count data across 2,503 genes, covering more than 95% of the S. aureus 6850 genome (9). The distribution of raw read counts following the rank ordering of genes from scantily to highly abundant revealed a distribution where most genes had counts of between 100 and 10,000 for the EX and AC conditions (see Fig. S1A in the supplemental material). However, in the case of CH data sets, a high number of genes were scantily covered or completely lacked coverage (see Fig. S1A in the supplemental material). Since the initial numbers of reads before removal of those originating from the host were similar between the different conditions (see Table S1 in the supplemental material), the poor coverage of CH samples could be attributed to the moderately lower bacterial numbers observed during this phase of infection (6) and/or to a much lower physiological activity of S. aureus during this phase of infection. To normalize each replicate within each condition against gene length and differing library sizes, the commonly used RPKM (reads per kilobase per million of mapped reads) method was applied. However, we observed that while the distribution of the RPKM data gave a picture comparable to that of the raw data for EX and AC conditions, the values for the scantily abundant genes of the CH condition were severely overinflated after RPKM normalization (see Fig. S1B in the supplemental material). These overinflated gene expression values can have a discordantly confounding effect on the downstream interpretation of differential gene expression. To overcome this problem, all replicates for each condition were random resampled to equal the smallest library size of 26,251 reads, which produced comparable distributions for further analysis (see Fig. S1C and Table S2 in the supplemental material).
Global gene expression analysis of S. aureus under in vitro growth and in vivo infection.
Clustering and ordination were used to determine the similarity among the biological replicates within each condition as well as the similarities between the transcriptional profiles of S. aureus under the different conditions. The dendrogram depicted in Fig. 1A revealed that all replicates within each condition grouped together, underlining the high reproducibility of the method. Permutational multivariate analysis of variance (PERMANOVA) showed that the clusterings of gene expression data for the three conditions were indeed significantly different from one another (P < 0.05) (see Table S3 in the supplemental material). The ordination plot generated by principal coordinate analysis (PCoA), which orders samples according to their global similarity based on expression data, shows that the global patterns of the transcriptomes were highly different between in vitro (EX) and in vivo (AC and CH) conditions, as indicated by the first principal component (PCO1) axis, which explains 73.9% of the total variation (Fig. 1B). The second principal component (PCO2) axis, which explains the variation between the in vivo transcriptomes (AC versus CH), covered only 12.3% of the total variation (Fig. 1B). This was further confirmed by performing pairwise comparison of AC versus EX (Fig. 1C) and AC versus CH (Fig. 1D) transcriptomes, which indicated that differences in gene expression were more extensive between S. aureus grown in vitro (EX) and in vivo AC (Fig. 1C) than between the in vivo AC and CH conditions (Fig. 1D). A circular ideogram plotting the mean abundance of transcripts at the genomic position of S. aureus strain 6850 is shown in Fig. S2 in the supplemental material.
FIG 1 .

Hierarchical clustering dendrogram (A) and two-dimensional PCoA plot (B) of the transcriptional data from S. aureus under EX, AC, and CH conditions. Bray-Curtis similarity scores were used to compare the different transcriptomes. Each shape represents one condition and each symbol one independent experimental set. (C) Pairwise scatter plots for the S. aureus transcripts during EX growth versus AC infection. Each symbol represents the mean of triplicate values. (D) Pairwise scatter plots for the S. aureus transcripts of AC versus CH infection. Each symbol represents the mean of triplicate values. The Pearson correlation coefficient (r) is indicated in each plot analysis.
Functional categorization of transcripts expressed by S. aureus during in vitro growth and in vivo infection.
Gene expression analysis based on the relative abundances of gene transcripts assigned to their respective clusters of orthologous groups (COG) showed that a large amount of sequence reads could be assigned to the COG class “Metabolism,” followed by “Information, storage and processing” in in vitro EX growth and “Cellular processes and signaling” in AC and CH phases in vivo (Fig. 2A). The pattern and distribution of transcripts among the COG categories are shown in Fig. 2B. While genes assigned to the category “Translation, ribosomal structure and biogenesis” were more highly expressed by S. aureus during in vitro EX growth, a higher number of transcripts encoding proteins classfied in “Post-translational modification, protein turnover, and chaperones” were detected in S. aureus during AC and CH infection. Regarding the categories associated with “Metabolism,” transcripts related to the category “Energy production and conversion” were considerably more abundant in S. aureus during in vitro EX growth, while transcripts related to the cateogry “Amino acid transport and metabolism” and “Inorganic transport and metabolism” were more abundant in S. aureus during AC and CH infection (Fig. 2B).
FIG 2 .

COG category overview of S. aureus gene expression. Mean total percentage abundances of transcripts assigned to major COG classes (A) and COG categories (B) during in vitro EX growth and during in vivo AC and CH infection are shown. The total abundance of transcripts of genes assigned to the genome of S. aureus strain 6850 is included as a reference (gray area in panel B).
Differential gene expression by S. aureus between in vitro and in vivo conditions.
Differential gene expression between pairs of conditions analyzed using a suite of algorithms (DESeq, EdgeR, and SAMseq) revealed that a total of 444 genes were differentially expressed by S. aureus between AC infection and in vitro EX growth; 180 genes were significantly more highly expressed during AC infection, and 264 genes were significantly more highly expressed during EX growth (Table 1). In contrast, only 51 genes were differentially expressed by S. aureus between in vivo CH and AC infection; specifically, 42 genes were expressed to higher levels during AC infection and only 9 genes during CH infection (Table 1). The complete list of the differentially expressed genes is provided in Table S4 in the supplemental material.
TABLE 1 .
Number of genes differentially expressed by S. aureus between in vitro EX growth and in vivo AC infection and between in vivo AC and CH infection
| Conditions testeda | Total no. of differentially expressed genesb |
No. of genes |
|||
|---|---|---|---|---|---|
| DESeq (P < 0.05, adjusted) | EdgeR (P < 0.05, FDR) | SAMseq (q < 0.05) | Differentially expressed | Significantly expressed | |
| AC/EX | 388 (388ER/208SS) | 559 (388DS/264SS) | 280 (208DS/264ER) | 444 | 180/264 |
| CH/AC | 22 (22ER/13SS) | 114 (22DS/42SS) | 52 (13DS/42ER) | 51 | 9/42 |
AC, in vivo acute bone infection (7 days); EX, in vitro growth to exponential phase; CH, in vivo chronic bone infection (28 days).
Gene expression was determined to be significant using the parametric algorithms DESeq (DS) and EdgeR (ER) (P < 0.05) and the nonparametric algorithm SAMSeq (SS) (q < 0.05). P values have been adjusted to take into account for multiple testing. The total numbers of genes identified as being differentially expressed by 2 or more of the methods and the number of genes significantly induced in each of the conditions are given. Results in parentheses are numbers of common significantly differentially expressed genes, with the corresponding algorithms.
(i) Differential expression of genes involved in metabolism.
The capacity of S. aureus to adapt its metabolism to the diverse and fluctuating environments is a prerequisite for survival during in vivo infection. We observed a number of transcripts encoding enzymes involved in the glycolytic pathway (glkA, pgi, and pfkA) expressed to a higher level in S. aureus during in vitro EX growth, suggesting that glucose is the major carbon source in this growth condition (Fig. 3A). The number of transcripts encoding glycolytic enzymes was significantly reduced in S. aureus isolated from infected bones and coincided with a large increase in transcripts encoding enzymes involved in the gluconeogenesis pathway (fbp, fda, gapA, pyc, and pckA) (Fig. 3A). During in vitro EX growth in a microaerobic environment, pyruvate originating from the glycolytic pathway was obviously reduced to lactic acid, as revealed by a high level of l-lactate dehydrogenase-encoding transcripts (ldh1) (Fig. 3A), indicating overflow metabolism as previously observed in aerobically growing S. aureus in an excess of glucose such as in rich medium (10). Interestingly, we found that many genes encoding secreted proteolytic enzymes, including a metalloprotease (aureolysin, encoded by aur), the serine protease V8 (sspA), two cysteine proteases (staphopain A [scpA] and staphopain B [sspB]), and five serine-like proteases (splABCEF), were largely expressed by S. aureus during AC and CH infection (Fig. 3B). It is possible that the primary role of these proteases is to break down host proteins to provide the bacterium with nutrients in the form of small peptides or amino acids.
FIG 3 .

Expression of genes related to glycolysis/gluconeogenesis, proteases, ADI, and stress responses by S. aureus under in vitro and in vivo conditions. Expression of genes encoding proteins involved in glycolysis/gluconeogenesis (A), those with protease activity (B), those involved in the ADI pathway (C), and those involved in the stress response (D) by S. aureus during EX, AC, and CH growth is shown. Each bar represents the mean and standard deviation (SD) of triplicates. n.s, the differences between AC and EX mean values and between CH and AC mean values are not statistically significant.
Under aerobic conditions, a large proportion of the ATP in microorganisms is synthetized by membrane-bound complexes through oxidative phosphorylation. ATP synthesis from ADP and Pi is catalyzed by the ATP synthase complex (FoF1 ATPase) and is driven by the proton gradient generated by respiration. This pathway was used by S. aureus only during in vitro growth, since the transcripts of important components of this complex (atpA, -B, -C, -D, -E, -F, and -G) were much more abundant in S. aureus during in vitro EX growth than during AC and CH infection (see Table S4 in the supplemental material). In the anaerobic environment encountered in the bones, S. aureus needs an alternative pathway to generate energy. In this regard, arginine may serve as an alternative energy source for S. aureus under anaerobic conditions through its metabolism via the arginine deiminase (ADI) pathway, encoded by the arc operon (11). The ADI pathway encompasses three reactions catalyzed by arginine deiminase (arcA, also called adi in S. aureus strain 6850), ornithine carbamoyl transferase (arcB), and carbamate kinase (arcC), resulting in the conversion of arginine into ornithine, ammonia, and CO2, with the concomitant production of ATP. The transcriptional data indicate that this pathway, including an arginine/ornithine antiporter (arcD), and a transcriptional regulator (arcR), was predominantly induced by S. aureus during CH infection (Fig. 3C).
(ii) Differential expression of genes involved in response to stress.
Survival of S. aureus in the host is dependent on evasion of the host immune defense, including mechanisms of microbial killing by phagocytes. These killing mechanisms involve the production of reactive oxygen species (superoxide anions, hydrogen peroxide, and hydroxyl radicals) and nitric oxide. Protection of S. aureus against such oxidative stress is mediated mainly by the PerR regulon, which comprises catalase (katA), alkylhydroperoxide reductase (ahpCF), the general stress protein Dps (dps), and thioredoxin reductase (trxB) (12). These detoxifying proteins, in addition to superoxide dismutase (sodA), were expressed by S. aureus to a much higher level during AC and CH infection than during in vitro EX growth (Fig. 3D).
Proteins that have been modified by reactive oxygen and nitrogen species generated during infection are generally removed by the proteasome system in order to prevent metabolic disturbances (13). However, under severe oxidative conditions, such as those encountered by S. aureus in infected tissue, proteins tend to unfold and aggregate due to the exposure of patches of hydrophobic amino acids, thus becoming less susceptible to proteolytic cleavage (13). Hsp100/Clp ATPases constitute a family of closely related proteins that catalyze the unfolding, disassembly, and disaggregation of oxidized proteins. Clp ATPases are important components of the stress response in S. aureus (14). The transcriptome data revealed that the level of expression of the three genes (clpC, clpL, and clpB) encoding Clp ATPases was significantly higher in S. aureus during AC and CH infection than during in vitro EX growth (Fig. 3D). In Escherichia coli, ClpB cooperates with the DnaK chaperone system to solubilize larger protein aggregates (15). Likewise, we found that dnaK transcripts were significantly more abundant in S. aureus growing in infected tissue (AC and CH) than in culture medium (EX) (Fig. 3D).
When microorganisms face environments with nutrient limitation and amino acid starvation, such as those encountered during in vivo infection, a coordinated program known as the stringent response is induced (16). Amino acid starvation results in the activation of the ribosome-associated enzyme RelA, which is the major synthetase of the small-molecule effectors guanosine tetra- and pentaphosphates [(p)ppGpp] (16). (p)ppGpp levels are also controlled by the spoT gene product, a bifunctional enzyme having both a regulated hydrolase activity for degrading (p)ppGpp and a weak synthetase activity (16). Binding of (p)ppGpp to RNA polymerase modulates the transcription of hundreds of genes and remodels the bacterial physiology (16). We found that the genes encoding RelA/SpoT were significantly expressed only during CH infection (Fig. 3D), further confirming the severe nutrient limitation faced by S. aureus in chronically infected tissue.
(iii) Differential expression of genes required for tissue colonization and subversion of the host defense.
The initial step of S. aureus infection is to attach to the host tissue. For this purpose, S. aureus strains express surface receptors designated MSCRAMMs (microbial surface components recognizing adhesive matrix molecules) that interact with proteins of the extracellular matrix (17). Binding to fibronectin, elastin, and fibrinogen seems to be the major mechanism of S. aureus for infection of bone tissue, since the transcripts of genes for fibronectin-binding protein A (fnbA), elastin-binding protein (ebpS), and fibrinogen-binding clumping factor A (clfA) were highly abundant in S. aureus during AC and CH infection (Fig. 4). The gene encoding the extracellular adherence protein Map (also known as Eap), which has been shown to bind to cartilage (18), was significantly expressed by S. aureus during the AC phase of infection (Fig. 4).
FIG 4 .
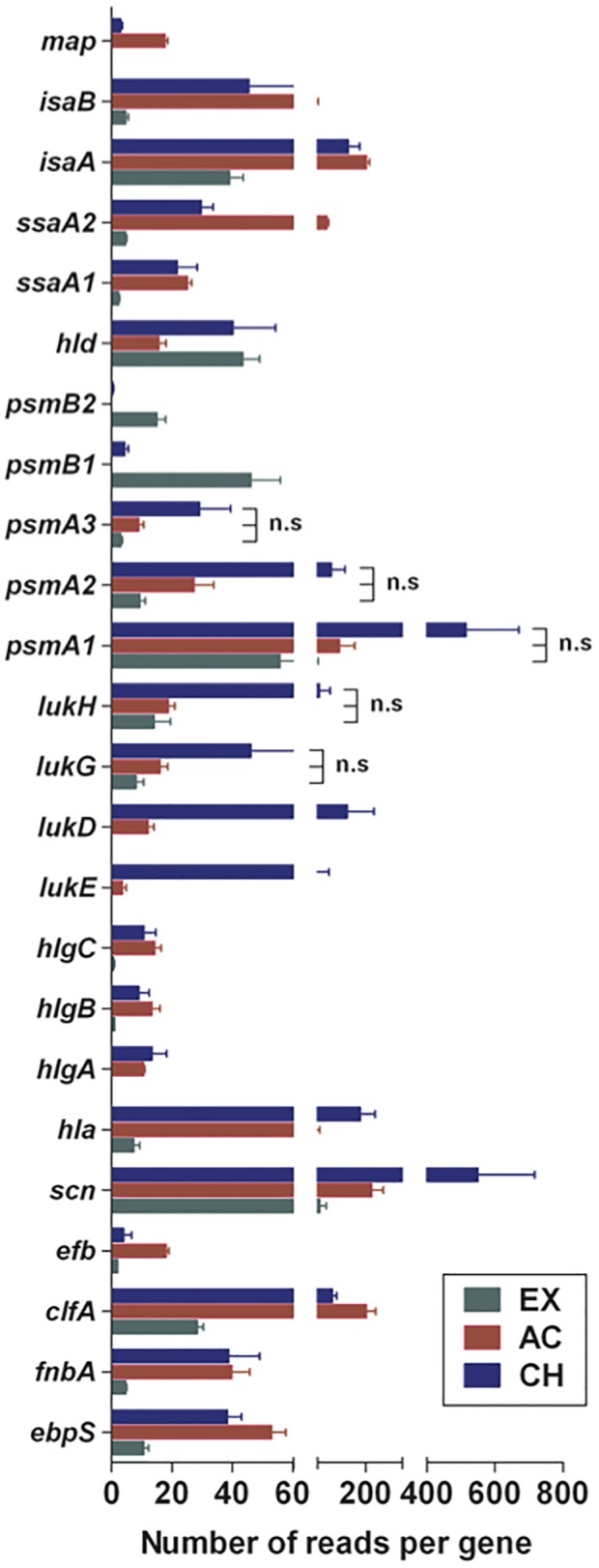
Expression of genes encoding virulence factors by S. aureus under in vitro and in vivo conditions. Expression of genes encoding proteins involved in pathogenesis by S. aureus during EX, AC, and CH growth is shown. Each bar represents the mean and SD of triplicates. n.s, the differences between AC and EX mean values and between CH and AC mean values are not statistically significant.
To survive inside the host and maintain a persistent infection, S. aureus uses a variety of mechanisms to evade or overcome the host immune defenses, especially the innate immune system (19). The staphylococcal complement inhibitor (SCIN), which interferes with all complement activation pathways (the lectin, classical, and alternative pathways) by blocking C3 convertases, is the most efficient complement inhibitor (20). We found that the gene encoding SCIN (scn) was significantly more highly expressed by S. aureus during AC and CH infection than during in vitro EX growth (Fig. 4).
S. aureus secretes numerous exotoxins that directly destroy phagocytic cells, representing a very efficient way to evade phagocytic-mediated killing (21). These exotoxins include pore-forming toxins such as α-hemolysin and the bi-component γ-hemolysin (HlgACB), LukGH (also known as LukAB) and LukED, β-hemolysin, and the phenol-soluble modulins (PSMs) (21). The gene encoding the single-component α-hemolysin (hla) was highly expressed by S. aureus during AC and CH infection (Fig. 4). The genes encoding LukGH (lukG and -H) and LukED (lukE and -D) were also expressed to a greater extent by S. aureus during in vivo infection but predominately during the CH phase (Fig. 4). PSMs are small peptide toxins known to activate, attract, and lyse neutrophils (22) as well as to mediate S. aureus phagosomal escape in both professional and nonprofessional phagocytes (23). Depending on the size, PSMs are classified into two groups. The smaller α-type PSMs (PSMα1, PSMα2, PSMα3, and PSMα4), with a length of 20 to 30 amino acids, are considered the most toxic PSMs, whereas the larger β-type PSMs (PSMβ1 and PSMβ2), approximately 44 amino acids in length, seem to have different functions (22). Interestingly, the genes psmA1, -2, and -3, encoding PSMα1 to -3, were highly expressed by S. aureus during AC and CH infection, although to a much greater extent during the latter. On the other hand, the genes encoding PSMβ1 (psmB1) and PSMβ2 (psmB2) were expressed by the bacteria only during in vitro EX growth (Fig. 4), indicating that PSMβ may not be required by S. aureus to survive within the bone tissue.
(iv) Differential expression of genes involved in iron homeostasis.
Iron uptake is a critical event for S. aureus survival within the host, as the host limits iron availability as part of its innate defense against invading pathogens (24). The gene encoding ferritin (ftn), which is responsible for mobilizing iron from intracellular reserves when external iron supplies are limited (25), was highly expressed by S. aureus during the AC phase and at lower levels during the CH phase of infection (see Table S4 in the supplemental material). S. aureus has evolved several sophisticated iron uptake systems, including the cell wall-based iron acquisition Isd pathway (26). Within this pathway, the genes encoding the cell wall-anchored heme-binding protein IsdA and the hemoglobin-binding protein IsdB were more extensively expressed by S. aureus during the CH phase of infection (see Table S4 in the supplemental material).
(v) Differential expression of genes encoding two-component regulatory systems (TCS).
Two-component regulatory systems enable bacteria to sense their environment and respond appropriately by coordinating gene expression. The expression of most S. aureus virulence factors is directly or indirectly controlled by diverse regulators, such as agr, sarA, and sae (27). The transcription data showed high expression of agrA, -B, and -C by S. aureus during in vitro EX growth and low expression during AC and CH infection (see Table S5 in the supplemental material). In contrast, the accessory regulators sarA and -R and saeR and -S were expressed by S. aureus to a greater level in infected bones (see Table S5 in the supplemental material). The VicR/VicK (also known as WalK/WalR and YycG/YycF) regulatory system was also more highly expressed by S. aureus during AC and CH infection (see Table S5 in the supplemental material).
Expression of regulatory small RNAs by S. aureus during in vitro growth and in vivo infection.
In addition to the classical transcriptional regulatory systems, noncoding small regulatory RNAs (sRNAs) seem to play important roles in the posttranscriptional regulation of many genes in S. aureus (28). The 15 most highly expressed sRNAs of the well-known and characterized sRNAs in the Repository of Bacterial Small Regulatory RNA (BSRD) database are shown in Table S6 in the supplemental material. The sRNAs more highly induced by S. aureus under all conditions tested were sRNAs with housekeeping functions, including the 4.5s RNA or ffs (ssao445.1) component of the signal recognition particle (SRP), the RNase P (ssao1419.1) responsible for processing of tRNAs, and the transfer-messenger RNA SsrA (ssao789.1), which rescues stalled ribosomes during translation of defective mRNAs. Among the 15 sRNAs most highly expressed by S. aureus during infection was RsaC (ssao624.1), which was expressed during the AC phase (see Table S6 in the supplemental material). The sRNAs corresponding to the T-box riboswitch (ssao357.2) involved in the regulation of metB, encoding cystathionine gamma-synthase, and that (ssao1688.1) involved in the regulation of thrS, encoding threonyl-tRNA synthetase, were highly expressed by S. aureus during both AC and CH phases of infection (see Table S6 in the supplemental material). The S-box riboswitch senses S-adenosylmethionine and regulates methionine metabolism in response to methionine starvation (29). The S-box (ssao1819.1) leader of metK, encoding S-adenosylmethionine synthase, and the S-box (ssao813.1) leader of metN2, encoding a methionine import ATP-binding protein, were among the 15 sRNAs most highly expressed by S. aureus during CH infection (see Table S6 in the supplemental material). This is in accordance with the assumption of nutrients starvation during the CH phase of bone infection.
Expression of hypothetical proteins by S. aureus during in vitro growth and in vivo infection.
Many genes encoding proteins with unknown function were expressed to a higher level by S. aureus during AC and CH infection than during in vitro EX growth (see Table S4 in the supplemental material). These genes are of particular interest because they are likely to be important for bacterial survival during the specific stages of osteomyelitis but no obvious function has yet been assigned.
Confirmation of selected S. aureus in vivo-expressed genes in human patients.
The expression of key genes from different functional categories found to be highly expressed by S. aureus during in vivo infection in the experimental murine model was then confirmed by qRT-PCR in infected bone samples isolated from human patients. To this end, we collected clinical specimens from patients with subacute or chronic S. aureus infection involving soft tissue and bones and used these samples to perform qRT-PCR analysis of specific genes after direct extraction of RNA (see Table S7 in the supplemental material). Similar to what was observed in the murine osteomyelitis model, the S. aureus genes encoding the cysteine protease staphopain B (Fig. 5A), clumping factor A (Fig. 5C), DnaK chaperon (Fig. 5D), arginine deiminase (Fig. 5E), carbamate kinase (Fig. 5F), and a manganese ABC transporter (Fig. 5G) were more highly expressed by S. aureus in the infected human tissue than in S. aureus cultured in vitro in rich medium. Also comparable to the murine system was the behavior of the gene encoding the l-lactate dehydrogenase, whose expression was lower in S. aureus during in vivo bone infection (Fig. 5B). We also assessed the global resemblance between the changes in gene expression of S. aureus in the different human samples and those of S. aureus isolated from murine bones during AC and CH infection using the Bray-Curtis similarity index. The dendrogram of the Bray-Curtis distance matrix (Fig. 5H) revealed that the expression of the selected genes by S. aureus in patients 2 (P-2) and 3 (P-3) cluster very closely with the expression of those genes by S. aureus isolated from chronic murine osteomyelitis (~80% similarity). The respective S. aureus strains isolated from P-2 and P-3 were of the same spa type (t009) and therefore genetically closely related (see Table S7 in the supplemental material). The gene expression pattern of S. aureus in patients 4 (P-4) and 5 (P-5) exhibited around 60% similarity with the expression of genes by S. aureus in both AC and CH murine infection, whereas the gene expression of patient 1 (P-1) was the most divergent (only 40% similarity with the other data sets) (Fig. 5H). Interestingly, the S. aureus strain infecting P-1 was an MRSA strain, whereas the other strains were all methicillin-susceptible S. aureus (MSSA) strains (see Table S7 in the supplemental material).
FIG 5 .

Confirmation of selected S. aureus in vivo-expressed genes in clinical samples by qRT-PCR. Changes (fold) in expression of sspB (A), ldh1 (B), clfA (C), dnaK (D), adi (E), arcC (F), and the gene encoding the manganese ABC transporter (RSAU_000587) (G) in S. aureus in infected human tissue (circles) were determined by qRT-PCR, and those in murine bones (squares) were determined by RNA-Seq. The expression of the respective gene by S. aureus grown in vitro in rich medium was used as a reference to calculate the changes. Each circle represents one individual patient, and each square represents one independent experimental set in the murine system, with black squares representing AC infection and white squares CH infection. (H) Hierarchical clustering dendrogram showing the change (fold) in gene expression data determined by Bray-Curtis similarity scores. P-1 to P-5, individual patients; AC-1 to AC-3, individual data sets from murine AC bone infection; CH-1 to CH-3, individual data sets from murine CH bone infection.
DISCUSSION
In this study, we characterized on a genome-wide scale the gene expression profile of S. aureus during the acute (7 days) and chronic (28 days) phases of hematogenous osteomyelitis in an experimental murine model that closely mimics the natural infection in humans (6). As the ultimate aim is to bring these findings closer to clinics, we confirmed the expression of a selected set of S. aureus in vivo-expressed genes in the murine system in clinical specimens from five patients with chronic staphylococcal osteomyelitis. Our results show that the variation in gene expression was more extensive when we compared S. aureus grown in vitro versus S. aureus in infected tissue in vivo than when we compared S. aureus in vivo during AC versus CH phases of infection. These observations suggest that S. aureus required a higher number of genes for adjusting to the host environment than for longstanding persistence within the infected tissue.
Acquisition of nutrients and metabolism within the host is a fundamental aspect of S. aureus pathogenesis. The shift from glycolysis to gluconeogenesis observed in S. aureus transcriptional response during the transition from the in vitro to the in vivo environment indicates a lack of glucose availability in bone tissue. The lack of easy-to-degrade nutrients during bone infection forces the use of alternative carbon sources. In this regard, S. aureus has evolved unique mechanisms to maximize the harvesting of nutrients from host resources. Thus, we found that a number of genes encoding secreted proteolytic enzymes (aur, sspA, sspB, scpA, and splABCEF) were expressed by S. aureus only during infection. The gene sspB, selected as representative of this group of proteolytic enzymes, was also highly expressed by S. aureus in infected human tissue. Similar to our results, the genes encoding proteases were reported to be highly expressed by S. aureus strain USA300 in vivo in a murine model of kidney infection (5). It is possible that S. aureus releases these proteases to obtain small peptides from the host milieu. However, these enzymes have also been shown to play a role in pathogenesis by inducing tissue destruction to facilitate bacterial dissemination (30). The metabolic adjustment of S. aureus to the bone environment contrasts with that reported in the lungs, where its growth was primarily driven by carbohydrates, as indicated by the upregulation of glycolysis and the downregulation of pathways for gluconeogenesis and amino acid catabolism (4). These observations underscore the difference in nutrient availability in the different anatomical locations within the host and thus the bacterial need for multiple adaptive strategies for survival during in vivo infection.
The transcriptomic profile also indicated that the aerobic electron transfer chain seems to be repressed in S. aureus during bone infection. This is a critical issue, since the respiratory ATP synthesis pathway has been proposed as a potential antibiotic target against S. aureus and small-molecule inhibitors with diarylquinoline scaffold have been shown to inhibit the growth of S. aureus in the planktonic state as well as in metabolically resting bacteria grown in a biofilm culture (31). However, the fact that this pathway is not used by S. aureus during bone infection may hinder the antimicrobial effect of these inhibitors for the treatment of S. aureus osteomyelitis. Interestingly, the ADI pathway, encoded by the arc operon, was induced by S. aureus predominantly during the CH phase of experimental bone infection and was highly expressed by S. aureus in all human samples. This pathway enables S. aureus to use arginine as a source of energy under anaerobic conditions and is generally induced when other sources of energy such as glucose are absent (11). In addition to its role in generating ATP under anaerobic conditions, the ADI pathway can also play a role in maintaining pH homeostasis during infection, since the ammonia generated during this process can neutralize acids generated by the fermentative metabolism (32). Because l-arginine may also serve as a substrate for the production of antimicrobial nitric oxide (NO) by the nitric oxide synthase in classically activated macrophages (M1) (33), the use of the ADI pathway may help S. aureus to evade this antimicrobial mechanism by consuming arginine, as shown for other pathogens (34).
Iron acquisition is fundamental for S. aureus to survive within the host (26). The in vivo transcriptional profile suggests that S. aureus may use iron from its internal stores during AC osteomyelitis and may scavenge iron from the host by acquiring heme complexes during CH osteomyelitis.
The transition from acute to chronic infection seems to be associated with the induction of the stringent response, since the RelA/SpoT system was significantly expressed by S. aureus only during CH infection. The stringent response is a bacterial adaptation to starvation and is essential for S. aureus viability (35). The isolation of an S. aureus strain harboring a mutation in relA from a patient with a very difficult-to-treat chronic infection supports the relevance of the stringent response for S. aureus in vivo persistence (36).
Important sets of genes expressed by S. aureus during in vivo infection were those aiming to interfere with the immune defense of the host. In this regard, we found a high level of expression of genes encoding proteins (e.g., SCIN) that interfere with the complement system and thus hamper bacterial phagocytic uptake. Also, genes encoding toxins that can kill host immune cells, such as α-hemolysin, γ-hemolysin (HlgAB and HlgCB), and leukocidins LukED and LukAB, as well as genes encoding PSMs, were highly expressed by S. aureus during bone infection. In this regard, LukED was recently reported to contribute to the lethality observed upon S. aureus bloodstream infection (37), and α-hemolysin has been implicated in the pathogenesis of lethal pneumoniae (38), severe skin infections (39), and brain abscesses (40).
The vast array of S. aureus virulence determinants are very tightly controlled by several regulators, which include the two-component regulatory systems agrAC and saeRS as well as the sarA gene family. Investigation of the regulatory networks coordinating the expression of S. aureus virulence factors has essentially been limited to in vitro culture conditions because of the difficulty of monitoring the activity of regulatory networks in the infected host. This is complicated further by the fact that one target gene can be under the control of several regulators to ensure that specific factors are produced only when required during the infection process. The regulatory networks expressed by S. aureus during in vivo infection remain largely unclear. In this study, we found that agr was highly expressed by S. aureus during in vitro EX growth but at significantly reduced levels during bone infection. Interestingly, agr-defective mutants have been shown to be selected in patients with chronic infections (41). In contrast, the loci encoding the saeRS system were expressed at very low levels by S. aureus during in vitro EX growth and were significantly upregulated during bone infection, mainly during the AC phase of infection. In line with this observation, Date et al. (5) reported agrC to be downregulated and saeR upregulated by S. aureus USA300 in the kidneys of infected mice. The higher expression of the sarA locus may be responsible for the high expression level of genes encoding fibronectin- and fibrinogen-binding proteins as well as those encoding α-, β-, and δ-toxins observed here during AC and CH infection in vivo (42). The VicR/VicK (also known as WalK/WalR and YycG/YycF) regulatory system was also more highly expressed by S. aureus during AC and CH infection. This two-component system regulates expression of genes encoding proteins involved in modulating the cell wall structure, such as lytM, encoding a major autolysin, as well as ssaA and isaA, which encode proteins potentially involved in peptidoglycan hydrolysis (43). The genes encoding these proteins were all expressed to a higher extent by S. aureus during bone infection.
A total of 42 genes exhibited lower expression levels in S. aureus during CH than during AC infection. These included genes encoding stress-related proteins (ahpF and katA), most probably reflecting the shift from a highly oxidative/nitrosative environment imposed by the inflammatory response during the AC phase of infection to a less harsh environment dominated mainly by tissue repair processes during the CH phase of infection. Other genes expressed by S. aureus to a significantly lower extent during the CH versus AC phase of osteomyelitis comprised genes encoding proteins involved in DNA synthesis and cell division (cvfA, atl, lytM, and pyrR) or amino acid metabolism (dhoM, gltA, glnA, thrBC, and ald2). The lower expression of these genes suggests a drop in the bacterial multiplication rate and amino acid metabolism during the CH phase of infection.
Small regulatory RNAs (sRNAs) represent an additional level of posttranscriptional gene regulation that allows the pathogen to adapt to its metabolic needs during infection and to express virulence genes (44). They can be encoded as part of the mRNA that they regulate (cis-acting RNAs), such as riboswitches or 5′ untranslated regions (UTRs), or as independent transcripts (trans-acting RNAs) (45). Most sRNAs exert their action either by base-pairing with their mRNA target, modulating its translation and/or stability, or by binding to proteins, antagonizing their function (45). In recent years, a number of sRNAs have been identified in S. aureus (28); however, a specific biological role for most of them has yet to be assigned. Whereas sRNAs with housekeeping functions, including the 4.5s RNA encoded by the ffs gene, the RNase P, and SsrA RNA, were highly expressed by S. aureus under all three conditions, other sRNAs were more highly expressed by S. aureus during in vivo infection than during in vitro growth. As examples, rsaC, the function of whose product is still unclear, the T box involved in the regulation of metB encoding cystathionine γ-synthase involved in the synthesis of methionine, and the T box regulating thrS, which encodes threonyl-tRNA synthetase, a key enzyme of protein biosynthesis which charges tRNA molecules with threonine, were among the sRNAs most highly expressed by S. aureus during the AC phase of infection. The sRNAs expressed by S. aureus during infection may provide good therapeutic targets. In fact, inhibitors of threonyl-tRNA synthetase, regulated by the T box/thrs, are currently being investigated as potential therapeutic agents (46). Two S-box riboswitches, metK (encoding S-adenosylmethionine synthase) and metN2 (encoding methionine import ATP-binding protein, which regulates methionine transport), were among the most highly expressed sRNAs by S. aureus during the CH phase of infection. S-Adenosylmethionine serves as a methyl group donor in a variety of cellular processes and is the precursor molecule in polyamine synthesis, and its expression is induced in response to starvation for methionine (47). The S box has been reported to play an important role in the pathogenicity of S. aureus and consequently has been proposed as a potential target for new anti-infectives (48).
In summary, this is the first study reporting the transcriptional response at a whole-genome level of S. aureus during acute and chronic phases of experimental osteomyelitis. Our results indicate that during the acute phase of infection, S. aureus experienced massive transcriptional remodeling to adapt to the nutrient availability in the tissue and to the biological pressure imposed by the host immune system. During the chronic phase, S. aureus switched its transcriptional response from a proliferative mode to a survival mode that enabled persistence. In this regard, chronic and antibiotic refractory staphylococcal infections have been linked to the emergence of a subpopulation of S. aureus with a slow-growing small-colony-variant (SCV) phenotype (49, 50). SCVs can be regarded as a bacterial strategy for survival and persistence in a harsh environment (49, 50). The transcriptional response of S. aureus during the chronic phase of osteomyelitis described in our study parallels many features of SCVs. Thus, the global transcriptional profile obtained in this study may be the sum of independent responses of different S. aureus subpopulations present in the infected bone. It would be therefore of great interest to find a way to discriminate the transcriptional response of the SCVs from that of the original parental bacterial population during in vivo infection.
MATERIALS AND METHODS
Ethics statement.
Ethical approval for the experiments, including those using human samples, was obtained from the Ethy Kommission der Ärztekammer Westfalen-Lippe und der Medizinische Fakultät der Westfälischen Wilhelms-Universität (permit 2014-376-f-S).
Animal experiments were performed in strict accordance with the German regulations of the Society for Laboratory Animal Science (GV-SOLAS) and the European Health Law of the Federation of Laboratory Animal Science Associations (FELASA). All experiments were approved by the ethical board Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit, Oldenburg, Germany (permit 33.9-42502-04-10/0296).
Bacteria.
S. aureus strain 6850, originally isolated from a patient with a skin abscess that progressed to staphylococcal bacteremia, hematogenous osteomyelitis, septic arthritis, and multiple systemic abscesses, was used in this study (8, 9). Bacteria were cultivated in brain heart infusion (BHI) medium (Roth, Karlsruhe, Germany) under microaerobic conditions using a 5:1 flask volume ratio and incubated on a shaker at 37°C and 125 rpm. As a reference for the downstream RNA-Seq analyses, S. aureus was harvested during the exponential (EX) (optical density at 600 nm [OD600] = 0.4) growth phase.
Mice and infection model.
Pathogen-free C57BL/6 10-week-old mice were obtained from Harlan-Winkelmann (Borchen, Germany) and infected with 106 CFU of S. aureus in 150 µl of phosphate-buffered saline (PBS) via a lateral tail vein as previously described (6). Three independent sets of 8 to 10 mice were sacrificed by CO2 asphyxiation at day 7 (AC) and day 28 (CH) of infection; the tibiae from each animal were removed by dissection, and the RNA was stabilized by incubation in 2 ml of RNAlater solution (Ambion) overnight at 4°C.
Bacterial extraction from infected bones using antibody-bound magnetic beads.
RNA-stabilized samples were homogenized in 2 ml of sterile PBS supplemented with 1% β-mercaptoethanol (Sigma), and tissue debris was removed by centrifugation at 200 × g for 5 min at 4°C. Magnetic beads bound to anti-S. aureus IgG antibodies were employed to extract S. aureus from the bone homogenates prior to RNA extraction. Purified rabbit anti-S. aureus IgG antibodies were coupled to sheep anti-rabbit IgG conjugated magnetic Dynabeads (1 µg/107 beads; M280; Invitrogen), washed with sterile PBS supplemented with 0.1% bovine serum albumin (BSA), and resuspended in 1 ml of sterile PBS–0.1% BSA. A volume of 50 µl (~107 beads) was added to the bone homogenates, and the mixture was incubated with gentle agitation at room temperature for 30 min. The beads were subsequently pelleted in a magnetic rack (NEB), washed with 1 ml of sterile PBS–0.1% BSA, treated with 100 µl of lysostaphin (5 µg/ml) (Sigma) for 10 min at room temperature, and subjected to RNA extraction. In vitro-grown S. aureus was also subjected to antibody–magnetic-bead separation to minimize the effects of sample preparation and processing on downstream comparative RNA-Seq data analyses.
Total RNA extraction, purification, and mRNA enrichment.
For extraction of total RNA, S. aureus cells isolated from in vitro cultures and infected bones were resuspended in 1 ml of cold RLT (guanidine thiocyanate) buffer (Qiagen) supplemented with 1% β-mercaptoethanol (Sigma) and transferred to lysing matrix B tubes (MP Biomedicals) on ice. Samples were disrupted using the FastPrep-24 instrument (MP Biomedicals) at an intensity of 5.5 for 40 s. Samples were returned to ice for 4 min, disrupted further using the same settings, and then centrifuged at 13,500 × g for 10 min at 4°C. The supernatant was subsequently transferred to 1.5-ml RNase-free Biopur centrifuge tubes (Eppendorf), and the RNA was extracted using the RNeasy minikit according to the manufacturer’s instructions, including optional DNase treatment on the column (Qiagen). RNA was eluted with nuclease-free water (Ambion) and concentrated by ethanol precipitation using standard procedures.
To remove any possibly contaminating DNA, a further DNase treatment step was performed on all samples using the Turbo DNA-free kit (Invitrogen), and the samples were concentrated by ethanol precipitation. The mRNA was subsequently enriched from the samples using Terminator 5′-phosphate-dependent exonuclease (Epicentre) according to the manufacturer’s instructions. Samples were further concentrated by ethanol precipitation and re-eluted in nuclease-free water (Ambion). RNA integrity and quantity were measured using the Agilent 2100 Bioanalyzer (Agilent Technologies) and NanoDrop 1000 spectrophotometer (Thermo Scientific).
For extraction of total RNA from clinical specimens, bones were homogenized in 1 ml of lysis buffer (RNAPro solution; MP Biomedicals) supplemented with 1% β-mercaptoethanol and further disrupted using lysing matrix B beads (MP Biomedicals) in a FastPrep-24 instrument. Cell lysates were then centrifuged at 13,500 × g for 2 min at 4°C, and total RNA was extracted from all samples using the RNeasy minikit (Qiagen). To eliminate DNA contamination, each RNA sample was treated with 2.5 µl of DNase I (Qiagen) and incubated for 30 min at room temperature. Following this, RNA samples were cleaned up and concentrated using the RNeasy MinElute cleanup kit (Qiagen).
Illumina library generation and sequencing.
Libraries were generated using the ScriptSeq v2 RNA-Seq library preparation kit (Epicentre) and purified using the Minelute PCR purification kit (Qiagen) according to the manufacturer’s instructions. Libraries were sequenced on the Illumina HiSeq 2500 platform using the TruSeq S.R. cluster kit, v3-cBot-HS (Illumina). Three libraries were multiplexed per lane (12 pM/library) and sequenced to 58 cycles in one direction. Each library produced between 53 and 105 million reads of 50 nucleotides (nt).
RNA-Seq data processing.
Sequence reads were preprocessed for quality and trimmed using a combination of in-house Ruby scripts and open-source tools (http://wiki.bioinformatics.ucdavis.edu/index.php/Trim.pl and http://www.mothur.org/). Reads matching rRNA sequences were identified and filtered by BLAST searching against the ribosomal genes annotated in the genome of the S. aureus strain SAU6850 (9). Reads were collapsed into unique representatives, associated to their copy numbers, and mapped to the S. aureus strain 6850 genome sequence using BLAST (51). In addition, the small fraction of noncoding mRNA reads (from 0.01 to 7.3% of the total generated reads per library) was further BLAST searched against the BSRD database (http://kwanlab.bio.cuhk.edu.hk/BSRD), where sequence matches of >28 nt with >98% sequence homology and an E value of 0.001 were considered. Raw mapped read counts were then either normalized as the numbers of reads per kb of transcript per million reads mapped (RPKM) (52) or randomly resampled to the smallest library size of 26,251 reads, taking the average from 999 resampling events (using an in-house Perl script).
qRT-PCR in clinical samples.
Total RNA was isolated from S. aureus-infected clinical specimens, and the expression of seven genes (adi, arcC, clfA, dnaK, RSAU_000587, ldh1, and sspB), as well as two housekeeping genes, gyrB and tpiA, was quantified by qRT-PCR. RNA samples were reverse transcribed and amplified with a SensiFAST SYBR No-ROX one-step kit using the LightCycler 480 real-time PCR system. Thermal cycling conditions consisted of reverse transcription for 15 min at 45°C and activation of polymerase for 5 min at 95°C, followed by 40 cycles of 20 s at 95°C, 20 s at 58°C, and 20 s at 72°C, which correspond to denaturation, annealing, and extension, respectively. Primer sequences (see Table S8 in the supplemental material) were designed for each gene as described above. The S. aureus strain isolated from each human sample (n = 5) (see Table S7 in the supplemental material) and grown to an OD600 of 0.4 to 0.5 (EX growth) was used as a reference for quantitative RT-PCR analyses. Values were normalized against the values for the housekeeping genes gyrB and tpiA, and the relative copy numbers for each gene were estimated using the Pfaffl equation (53). Values were expressed as the change (fold) between the expression of the specific gene in human tissue and the expression in in vitro exponential-growth-phase samples.
Statistical analysis.
Statistical analyses were carried out using both PRIMER (v.6.1.6, PRIMER-E; Plymouth Marine Laboratory) and the free software R package for statistical computing and graphics (http://www.r-project.org). To assess the global similarity between replicates within each growth phase (EX, AC, and CH), a sample-similarity matrix was generated using the Bray-Curtis coefficient, and the transcriptome profiles were compared by both principal coordinates analysis (PCoA) and group-average agglomerative hierarchical clustering. A permutational multivariate analysis of variance (PERMANOVA) (54) was used to determine the statistical significance of the global transcriptional profile of independent conditions (EX, AC, and CH). For the one-way PERMANOVA, where condition was the main factor, the resemblance matrix was derived following the Bray-Curtis algorithm using type III (partial) sums of squares with a fixed effects sum to zero for mixed terms. Exact P values were generated using unrestricted permutation of raw data. Pseudo-F statistic and generated P values were reported for each pair of conditions where differences in gene expression were observed. Further pairwise tests were also conducted using PERMANOVA. Monte Carlo tests were undertaken in the pairwise test function in PERMANOVA if low permutations were obtained. The Monte Carlo (P) value is better suited and more reliable when there are not enough possible permutations. The conditions were considered significantly different if the P value fell below 0.05.
To assess differential gene expression between conditions, univariate algorithms were applied to the rerandomized data after genes for which transcripts levels were below 0.05% (<14 copies per 26,251 reads) had been filtered out. Considering that there is no current consensus on which algorithm to use for determination of differential expression in RNA-Seq data, three robust algorithms—DESeq and EdgeR, assuming an NB distribution, and SAMseq, assuming a distribution-free condition (all in Bioconductor, R package)—were applied, and a gene was considered significantly different in expression between a pair of conditions if the P value fell to <0.05 in any 2 of the 3 algorithm methods used (EdgeR, DESeq, and SAMseq). All P values were corrected for multiple testing using default parameters.
RNA sequence accession number.
The RNA sequencing reads have been deposited at the European Nucleotide Archive under the accession number PRJEB6003 (http://www.ebi.ac.uk/ena/data/view/PRJEB6003).
SUPPLEMENTAL MATERIAL
Rank-ordered distributions of S. aureus gene expression levels under in vitro and in vivo conditions. Download
Visualization of the global expression of S. aureus during in vitro growth (EX) and AC and CH phases of infection. Download
Summary of data filtering for replicated RNA-Seq experiments.
Raw, RPKM, and resampled transcript numbers from S. aureus during EX (EX1 to -3), AC (AC1 to -3), and CH (CH1 to -3) conditions.
Comparison of global gene expression profiles between conditions using permutational multivariate analysis of variance (PERMANOVA).
Transcript abundances of genes expressed by S. aureus during in vitro growth (EX) and AC and CH phases of infection.
Transcript abundances of genes encoding two-component systems in S. aureus during EX in vitro growth or in vivo AC and CH infection.
The top-15-ranked (most highly expressed) noncoding regions in S. aureus during in vitro growth (EX) and AC and CH phases of infection.
Description of human samples.
Primers used for RT-PCR in human samples.
ACKNOWLEDGMENTS
Financial support was provided by the Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung-BMBF) “Medizinische Infektionsgenomik” (grant 01KI1009B), by the Helmholtz International Graduate School for Infection Research under contract number VH-GS-202, and by DFG (Transregio34/C12).
We thank the sequencing platform at the HZI for sequencing support, in particular Robert Geffers and Michael Jarek, as well as Jennifer Geraci for providing support during the processing of the clinical specimens and Iris Plumeier for support in spa typing.
Footnotes
Citation Szafranska AK, Oxley APA, Chaves-Morenob D, Horsta SA, Roßlenbroich S, Peters G, Goldmann O, Rohde M, Sinha B, Pieper DH, Löffler B, Jauregui R, Wos-Oxley ML, Medina E. 2014. High-resolution transcriptomic analysis of the adaptive response of Staphylococcus aureus during acute and chronic phases of osteomyelitis. mBio 5(6):e01775-14. doi:10.1128/mBio.01775-14.
REFERENCES
- 1. Grundmann H, Aires-de-Sousa M, Boyce J, Tiemersma E. 2006. Emergence and resurgence of methicillin-resistant Staphylococcus aureus as a public-health threat. Lancet 368:874–885. 10.1016/S0140-6736(06)68853-3. [DOI] [PubMed] [Google Scholar]
- 2. Lowy FD. 2012. Staphylococcus aureus infections. N. Engl. J. Med. 339:520–532. 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 3. Yarwood JM, McCormick JK, Paustian ML, Kapur V, Schlievert PM. 2002. Repression of the Staphylococcus aureus accessory gene regulator in serum and in vivo. J. Bacteriol. 184:1095–1101. 10.1128/jb.184.4.1095-1101.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chaffin DO, Taylor D, Skerrett SJ, Rubens CE. 2012. Changes in the Staphylococcus aureus transcriptome during early adaptation to the lung. PLoS One 7:e41329. 10.1371/journal.pone.0041329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Date SV, Modrusan Z, Lawrence M, Morisaki JH, Toy K, Shah IM, Kim J, Park S, Xu M, Basuino L, Chan L, Zeitschel D, Chambers HF, Tan MW, Brown EJ, Diep BA, Hazenbos WL. 2014. Global gene expression of methicillin-resistant Staphylococcus aureus USA300 during human and mouse infection. J. Infect. Dis. 209:1542–1550. 10.1093/infdis/jit668. [DOI] [PubMed] [Google Scholar]
- 6. Horst SA, Hoerr V, Beineke A, Kreis C, Tuchscherr L, Kalinka J, Lehne S, Schleicher I, Köhler G, Fuchs T, Raschke MJ, Rohde M, Peters G, Faber C, Löffler B, Medina E. 2012. A novel mouse model of Staphylococcus aureus chronic osteomyelitis that closely mimics the human infection: an integrated view of disease pathogenesis. Am. J. Pathol. 181:1206–1214. 10.1016/j.ajpath.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 7. Lew DP, Waldvogel FA. 2004. Osteomyelitis. Lancet 364:369–379. 10.1016/S0140-6736(04)16727-5. [DOI] [PubMed] [Google Scholar]
- 8. Vann JM, Proctor RA. 1987. Ingestion of Staphylococcus aureus by bovine endothelial cells results in time- and inoculum-dependent damage to endothelial cell monolayers. Infect. Immun. 55:2155–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fraunholz M, Bernhardt J, Schuldes J, Daniel R, Hecker M, Sinha B. 2013. Complete genome sequence of Staphylococcus aureus 6850, a highly cytotoxic and clinically virulent methicillin-sensitive strain with distant relatedness to prototype strains. Genome Announc. 1:e00775-13. 10.1128/genomeA.00775-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fuchs S, Pané-Farré J, Kohler C, Hecker M, Engelmann S. 2007. Anaerobic gene expression in Staphylococcus aureus. J. Bacteriol. 189:4275–4289. 10.1128/JB.00081-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cunin R, Glansdorff N, Piérard A, Stalon V. 1986. Biosynthesis and metabolism of arginine in bacteria. Microbiol. Rev. 50:314–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Horsburgh MJ, Clements MO, Crossley H, Ingham E, Foster SJ. 2001. PerR controls oxidative stress resistance and iron storage proteins and is required for virulence in Staphylococcus aureus. Infect. Immun. 69:3744–3754. 10.1128/IAI.69.6.3744-3754.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Davies KJ. 2001. Degradation of oxidized proteins by the 20S proteasome. Biochimie 83:301–310. 10.1016/S0300-9084(01)01250-0. [DOI] [PubMed] [Google Scholar]
- 14. Frees D, Gerth U, Ingmer H. 2014. Clp chaperones and proteases are central in stress survival, virulence and antibiotic resistance of Staphylococcus aureus. Int. J. Med. Microbiol. 304:142–149. 10.1016/j.ijmm.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 15. Mogk A, Tomoyasu T, Goloubinoff P, Rüdiger S, Röder D, Langen H, Bukau B. 1999. Identification of thermolabile Escherichia coli proteins: prevention and reversion of aggregation by DnaK and ClpB. EMBO J. 18:6934–6949. 10.1093/emboj/18.24.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Potrykus K, Cashel M. 2008. (p)ppGpp: still magical? Annu. Rev. Microbiol. 62:35–51. 10.1146/annurev.micro.62.081307.162903. [DOI] [PubMed] [Google Scholar]
- 17. Clarke SR, Foster SJ. 2006. Surface adhesins of Staphylococcus aureus. Adv. Microb. Physiol. 51:187–224. 10.1016/S0065-2911(06)51004-5. [DOI] [PubMed] [Google Scholar]
- 18. Hansen U, Hussain M, Villone D, Herrmann M, Robenek H, Peters G, Sinha B, Bruckner P. 2006. The anchorless adhesin Eap (extracellular adherence protein) from Staphylococcus aureus selectively recognizes extracellular matrix aggregates but binds promiscuously to monomeric matrix macromolecules. Matrix Biol. 25:252–260. 10.1016/j.matbio.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 19. Foster TJ. 2005. Immune evasion by staphylococci. Nat. Rev. Microbiol. 3:948–958. 10.1038/nrmicro1289. [DOI] [PubMed] [Google Scholar]
- 20. Rooijakkers SH, Ruyken M, Roos A, Daha MR, Presanis JS, Sim RB, van Wamel WJ, van Kessel KP, van Strijp JA. 2005. Immune evasion by a staphylococcal complement inhibitor that acts on C3 convertases. Nat. Immunol. 6:920–927. 10.1038/ni1235. [DOI] [PubMed] [Google Scholar]
- 21. Vandenesch F, Lina G, Henry T. 2012. Staphylococcus aureus hemolysins, bi-component leukocidins, and cytolytic peptides: a redundant arsenal of membrane-damaging virulence factors? Front. Cell. Infect. Microbiol. 2:12. 10.3389/fcimb.2012.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Peschel A, Otto M. 2013. Phenol-soluble modulins and staphylococcal infection. Nat. Rev. Microbiol. 11:667–673. 10.1038/nrmicro3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grosz M, Kolter J, Paprotka K, Winkler AC, Schäfer D, Chatterjee SS, Geiger T, Wolz C, Ohlsen K, Otto M, Rudel T, Sinha B, Fraunholz M. 2014. Cytoplasmic replication of Staphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin α. Cell. Microbiol. 16:451–456. 10.1111/cmi.12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hammer ND, Skaar EP. 2011. Molecular mechanisms of Staphylococcus aureus iron acquisition. Annu. Rev. Microbiol. 65:129–147. 10.1146/annurev-micro-090110-102851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Andrews SC, Robinson AK, Rodríguez-Quiñones F. 2003. Bacterial iron homeostasis. FEMS Microbiol. Rev. 27:215–237. 10.1016/S0168-6445(03)00055-X. [DOI] [PubMed] [Google Scholar]
- 26. Maresso AW, Schneewind O. 2006. Iron acquisition and transport in Staphylococcus aureus. Biometals 19:193–203. 10.1007/s10534-005-4863-7. [DOI] [PubMed] [Google Scholar]
- 27. Bronner S, Monteil H, Prévost G. 2004. Regulation of virulence determinants in Staphylococcus aureus: complexity and applications. FEMS Microbiol. Rev. 28:183–200. 10.1016/j.femsre.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 28. Felden B, Vandenesch F, Bouloc P, Romby P. 2011. The Staphylococcus aureus RNome and its commitment to virulence. PLoS Pathog. 7:e1002006. 10.1371/journal.ppat.1002006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grundy FJ, Henkin TM. 2003. The T box and S box transcription termination control systems. Front. Biosci. 8:d20–d31. 10.2741/908. [DOI] [PubMed] [Google Scholar]
- 30. Potempa J, Pike RN. 2009. Corruption of innate immunity by bacterial proteases. J. Innate Immun. 1:70–87. 10.1159/000181144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Balemans W, Vranckx L, Lounis N, Pop O, Guillemont J, Vergauwen K, Mol S, Gilissen R, Motte M, Lançois D, De Bolle M, Bonroy K, Lill H, Andries K, Bald D, Koul A. 2012. Novel antibiotics targeting respiratory ATP synthesis in Gram-positive pathogenic bacteria. Antimicrob. Agents Chemother. 56:4131–4139. 10.1128/AAC.00273-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cotter PD, Hill C. 2003. Surviving the acid test: responses of gram-positive bacteria to low pH. Microbiol. Mol. Biol. Rev. 67:429–453. 10.1128/MMBR.67.3.429-453.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mills CD. 2012. M1 and M2 Macrophages: Oracles of health and disease. Crit. Rev. Immunol. 32:463–488. 10.1615/CritRevImmunol.v32.i6.10. [DOI] [PubMed] [Google Scholar]
- 34. Das P, Lahiri A, Lahiri A, Chakravortty D. 2010. Modulation of the arginase pathway in the context of microbial pathogenesis: a metabolic enzyme moonlighting as an immune modulator. PLoS Pathog. 6:e1000899. 10.1371/journal.ppat.1000899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gentry D, Li T, Rosenberg M, McDevitt D. 2000. The rel gene is essential for in vitro growth of Staphylococcus aureus. J. Bacteriol. 182:4995–4997. 10.1128/JB.182.17.4995-4997.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gao W, Chua K, Davies JK, Newton HJ, Seemann T, Harrison PF, Holmes NE, Rhee HW, Hong JI, Hartland EL, Stinear TP, Howden BP. 2010. Two novel point mutations in clinical Staphylococcus aureus reduce linezolid susceptibility and switch on the stringent response to promote persistent infection. PLoS Pathog. 6:e1000944. 10.1371/journal.ppat.1000944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alonzo F, III, Benson MA, Chen J, Novick RP, Shopsin B, Torres VJ. 2012. Staphylococcus aureus leucocidin ED contributes to systemic infection by targeting neutrophils and promoting bacterial growth in vivo. Mol. Microbiol. 83:423–435. 10.1111/j.1365-2958.2011.07942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bubeck Wardenburg J, Bae T, Otto M, Deleo FR, Schneewind O. 2007. Poring over pores: alpha-hemolysin and Pantón-Valentine leukocidin in Staphylococcus aureus pneumonia. Nat. Med. 13:1405–1406. 10.1038/nm1207-1405. [DOI] [PubMed] [Google Scholar]
- 39. Kennedy AD, Bubeck Wardenburg J, Gardner DJ, Long D, Whitney AR, Braughton KR, Schneewind O, DeLeo FR. 2010. Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J. Infect. Dis. 202:1050–1058. 10.1086/656043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kielian T, Cheung A, Hickey WF. 2001. Diminished virulence of an alpha-toxin mutant of Staphylococcus aureus in experimental brain abscesses. Infect. Immun. 69:6902–6911. 10.1128/IAI.69.11.6902-6911.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shopsin B, Drlica-Wagner A, Mathema B, Adhikari RP, Kreiswirth BN, Novick RP. 2008. Prevalence of agr dysfunction among colonizing Staphylococcus aureus strains. J. Infect. Dis. 198:1171–1174. 10.1086/592051. [DOI] [PubMed] [Google Scholar]
- 42. Heinrichs JH, Bayer MG, Cheung AL. 1996. Characterization of the sar locus and its interaction with agr in Staphylococcus aureus. J. Bacteriol. 178:418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dubrac S, Msadek T. 2004. Identification of genes controlled by the essential YycG/YycF two-component system of Staphylococcus aureus. J. Bacteriol. 186:1175–1181. 10.1128/JB.186.4.1175-1181.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Storz G, Vogel J, Wassarman KM. 2011. Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell 43:880–891. 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barciszewski J, Volker A. 2003. Noncoding RNAs: molecular biology and molecular medicine. Kluwer Academic/Plenum Publishers, New York, NY. [Google Scholar]
- 46. Hurdle JG, O’Neill AJ, Chopra I. 2005. Prospects for aminoacyl-tRNA synthetase inhibitors as new antimicrobial agents. Antimicrob. Agents Chemother. 49:4821–4823. 10.1128/AAC.49.12.4821-4833.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Parveen N, Cornell KA. 2011. Methylthioadenosine/S-adenosylhomocysteine nucleosidase, a critical enzyme for bacterial metabolism. Mol. Microbiol. 79:7–20. 10.1111/j.1365-2958.2010.07455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bao Y, Li Y, Jiang Q, Zhao L, Xue T, Hu B, Sun B. 2013. Methylthioadenosine/S-adenosylhomocysteine nucleosidase (Pfs) of Staphylococcus aureus is essential for the virulence independent of LuxS/AI-2 system. Int. J. Med. Microbiol. 303:190–200. 10.1016/j.ijmm.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 49. Proctor RA, von Eiff C, Kahl BC, Becker K, McNamara P, Herrmann M, Peters G. 2006. Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat. Rev. Microbiol. 4:295–305. 10.1038/nrmicro1384. [DOI] [PubMed] [Google Scholar]
- 50. Tuchscherr L, Medina E, Hussain M, Völker W, Heitmann V, Niemann S, Holzinger D, Roth J, Proctor RA, Becker K, Peters G, Löffler B. 2011. Staphylococcus aureus phenotype switching: an effective bacterial strategy to escape host immune response and establish a chronic infection. EMBO Mol. Med. 3:129–141. 10.1002/emmm.201000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and psi-blast: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5:621–628. 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 53. Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Anderson MJ. 2001. A new method for non-parametric multivariate analysis of variance. Austral. Ecol. 26:32–46. 10.1111/j.1442-9993.2001.01070.pp.x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Rank-ordered distributions of S. aureus gene expression levels under in vitro and in vivo conditions. Download
Visualization of the global expression of S. aureus during in vitro growth (EX) and AC and CH phases of infection. Download
Summary of data filtering for replicated RNA-Seq experiments.
Raw, RPKM, and resampled transcript numbers from S. aureus during EX (EX1 to -3), AC (AC1 to -3), and CH (CH1 to -3) conditions.
Comparison of global gene expression profiles between conditions using permutational multivariate analysis of variance (PERMANOVA).
Transcript abundances of genes expressed by S. aureus during in vitro growth (EX) and AC and CH phases of infection.
Transcript abundances of genes encoding two-component systems in S. aureus during EX in vitro growth or in vivo AC and CH infection.
The top-15-ranked (most highly expressed) noncoding regions in S. aureus during in vitro growth (EX) and AC and CH phases of infection.
Description of human samples.
Primers used for RT-PCR in human samples.