

ABSTRACT
Phylodynamic analysis of genome-wide single-nucleotide polymorphism (SNP) data is a powerful tool to investigate underlying evolutionary processes of bacterial epidemics. The method was applied to investigate a collection of 65 clinical and environmental isolates of Vibrio cholerae from Haiti collected between 2010 and 2012. Characterization of isolates recovered from environmental samples identified a total of four toxigenic V. cholerae O1 isolates, four non-O1/O139 isolates, and a novel nontoxigenic V. cholerae O1 isolate with the classical tcpA gene. Phylogenies of strains were inferred from genome-wide SNPs using coalescent-based demographic models within a Bayesian framework. A close phylogenetic relationship between clinical and environmental toxigenic V. cholerae O1 strains was observed. As cholera spread throughout Haiti between October 2010 and August 2012, the population size initially increased and then fluctuated over time. Selection analysis along internal branches of the phylogeny showed a steady accumulation of synonymous substitutions and a progressive increase of nonsynonymous substitutions over time, suggesting diversification likely was driven by positive selection. Short-term accumulation of nonsynonymous substitutions driven by selection may have significant implications for virulence, transmission dynamics, and even vaccine efficacy.
IMPORTANCE
Cholera, a dehydrating diarrheal disease caused by toxigenic strains of the bacterium Vibrio cholerae, emerged in 2010 in Haiti, a country where there were no available records on cholera over the past 100 years. While devastating in terms of morbidity and mortality, the outbreak provided a unique opportunity to study the evolutionary dynamics of V. cholerae and its environmental presence. The present study expands on previous work and provides an in-depth phylodynamic analysis inferred from genome-wide single nucleotide polymorphisms of clinical and environmental strains from dispersed geographic settings in Haiti over a 2-year period. Our results indicate that even during such a short time scale, V. cholerae in Haiti has undergone evolution and diversification driven by positive selection, which may have implications for understanding the global clinical and epidemiological patterns of the disease. Furthermore, the continued presence of the epidemic strain in Haitian aquatic environments has implications for transmission.
INTRODUCTION
Vibrio cholerae is one of the oldest and well-recognized pathogens of humans, yet there is much to be learned regarding transmission and evolution of cholera, the disease for which it is the causative agent. The interplay between person-to-person and environmental transmission remains to be explained in full detail, especially in the context of the evolutionary response of the bacterium to environmental and host-driven selective pressures. Mechanisms for these interactions have been proposed with variable viewpoints (1–3).
In October 2010, a case of cholera caused by the toxigenic Vibrio cholerae O1 El Tor biotype was reported in Mirebalais, Haiti (4), making it the first case in Haiti that was officially identified by U.S. public health authorities since the mid-19th century (5). Pulsed-field electrophoresis (PFGE), variable nucleotide tandem repeat (VNTR), and sequence data identified a clonal relationship consistent with point-source introduction, with the Nepalese garrison on UN peacekeeping duty in the aftermath of the January 2010 earthquake implicated as the putative source (6). As of May 2014, 703,510 cases and 8,562 deaths had been conservatively reported (5). The outbreak strain of V. cholerae has genetic characteristics of a “hybrid” or “altered” V. cholerae strain linked with increased virulence (3, 7). The epidemic provided a unique opportunity to investigate clinical and environmental strains of V. cholerae over time to gain an understanding of the evolutionary dynamics of the microorganism (8, 9). This dynamic was investigated using phylodynamic analysis of microbial genome data in the context of the demographic history of the pathogen (2, 10–12).
Historically, the genetic diversity of V. cholerae has not been recognized, with the first six pandemics ascribed to a classical biotype, with a genetic shift resulting in the El Tor biotype responsible for the ongoing seventh pandemic (13). The emergence of V. cholerae serogroup O139 strains in the early 1990s and “altered” or “hybrid” strains in the last decade, which are believed to be sublineages within a global expansion of the seventh cholera pandemic, demonstrate the ongoing genomic plasticity of this pathogen to yield new and potentially more successful variants through point mutations, DNA rearrangements, and horizontal gene transfer (1, 14). Interestingly, Bayesian molecular clock analysis has shown that bacterial populations, including strains of V. cholerae and methicillin-resistant Staphylococcus aureus, are measurably evolving even over relatively short time scales ranging from a few months to a few years (2, 15–17). However, those underlying factors that drive the emergence and evolution of novel microbial variants remain uncertain. For example, recent studies showed that V. cholerae in aquatic environments is naturally competent and in the presence of chitin is subject to significant selective pressure (13, 18). Selection in the human gut, related to acquired immunity, has also been considered an evolutionary driver, specifically in the shift from the Inaba to Ogawa serotypes observed in 2012 in the Artibonite region of Haiti (19). Also, a conceptual framework has been proposed in which V. cholerae evolution is driven by shift/drift cycles. This posits that transition between the sixth and seventh pandemic strains resulted from accumulation of base pair mutations (shift), while transition among V. cholerae O1 strains during the current pandemic arose from short-term changes with horizontal acquisition of genomic islands (drift) (1).
In this study, we observed short-term accumulation of nucleotide substitutions in the V. cholerae genome that may have a more significant evolutionary impact than previously considered. Throughout the first 2 years of the epidemic in Haiti, it appears V. cholerae diversified, accumulating a significant number of genomic polymorphisms driven by an increasing frequency of nonsynonymous mutations along the lineages successfully propagating over time. Furthermore, the environment is concluded to serve as a reservoir of diverse V. cholerae strains in Haitian waterways, including toxigenic V. cholerae O1 El Tor and non-O1/O139 strains and a novel nontoxigenic O1 El Tor-like strain.
RESULTS
A total of 28 clinical isolates and nine environmental isolates were recovered from 2010 to 2012 in this study (see Table S1 in the supplemental material). The 37 isolates were confirmed using serology, the mismatch amplification mutation assay (MAMA), and PCR for ompW and virulence genes toxR, tcpA, ctxA, rstA, and rstC (20). Among the nine environmental isolates that were characterized, four were toxigenic V. cholerae O1 strains, one was a nontoxigenic serogroup O1 strain, and four were non-O1/O139 strains. Whole genome sequences (WGS) were determined, and an additional 28 WGS previously reported by Katz et al. (9) were downloaded from the Sequence Read Archive (SRA) database for a total of 56 clinical V. cholerae O1 isolates from four Haitian regions: Artibonite, Ouest, Sud-Est, and Sud (see Fig. S1 in the supplemental material). After sequencing using the Illumina HiSeq 2000 platform, the reads were mapped to V. cholerae 2010EL-1786, a closed reference genome (GenBank accession no. NC_016445.1 and NC_016446.1) representing the early 2010 Haitian clone. De novo assemblies of all WGS were constructed.
Phylogenetic relationships between clinical and environmental V. cholerae isolates.
Monthly environmental surveillance for V. cholerae at selected stations in waterways of the Ouest region was initiated in April 2010. An initial maximum likelihood (ML) phylogenetic analysis of the 56 clinical and 4 environmental toxigenic V. cholerae O1 biotype El Tor strains indicated a close relationship with the Haitian epidemic 2010EL-1786 closed genome (see Fig. S2 in the supplemental material). Pairwise high-quality SNP (hqSNP) differences between the 4 environmental and 13 clinical toxigenic V. cholerae O1 strains isolated in 2012 ranged from 0 to 11 (median of 4).
The relationship between Haitian and historical V. cholerae strains is shown in the global phylogeny (Fig. 1), including de novo assembled genomes from the five environmental isolates (four non-O1/O139 isolates and one nontoxigenic O1 isolate) and a representative sample of published V. cholerae O1 and non-O1 strains (9, 21). This phylogeny also includes an environmental strain recently isolated in Haiti (2012EL-1759) that is highly similar to the O395 strain. Given the significant diversity between V. cholerae O1 and non-O1 genomes, the 60 closely related toxigenic V. cholerae O1 biotype El Tor strains from our study were collapsed and are represented in the phylogeny by the 2010EL-1786 reference sequence (Fig. 1), which closely clusters with other members of the El Tor biotype (N16961 Bangladesh, 1975 and CIRS101 Bangladesh, 2002). The four V. cholerae non-O1/O139 strains were included in the global phylogeny, suggesting a panmictic population of V. cholerae non-O1/O139 lineages present in the aquatic environment of Haiti. One of the isolates was nontoxigenic (ctx negative) V. cholerae O1 2012Env-9, a ctx-negative classical tcpA-positive strain that clustered basally to the V. cholerae O395 and O1 El Tor strains. Moreover, only 90.9% of 10 million reads mapped to the 2010EL-1786 genome, compared to an average of 96.52% among other toxigenic clinical and environmental isolates. Mapping to the reference strain confirmed the absence of the ctx phage in 2012Env-9.
FIG 1 .

Maximum likelihood (ML) tree of V. cholerae environmental and clinical strains. The ML tree was inferred from ProgressiveMauve alignment of four environmentally sampled non-O1/O139 isolate sequences (designated by green coloring and marked with an asterisk), one environmentally sampled nontoxigenic O1 sequence, and 27 reference sequences. The 60 closely related toxigenic O1 biotype El Tor strains from our study are collapsed and represented by the 2010EL-1786 reference strain, which clusters with other members of the El Tor lineage. The four non-O1/O139 strains were dispersed throughout the global phylogeny, indicating a panmictic population of non-O1/O139 strains in the environment. The phylogeny was constructed with RAxML, using the GTR nucleotide substitution model and 1,000 bootstrap replicates. Bootstrap values for branches are not indicated but were all above 90%.
Phylodynamics of toxigenic V. cholerae O1 strains in Haiti.
The 56 clinical and four environmental toxigenic V. cholerae O1 isolates were further analyzed by phylodynamic methods. A total of 68 genome-wide high-quality single nucleotide polymorphisms (hqSNPs) were identified (see Table S2 in the supplemental material), and pairwise hqSNP differences were compared (see Table S3 in the supplemental material). Most (59/68 [87%]) of the hqSNPs occurred in coding regions and included 33 nonsynonymous changes. As expected, the nucleotide diversity among clinical isolates collected in 2010, shortly after the first outbreak strain was isolated, was relatively low (0.011 nucleotide substitution/hqSNP site) but was already nine times larger (0.098 nucleotide substitution/hqSNP site) in 2011. Diversity decreased slightly in 2012 (0.073 nucleotide substitution/hqSNP site), although it was seven times larger than at the beginning of the epidemic (Table 1). Overall, the evolutionary rate (i.e., molecular clock) was estimated at 1.67 × 10−6 (95% highest posterior density [HPD], 4.50 × 10−7 to 3.34 × 10−6) hqSNP genome−1 year−1 or ~6.8 (95% HPD, 2 to 14) hqSNP year−1 in the core genome, consistent with previous estimates (2).
TABLE 1 .
Frequency of isolates by year and diversity/divergence estimates
| Yr | Frequency according to: |
Totalb | Diversityc | Divergenced | Within-group hqSNP differencee |
|
|---|---|---|---|---|---|---|
| UF-EPIa | Katz et al. (9) | |||||
| 2010 | 9 | 13 | 22 | 0.011 (0.004) | 0.636 (0.219) | |
| 2011 | 14 | 14 | 0.098 (0.016) | 0.056 (0.008) | 5.868 (0.855) | |
| 2012 | 23 | 1 | 24 | 0.073 (0.018) | 0.058 (0.018) | 3.978 (0.866) |
| Total | 32 | 28 | 60 | |||
The isolates listed were sampled by the University of Florida Emerging Pathogen Institute (UF-EPI).
Four non-O1/O139 isolates are not listed and were not included in the diversity/divergence estimates.
Diversity estimates of the mean evolutionary diversity within year-specific subpopulations. The number of nucleotide substitutions per hqSNP site from mean diversity calculations within subpopulations is shown with the standard error obtained by bootstrap estimates (1,000 replicates) in parentheses.
Divergence estimates and standard errors (in parentheses) of average evolutionary divergence compared to sequence sampled during the first epidemic outbreak in 2010. The number of nucleotide substitutions per hqSNP site from averaging over all sequence pairs within groups is shown.
Mean within-group hqSNP differences and standard errors (in parentheses) by collection year.
Likelihood mapping analysis showed that sequences contained, overall, high phylogenetic signal, and recombination was not detected (22, 23). Therefore, an evolutionary and demographic history of these V. cholerae O1 strains in Haiti could be further studied by analyzing genome-wide hqSNPs with Bayesian coalescent-based models (see Materials and Methods). In agreement with previous findings, V. cholerae adhered to a strict molecular clock, with the time of the most recent common ancestor (TMRCA) dating back to July 2010 (95% HPD, March to October 2010) (see Table S4 in the supplemental material), encompassing the 2010 Nepalese epidemic and the onset of the outbreak in Haiti according to official records (6, 9, 24). Phylogenetic relationships among cholera isolates were represented by a DensiTree showing superimposition of trees in the Bayesian posterior distribution (Fig. 2). DensiTrees allow the depiction of phylogenetic uncertainty and are a powerful alternative to a maximum clade credibility (MCC) or ML tree, which is inferred by heuristic algorithms and may not necessarily represent the most likely evolutionary history of the sampled sequences (25). Despite some variability, tree topologies in the posterior distribution consistently showed each toxigenic environmental V. cholerae O1 strain isolated in 2012 clustering and always branching out from clinical isolates sampled at the same time point (Fig. 2). This demonstrates the close relationship of the clinical and environmental strains. Moreover, the staircase morphology of the V. cholerae O1 Bayesian phylogenies is indicative of measurably evolving populations (17) characterized by expansion, diversification, and bottlenecks. In general, strains sampled at a specific time point are more closely related than strains sampled at a different time point (Fig. 2). Taken together with the change in hqSNP diversity over time (Table 1), the observed pattern is consistent with progressively shifting V. cholerae populations, where microbial strains circulating in a given year are replaced the following year through population bottlenecks. At least two major bottlenecks can be observed in the DensiTree: the first one leading to the replacement of the homogeneous variants circulating in 2010 with a more heterogeneous population in 2011 and the second one from 2010 and 2011 to 2012, with 19 of 24 (79.2%) strains isolated in 2012 located within the same clade with high posterior support (Fig. 2). A similar topological feature was observed in genealogies of seasonal epidemics of fast-evolving viruses, such as influenza virus (12), and Mutreja et al. in 2011 described a staircase topology marking transitions between cholera pandemic waves that have occurred since the 1960s (2). Here, however, we found an analogous evolutionary pattern within a localized epidemic over a short 2-year time span.
FIG 2 .

DensiTree of posterior distribution of trees from Bayesian phylogenetic analysis of 60 V. cholerae O1 isolates. A posterior distribution of trees was obtained from Bayesian phylogenetic analysis of genome-wide SNPs from 60 toxigenic V. cholerae O1 isolates using the GMRF skygrid model and strict molecular clock as implemented in BEAST 1.8.0. Tip dates for each node were assigned based on date of isolate collection, allowing the phylogeny to be scaled in time. DensiTree provides a visualization of the posterior distribution of trees by illustrating the frequency of node clustering to assess the support for clades and overall topology. Well-supported branches are indicated by solid colors, whereas webs represent little agreement. This is an alternative to presenting one “best” MCC tree.
The demographic history of V. cholerae during the Haitian epidemic was also studied by investigating changes in microbial effective population size (Ne)—a measure of genetic diversity representing the number of genomes effectively contributing to the next generation. Daily case counts for the Ouest Department (excluding Port-au-Prince), where a large proportion of isolates were collected, were obtained from daily reports to the Haitian Ministry of Public Health and Population (26). Aggregated incidence data were then covisualized with a Gaussian Markov random field (GMRF) skygrid plot showing Ne trajectory over time (Fig. 3, top panel) (25). The GMRF skygrid yielded the best-fitting model, based on Bayes factor comparison of marginal likelihood estimates from path sampling and stepping-stone analysis implemented in BEAST v. 1.8.0 (see Table S4 in the supplemental material) (27–29). By using epidemiological data, the V. cholerae population demographic history may be described as a point source introduction of a strain followed by epidemic spread. The skygrid plot shows an initial peak in Ne occurring during October 2010 to January 2011, leveling off as the epidemic progressed. Changes in Ne are reflected by epidemiological data showing an initial exponential increase in reported cases for the Ouest Department followed by stabilization and episodic fluctuation (Fig. 3, bottom panel).
FIG 3 .

Effective population size (Ne) estimates from Bayesian phylogenetic analysis and epidemiological case counts for the coinciding period. The blue line and the gray upper and lower bounds represent, respectively, median and 95% high posterior density (95% HPD) interval estimates of Ne over time. The bottom panel shows the number of cumulative (shaded area) and incident (histogram) cholera cases from October 2010 to June 2013.
Analysis of toxigenic V. cholerae O1 selection.
The progressive emergence of new V. cholerae lineages during the first 2 years of the Haitian epidemic may be the result of random (neutral) genetic drift or positive selection. Previous studies showed that acquisition of the STX family of antibiotic resistance elements shaped the topology of the pandemic spread of cholera over the last six decades (2). Bayes empirical Bayes (BEB) analysis implemented in PAML identified 2 amino acid changes—a glycine-to-cysteine replacement in the gene coding for prolyl-tRNA synthetase and an alanine-to-valine replacement in the gene coding for periplasmic nitrate reductase NapA—as sites of possible positive selection (BEB posterior probabilities of 76.2% and 76.3%, respectively), although by comparing different ML-based branch-site selection models, the hypothesis of neutrality could not be rejected (30, 31). Given the limitations of these tests when applied to population data (32), we investigated further the accumulation of synonymous (dS) and nonsynonymous (dN) substitutions per hqSNP site along different subsets of internal branches of V. cholerae O1 Bayesian MCC trees (see Materials and Methods). In staircase phylogenies, in particular, backbone paths can be defined (one such path is shown by the highlighted branches of the MCC tree in Fig. 4A), representing major lineages propagating through time from root node to most recently sampled sequences. That is, along a backbone path, each ancestral node gives rise to a daughter lineage that quickly stops propagating and to another ancestor from which all subsequent lineages emerge. External branches were excluded from the analysis to avoid the confounding factor of transient polymorphisms segregating on lineages not successfully propagating through time (33). Accumulation of dN and dS substitutions along backbone paths showed marked differences (Fig. 4B). Throughout the epidemic, dS values continued to remain steady over time—not surprisingly, since the dS substitution rate is expected to be proportional to the neutral mutation rate (34). As the incidence of cholera cases decreased throughout 2011, dN changes kept increasing, and a significantly greater number of dN than dS substitutions was consistently observed (P < 0.001), characteristic of a heterogeneous microbial population undergoing emergence and subsequent fixation of variants driven by positive selection (34). This pattern was also observed when dN and dS values were assessed along all internal branches (see Fig. S3 in the supplemental material). Interestingly, ML reconstruction of ancestral sequences in the MCC tree showed several amino acid substitutions along backbone paths that marked population bottleneck events between 2010 and 2011 and 2012 (Fig. 4C) (35). Potential fitness contributions of the observed amino acid changes were evaluated by reviewing available V. cholerae protein structures in the Protein Data Bank. The amino acid change in the gene coding for thiol:disulfide interchange protein DbsA (TcpG) (4C branch 7) was the only one mapping on a protein with known structure. This protein plays an important role in the secretion of virulence factors (36, 37). We observed that the alanine-to-threonine substitution occurs in a β-sheet near the hydrophobic binding pocket (Fig. 4). While it is difficult without subsequent analysis to determine whether the change in charge and size at this position alters the ability to fold disulfide-bonded proteins and potentially affects fitness in vivo, it is known that inactivation of this gene results in avirulent mutants (36).
FIG 4 .
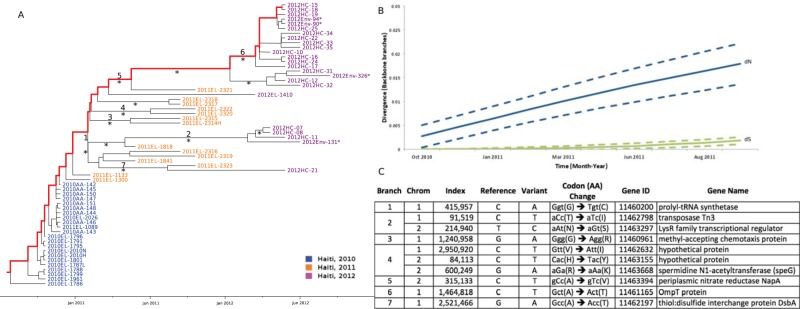
Bayesian MCC phylogeny and selection analysis along backbone paths. (A) Bayesian maximum clade credibility (MCC) tree of Haitian strains of V. cholerae with branch lengths scaled in time by enforcing a strict molecular clock. Strain labels are colored to indicate the time of sampling according to the legend in the figure. An asterisk along a branch marks subtending clades supported by a posterior probability of >0.75. An asterisk next to a node sequence name indicates an environmentally sampled isolate. One of the possible backbone paths, representing major lineages propagating over time from root node to most recently sampled sequence, is highlighted in red. Branch labels correspond to nonsynonymous mutations indicated in the table on panel C. (B) Mean nonsynonymous (dN) and synonymous (dS) divergence along backbone paths in a subsample of trees from the posterior distribution (see Materials and Methods) during the cholera epidemic in Haiti are represented by blue and green lines, respectively. Estimates 1 standard deviation above and below the mean dN or dS are represented by broken lines. The y axis represents the number of nucleotide (dN or dS) changes per (synonymous or nonsynonymous) SNP site. The x axis matches the time scale of the epidemic. Estimates were obtained by reconstructing and comparing ancestral sequences along V. cholerae trees sampled from the posterior distribution from the Bayesian analysis. (C) Inferred codon and amino acid changes in the V. cholerae genome (numbered according to the V. cholerae 2010EL-1786 reference strain) along internal branches of the O1 Haitian phylogeny. Branch numbers correspond to numbered branches in the MCC tree given in panel A.
DISCUSSION
In-depth characterization of the phylodynamic patterns of Haitian clinical and environmental V. cholerae isolated between October 2010 and August 2012 resulted in three findings. First, we observed diversity in both environmental nontoxigenic and toxigenic V. cholerae O1 strains in the Haitian aquatic environment, the latter showing a close phylogenetic relationship between these clinical and environmental strains. Second, during the short time span of the ongoing Haitian epidemic, V. cholerae O1 comprises a measurably evolving population characterized by genetic diversification (17). Last, as the epidemic progressed, a differential accumulation of dN and dS mutations indicative of positive selection was observed, a result that may have significant implications for understanding the evolutionary processes of V. cholerae.
Whole-genome hqSNP analysis showed that toxigenic V. cholerae O1 strains isolated from water samples collected in 2012 were highly related to clinical isolates sampled that year. The shared ancestry of clinical and environmental isolates was also evident in the phylogenetic tree, where environmental strains always originated from clinical lineages. Environmental samples collected in 2013 at the same sites yielded additional toxigenic V. cholerae O1 strains in the presence of low numbers of fecal coliforms (e.g., 80 CFU/100 ml) (38). Thus, the fact that V. cholerae, a natural inhabitant of aquatic ecosystems, is present in Haitian coastal, estuarine, and river systems makes it critical to undertake extensive monitoring of Vibrio populations (39).
Results from studies done in Bangladesh using VNTR typing have suggested clinical and environmental strains comprise distinct populations but with different V. cholerae populations detectible in different villages (40). The four toxigenic V. cholerae O1 Haitian environmental isolates in this study were found to be significantly similar to the clinical isolates. These findings indicate that there is an environmental presence of toxigenic V. cholerae O1 in the aquatic ecosystem of Haiti. This creates a dilemma since models for elimination of epidemic cholera in Haiti do not consider the environment as a reservoir of toxigenic V. cholerae O1 or as an environmental source of this pathogen (41). Infected individuals very likely continue to seed the aquatic environment, but by ignoring the aquatic environmental habitat of V. cholerae, this mode of transmission as a significant contributor to the ongoing epidemic will be inappropriately discounted.
Phylogenic analysis showed each environmental isolate branching from a different clinical isolate sampled at the same time point. However, it is not possible to determine whether mutations of clinical and environmental strains mostly occurred intrahost during infection or during subsequent replication cycles after release into the environment. Analyses of additional environmental and clinical isolates, collected longitudinally from different geographic areas, will help to further elucidate the transmission and population dynamics of these two components of the epidemic. In this study, we conclude V. cholerae non-O1/O139 is most likely naturally occurring in the Haitian aquatic environment. Interestingly, one of the strains (2012Env-32) is located on a monophyletic clade with the V. cholerae cp1110 O75 strain responsible for the 2010-to-2011 oyster-borne outbreak in Florida (Fig. 1) (42). Furthermore, a nontoxigenic (ctx ϕ-negative) V. cholerae O1 strain possessing the classical tcpA gene (2012Env-9) was also isolated, demonstrating the diversity of V. cholerae strains in Haiti. The diversity of strains and continued interaction with the microbiome of resident Haitian human with environmental bacteria and bacteriophages may give rise to further advantageous mutations or acquisition of plasmids coding for drug resistance, as there is some evidence that this has already occurred. A recent study identified a Haitian epidemic isolate that gained multidrug resistance through acquisition of an IncA/C plasmid that may have been derived from a species of Enterobacteriaceae (43). In the present study, we emphasize the impact of point mutations on the V. cholerae genome by illustrating an increasing accumulation of nucleotide diversity over the course of the epidemic, mainly driven by nonsynonymous substitutions.
Comparison of genomes of strains isolated during the first 2 years of the epidemic showed a progressive accumulation of polymorphisms among strains sampled during different time periods, characteristic of population expansion and diversification. In particular, toxigenic V. cholerae O1 strains isolated during 2010 represent a rather homogenous, compartmentalized, but not identical population, with sparse genomic variation and low genetic diversity. In 2011, a period of expansion and wider diversification occurred followed by an observed population bottleneck, with diversity decreasing between 2011 and 2012. This bottleneck was also reflected by a slight decrease in both Ne and reported case counts from March to April 2012 (Fig. 3).
To determine the factors driving evolution of V. cholerae, selection among toxigenic V. cholerae O1 variants was investigated. Such studies have employed assessment of dN/dS substitution ratios to investigate selection pressure driving the emergence and fixation of new genetic lineages (32). However, when applied to intraspecies (single population) data, dN/dS values tend to be insensitive to selection coefficients, and ratios of less than 1 are not necessarily a reliable indicator of negative selection (32). Moreover, transient polymorphisms segregating along external branches of a genealogy may bias dN/dS estimates toward values greater than 1, even in the absence of positive selection (33). To avoid these confounders, we used an alternative approach to estimate absolute rates of dN and dS substitutions for the internal branches and the backbone paths of the Bayesian phylogenies, which represent polymorphisms along successfully propagating lineages that tend to be fixed over time (33, 44). The analysis showed that at the putative onset of the epidemic in October 2010, evolution of V. cholerae was characterized by neutral genetic drift. Emergence and diversification of microbial variants in 2011 was marked by a progressive increase of nonsynonymous mutations and a steady number of synonymous mutations, as would be expected under positive selection.
Amino acid changes at locations of nonsynonymous mutations occurring along internal branches of the phylogeny between 2010 and 2011 and 2012 strains were further assessed for biological significance. However, the protein structures available for V. cholerae or homologs among other bacteria are limited. In one instance, it was possible to map an amino acid change to a known protein structure, the thiol:disulfide interchange protein, that can impact virulence. Prolyl-tRNA synthetase has also been identified as a putatively essential gene for V. cholerae, and it is likely that the LysR family transcriptional regulator plays a role in regulating the heme transport gene hutA (45). Overall, our results show that most of the amino acid replacements along the internal branches of the Haitian cholera phylogeny have been accumulating at a rate faster than would be expected with neutral genetic drift, implying an effect on fitness and warranting further analysis of these mutations to understand their biological and evolutionary impact.
Other limits must be kept in mind. First, genomic variation was assessed based only on hqSNPs, which restricted selection analysis to those sites. Insertions and deletions were also excluded, and only those genomic regions conserved in all isolates were considered. Therefore, more complex genomic rearrangements or horizontal gene transfer may also play a role in the evolutionary dynamics of these Haitian strains. Furthermore, the 2012 microbial population bottleneck and the evolutionary and demographic dynamics inferred from the V. cholerae genealogies of the isolates included in this study may not fully be representative of the Haitian populations of V. cholerae.
In conclusion, this phylodynamic analysis of Haitian V. cholerae isolates, the most extensive to date, shows that during the 2 years after the first report of cholera in Hispaniola, the Haitian V. cholerae O1 isolates examined in this study demonstrate measurable diversity. It is postulated that evolution through a progressive accumulation of nucleotide changes is occurring, driven mainly by positive selection. This phylogenetic information may prove useful for devising strategies to prevent and control cholera in Haiti (46). As cases of cholera in Haiti continue to occur, selection and diversity of both clinical and environmental V. cholerae isolates should be monitored for their potential influence on virulence, transmission dynamics, and vaccination strategy.
MATERIALS AND METHODS
Clinical and environmental sampling of V. cholera.
Thirty-two toxigenic V. cholerae O1 El Tor strains were isolated in Haiti during 2010 and 2012 (see Table S1 in the supplemental material). Nine of the V. cholerae O1 El Tor strains were isolated from patients at the St. Mark Hospital, Haiti, in 2010 as described previously (7). Nineteen of the strains isolated in 2012 were from stool samples from suspected cholera patients admitted to the Gressier Cholera Treatment Center. These samples were transported to the University of Florida Public Health Laboratory housed in the Christianville Foundation (a U.S.-based nongovernmental organization) in Haiti. Four toxigenic V. cholerae O1 El Tor strains, four V. cholerae non-O1/O139 strains, and a single nontoxigenic V. cholerae O1 strain were isolated from water samples collected at 15 stations comprising an environmental program for surveillance of selected rivers and estuaries in the Gressier region of Haiti, details of which are provided elsewhere (38). Samples were enriched in alkaline peptone water (APW) and plated onto thiosulfate-citrate-bile salts-sucrose (TCBS) agar. Suspected toxigenic V. cholerae colonies were confirmed using serology and PCR assays, as described previously (7). A total of 28 clinical isolates from Katz et al. were also included in the analyses (9). WGS data were downloaded from the SRA database. Figure S1 in the supplemental material shows the sampling locations where the isolates for this study were obtained.
Molecular analysis and hqSNP discovery.
Each of the V. cholerae isolates was subcultured from glycerol stock medium to L agar and incubated overnight at 37°C. A single colony was selected, transported to 3 ml L broth, and incubated overnight at 37°C with shaking (250 rpm). Genomic DNA (gDNA) was isolated from the pelleted bacteria using the Roche High Pure PCR kit (Roche Applied Science, Indianapolis, IN). The quality of the gDNA was determined by gel electrophoresis, and the quantity was determined using the Nanodrop 2000 (Fisher ND-2000), after which 5 µg of gDNA was sequenced on the Illumina HiSeq 2000 platform (Johns Hopkins University Center for Inherited Disease Research [CIDR]). PCR analysis of virulence genes (ompW, toxR, tcpA, ctxA, rstA, and rstC) was also done using previously published primers (20). MAMA-PCR was used to further discriminate between the classical (CL) and El Tor (ET) ctxB genes by detecting a polymorphism in nucleotide position 203 of the ctxB gene using a conserved forward primer (Fw-con, 5′ ACTATCTTCAGCATATGCACATGG 3′) and two allele-specific polymorphism detection primers, Rv-cla (5′ CCTGGTACTTCTACTTGAAACG 3′) and Rv-elt (5′ CCTGGTACTTCTACTTGAAACA 3′).
Genome-wide hqSNPs were identified using an optimized analysis pipeline implemented in the Galaxy platform (see Fig. S5 in the supplemental material) (47). After demultiplexing, single FASTQ output files of raw reads were filtered by length threshold of 30 and quality threshold of 30 using Sickle v. 1.2. Filtered FASTQ files were mapped to the V. cholerae 2010EL-1786 closed reference genome (accession no. NC_016445.1 and NC_016446.1) with Bowtie 2 v. 2.2.3 short read alignment software using a maximum insert size of 1,200, specifying read group, library, platform, and sample name and with default mapping settings to produce BAM files (48). Base quality scores were recalibrated, and realignment around insertions and deletions was performed using the Genome Analysis Toolkit v. 3.1.1, a framework for analyzing next-generation sequence data, using default settings (49). SNPs were called using FreeBayes Bayesian genetic variant detector v. 0.9.14 for haploid organisms (“ploidy 1”) by left aligning the indels (“left-align-indels”), requiring a minimum base quality of 20 (“min-base-quality”) and a minimum alternate fraction of 0.75 (“min-alternate-fraction”) (50). Putative SNPs were initially filtered by depth of coverage (<10), quality (<20), and genotype likelihood (<10) using VCFtools v. 0.1.10. SNP locations not conserved across all samples (ambiguous sites) were removed. The remaining hqSNPs were assessed and manually curated, based on mapping quality and distance from neighboring SNPs. FASTA alignments were generated for subsequent phylodynamic analysis. SnpEFF v. 3.6 was used to annotate hqSNPs specifically falling within genes using default settings (51). De novo assemblies were also constructed using the Velvet optimizer script, and contigs were ordered to the 2010EL-1786 reference using Mauve v. 2.3.1 (52, 53).
Phylogenetic analysis.
To assess phylogenetic relationships among the 60 V. cholerae O1 El Tor strains (56 clinical and 4 environmental), 68 genome-wide hqSNPs were used to estimate genetic distances (p-distances and distances estimated by maximum likelihood composite), and standard errors evaluated by bootstrapping (1,000 replicates), using the program MEGA 6 (54). Phylogenetic signal and potential recombination were tested by likelihood mapping analysis using TREEPUZZLE v. 5.2 using default settings (23, 55) and the pairwise homoplasy index (PHI) test for recombination using SplitsTree v. 4.10 using default settings (22, 56), respectively. For phylogenetic analyses, the HKY nucleotide substitution model was chosen as the best-fitting model by using the hierarchical likelihood ratio test as described by Swofford and Sullivan (57). Statistical support for internal branches of the ML trees was evaluated by bootstrapping (1,000 replicates) and the ML-based zero-branch-length test (57, 58). ML analyses were carried out using the software PAUP* v. 4b10 and RAxML v. 8.0 using the GTR + Γ nucleotide substitution model, ascertainment bias correction, and 1,000 bootstrap replicates.
To assess the global phylogeny of environmental and clinical V. cholerae isolates, a multiple alignment of de novo assemblies from environmental O1 (n = 1) and non-O1/O139 (n = 4) genomes and 21 representative reference genomes was constructed using ProgressiveMauve (59). Contigs or closed genomes for reference strains were downloaded from the National Center for Biotechnology Information GenBank database (Table 1). The 2010EL-1786 genome was used to represent the 60 toxigenic V. cholerae O1 El Tor isolates. Orthologous regions of the genome were used for comparison, and all ambiguous sites were removed. ML phylogenetic analysis was performed with RAxML v. 8.0, using the GTR + Γ substitution model with ascertainment bias correction and 1,000 bootstrap replicates.
Phylodynamic analysis.
Different coalescent-based demographic models for the 60 Haitian V. cholerae O1 genomes were evaluated using the Bayesian framework implemented in the program BEAST v. 1.8.0 (60). To depict the relative changes of the bacterial effective population size (Ne) over time, which can be interpreted as a measure of V. cholerae population diversity (the number of genomes effectively contributing to the next generation), five demographic models were compared: constant population size, exponential population size, Bayesian skyline plot (BSP), Bayesian skyride, and Gaussian Markov random field (GMRF) skygrid (25, 61, 62). The BSP assumes a piecewise constant model of Ne within predefined intervals in the tree, while the Bayesian skyride applies a temporal smoothing that does not evoke strong prior assumptions and noisy estimates as observed with the BSP model. The GMRF skygrid model parameterizes Ne and uses a GMRF prior to smooth the trajectory, resulting in improved performance over the BSP and Bayesian skyride. This prior is also independent of the genealogy allowing for better estimation of TMRCA. Katz et al. previously identified exponential growth as the best-fitting demographic model; however, skyride and skygrid models were not tested (21). For each coalescent model, calculations were performed using the HKY nucleotide substitution model and by enforcing either a strict or a relaxed molecular clock (with log-normal distributed rates). Three independent Markov chain Monte Carlo (MCMC) runs were carried out for 750 million generations, with sampling every 75,000 generations, for each model. Posterior probabilities were calculated using the program Tracer v. 1.6 (available from http://beast.bio.ed.ac.uk/), after appropriate 10% burn-in. Proper mixing of the MCMC was determined by visual examination of traces and evolutionarily stable strategy (ESS) values of >200 for each estimated parameter in Tracer v. 1.6. The marginal likelihoods for each model were obtained using path sampling and stepping-stone analyses (28, 63, 64). Marginal likelihood estimates of the different models were compared using Bayes factors, and the GMRF skygrid model enforcing a strict molecular clock was selected as the most appropriate representation of the population demographic history (27, 65, 66).
The posterior distribution of trees was viewed using DensiTree (67). DensiTree allows for visualization of a set of trees within a model space. Well-supported branches are illustrated by solid-colored areas, whereas webs represent little agreement. Maximum clade credibility (MCC) trees were inferred from the posterior distribution of the best-fitting model using the program TreeAnnotator implemented in the BEAST v. 1.8.0 package. Finally, epidemiological data were obtained from case reports in the Ouest Department (minus Port-au-Prince) compiled by the Ministere de la Sante Publique et de la Population (MSPP) for the time period from October 2010 to June 2013 (http://www.mspp.gouv.ht/site/index.php) and assessed in the context of the phylodynamic analysis.
Selection analysis.
The numbers of synonymous (dS) and nonsynonymous (dN) substitutions per hqSNP site along V. cholerae phylogenies were estimated by an empirical extension of the coalescent-based Bayesian molecular clock models (33). A codon alignment was obtained from the annotated hqSNPs for the 60 V. cholerae O1 isolates. A codon model with independent rates for each codon position was run using the GMRF skygrid model with a strict molecular clock implemented in BEAST v. 1.8.0 as previously described. A subsample of 200 trees was randomly selected from the posterior distribution obtained by BEAST and used to reestimate branch lengths proportional to either dS or dN substitutions. Point estimates of dN and dS over time were then plotted according to the method described by Lemey et al. (33). For each clock-like tree, calculations were performed, including all branches, as well as internal or backbone paths only. Backbone paths represent lineages propagating (i.e., effectively surviving) over time from root node to sequences sampled at the last time point. Specific amino acid changes along internal branches of the MCC and ML tree were inferred by ML reconstruction of ancestral sequences using the HKY model with the Baseml program implemented in the PAML v. 4.7 software package (35). Amino acid changes were assessed by visualization using the PyMOL v. 1.7.2 molecular graphics system for V. cholerae protein structures available in the Protein Data Bank (68, 69). Sites under potential positive selection (dN/dS ratio of >1) were further evaluated using a codon-based alignment that included all three codon positions of each hqSNP site in a coding region. Site-specific and branch/site-specific models implemented in the Codeml program of the PAML package were used to detect sites under positive selection (dN/dS ratio of >1), with a Bayes empirical Bayes P value of ≥0.9 indicating significance (30). The codon-based alignment was also tested with the Fast Unconstrained Bayesian Approximation (FUBAR) via an online server (http://www.datamonkey.org) using the MCC Bayesian tree and the HKY nucleotide substitution (70).
Ethics statement.
This study was reviewed by the University of Florida Institutional Review Board and deemed exempt because samples underwent deidentification.
SUPPLEMENTAL MATERIAL
Study sites. Blue dots designate the locations where clinical and environmental samples were collected by the University of Florida Emerging Pathogens Institute. Samples collected during 2010 were from the St. Marc Hospital in the Artibonite Department. Samples obtained during 2012 were from the Ouest Department. Clinical samples were collected from the Gressier Cholera Treatment Center, and environmental strains were isolated from samples collected in the surrounding Gressier region. Samples included in study from Katz et al. (9) are designated by black dots. The inset shows the locations of the departments on a Haiti country map. Download
Maximum likelihood (ML) tree of V. cholerae O1 strains. The ML tree was inferred from 68 genome-wide SNPs among the 60 V. cholerae O1 strains using the GTR nucleotide substitution model and ascertainment bias correction implement in RAxML. Tree was rooted using closely related strains from the Nepalese epidemic as an outgroup. Strain labels were colored to represent the sampling time according to the table in the figure. Branch lengths are scaled in nucleotide substitutions per SNP site according to the bar at the bottom. Values along the branches represent statistical support according to bootstrapping (1,000 replicates). Download
Mean nonsynonymous (dN) and synonymous (dS) divergence over the course of the V. cholerae epidemic in Haiti for internal branches. Blue and green lines represent, respectively, mean nonsynonymous (dN) and synonymous (dS) divergence along internal branches over the course of the V. cholerae epidemic in Haiti. Estimates 1 standard deviation above and below the mean dN or dS are represented by broken lines. The y axis represents the number of nucleotide (dN or dS) changes per (synonymous or nonsynonymous) SNP site. Estimates were obtained by reconstructing and comparing ancestral sequences along V. cholerae trees sampled from the posterior distribution obtained with the Bayesian analysis. Download
Three-dimensional structure of the thiol:disulfide interchange protein. The three-dimensional protein structure of the thiol:disulfide interchange protein DbsA (TcpG) was visualized in the PyMOL molecular graphics system obtained from the Protein Data Bank (accession no. 4DVC). The white circle represents the site of the alanine-to-threonine amino acid substitution (A169T) occurring near a β-sheet in the hydrophobic binding site region. This protein plays an important role in the secretion of virulence factors and represents one example of how point mutations could potentially affect pathogen success. Download
Bioinformatics analysis pipeline of V. cholerae genome-wide SNPs. Raw paired-end Illumina reads in the FASTQ format were filtered by Phred quality score using Sickle (https://github.com/najoshi/sickle). Filtered FASTQ files were then mapped to the V. cholerae 2010EL-1786 closed reference genome (accession no. NC_016445.1 and NC_016446.1) with Bow tie 2 short read alignment software using default settings to produce BAM files (48). Base quality scores were recalibrated, and realignment around insertions and deletions was performed using the Genome Analysis Toolkit, a framework for analyzing next-generation sequence data (49). We called SNPs using the FreeBayes Bayesian genetic variant detector for haploid organisms requiring a minimum base quality of 20 and a minimum alternate fraction of 0.75 (50). Putative SNPs were initially filtered by quality and likelihood using VCFtools, and SNP locations not represented among all samples were removed (71). The remaining high-quality SNPs were then assessed and manually curated based on mapping quality and distance from neighboring SNPs to remove clusters. A FASTA alignment of SNPs was generated for phylogenetic analysis. Finally, SnpEFF was used to annotate SNPs, and codons of synonymous and nonsynonymous mutations were abstracted (51). Selection analysis was conducted by tracing the annotated codons on the V. cholerae tree using the site-specific and branch/site-specific models implemented in PAML (30). The bioinformatics pipeline was constructed using the University of Florida High Performance Computing Center’s local instance of Galaxy (47). Download
List of sequenced cholera strains. Listed are the sample name, site of isolation, source (clinical versus environmental), date of sample, and serogroup. Accession numbers for genomes in the present study are to be determined (TBD).
List of single nucleotide polymorphisms (SNP) identified among the 56 clinical and 4 environmental toxigenic V. cholerae O1 isolates. Listed are the SNP sites, effect (noncoding, coding synonymous, and coding nonsynonymous), and gene location (if coding).
Pairwise SNP differences between the 56 clinical and 4 environmental toxigenic V. cholerae O1 isolates.
Bayesian coalescent estimates inferred from the SNPs of the Haiti V. cholerae O1 strains collected from 2010 to 2012. Shown are comparisons of molecular clock models, demographic models, the times of the most recent common ancestor (TMRCA), and the evolutionary rates for each model.
ACKNOWLEDGMENTS
This work was supported by NIH R01 grant AI097405.
We are grateful to the Genetic Resources Core Facility, Johns Hopkins Institute of Genetic Medicine, Baltimore, MD, for performing full genome sequencing of the V. cholerae isolates and assisting in the analyses.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the Centers for Disease Control and Prevention.
Footnotes
Citation Azarian T, Ali A, Johnson JA, Mohr D, Prosperi M, Veras NM, Jubair M, Strickland SL, Rashid MH, Alam MT, Weppelmann TA, Katz LS, Tarr CL, Colwell RR, Glenn Morris J, Jr, Salemib M. 2014. Phylodynamic analysis of clinical and environmental Vibrio cholerae isolates from Haiti reveals diversification driven by positive selection. mBio 5(6):e01824-14. doi:10.1128/mBio.01824-14.
REFERENCES
- 1. Chun J, Grim CJ, Hasan NA, Lee JH, Choi SY, Haley BJ, Taviani E, Jeon Y-S, Kim DW, Brettin TS, Bruce DC, Challacombe JF, Detter JC, Han CS, Munk AC, Chertkov O, Meincke L, Saunders E, Walters RA, Huq A, Nair GB, Colwell RR. 2009. Comparative genomics reveals mechanism for short-term and long-term clonal transitions in pandemic Vibrio cholerae. Proc. Natl. Acad. Sci. U. S. A. 106:15442–15447. 10.1073/pnas.0907787106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mutreja A, Kim DW, Thomson NR, Connor TR, Lee JH, Kariuki S, Croucher NJ, Choi SY, Harris SR, Lebens M, Niyogi SK, Kim EJ, Ramamurthy T, Chun J, Wood JL, Clemens JD, Czerkinsky C, Nair GB, Holmgren J, Parkhill J, Dougan G. 2011. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature 477:462–465. 10.1038/nature10392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Morris JG. 2011. Cholera—modern pandemic disease of ancient lineage. Emerg. Infect. Dis. 17:2099–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ivers LC, Walton DA. 2012. The “first” case of cholera in Haiti: lessons for global health. Am. J. Trop. Med. Hyg. 86:36–38. 10.4269/ajtmh.2012.11-0435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Centers for Disease Control and Prevention 2014. Travel notice: cholera in Haiti. Centers for Disease Control and Prevention, Atlanta, GA.. [Google Scholar]
- 6. Frerichs RR, Keim PS, Barrais R, Piarroux R. 2012. Nepalese origin of cholera epidemic in Haiti. Clin. Microbiol. Infect. 18:E158–E163. 10.1111/j.1469-0691.2012.03841.x. [DOI] [PubMed] [Google Scholar]
- 7. Ali A, Chen Y, Johnson JA, Redden E, Mayette Y, Rashid MH, Stine OC, Morris JG. 2011. Recent clonal origin of cholera in Haiti. Emerg. Infect. Dis. 17:699–701. 10.3201/eid1704.101973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grad YH, Waldor MK. 2013. Deciphering the origins and tracking the evolution of cholera epidemics with whole-genome-based molecular epidemiology. mBio 4(5):e00670-13. 10.1128/mBio.00670-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Katz LS, Petkau A, Beaulaurier J, Tyler S, Antonova ES, Turnsek MA, Guo Y, Wang S, Paxinos EE, Orata F, Gladney LM, Stroika S, Folster JP, Rowe L, Freeman MM, Knox N, Frace M, Boncy J, Graham M, Hammer BK, Boucher Y, Bashir A, Hanage WP, Van Domselaar G, Tarr CL. 2013. Evolutionary dynamics of Vibrio cholerae O1 following a single-source introduction to Haiti. mBio 4(4):e00398-13. 10.3391/mbi.2013.4.1.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Harris SR, Feil EJ, Holden MT, Quail MA, Nickerson EK, Chantratita N, Gardete S, Tavares A, Day N, Lindsay JA, Edgeworth JD, de Lencastre H, Parkhill J, Peacock SJ, Bentley SD. 2010. Evolution of MRSA during hospital transmission and intercontinental spread. Science 327:469–474. 10.1126/science.1182395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Salemi M, Lamers SL, Yu S, de Oliveira T, Fitch WM, McGrath MS. 2005. Phylodynamic analysis of human immunodeficiency virus type 1 in distinct brain compartments provides a model for the neuropathogenesis of AIDS. J. Virol. 79:11343–11352. 10.1128/JVI.79.17.11343-11352.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grenfell BT, Pybus OG, Gog JR, Wood JL, Daly JM, Mumford JA, Holmes EC. 2004. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 303:327–332. 10.1126/science.1090727. [DOI] [PubMed] [Google Scholar]
- 13. Faruque SM, Mekalanos JJ. 2012. Phage-bacterial interactions in the evolution of toxigenic Vibrio cholerae. Virulence 3:556–565. 10.4161/viru.22351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hasan NA, Choi SY, Eppinger M, Clark PW, Chen A, Alam M, Haley BJ, Taviani E, Hine E, Su Q, Tallon LJ, Prosper JB, Furth K, Hoq MM, Li H, Fraser-Liggett CM, Cravioto A, Huq A, Ravel J, Cebula TA, Colwell RR. 2012. Genomic diversity of 2010 Haitian cholera outbreak strains. Proc. Natl. Acad. Sci. U. S. A. 109:E2010–E2017. 10.1073/pnas.1207359109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gray RR, Tatem AJ, Johnson JA, Alekseyenko AV, Pybus OG, Suchard MA, Salemi M. 2011. Testing spatiotemporal hypothesis of bacterial evolution using methicillin-resistant Staphylococcus aureus ST239 genome-wide data within a Bayesian framework. Mol. Biol. Evol. 28:1593–1603. 10.1093/molbev/msq319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Prosperi M, Veras N, Azarian T, Rathore M, Nolan D, Rand K, Cook RL, Johnson J, Morris JG, Salemi M. 2013. Molecular epidemiology of community-associated methicillin-resistant Staphylococcus aureus in the genomic era: a cross-sectional study. Sci. Rep. 3:1902. 10.1038/srep01902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Drummond AJ, Pybus OG, Rambaut A, Forsberg R, Rodrigo AG. 2003. Measurably evolving populations. Trends Ecol. Evol. 18:481–488. 10.1016/S0169-5347(03)00216-7. [DOI] [Google Scholar]
- 18. Meibom KL, Blokesch M, Dolganov NA, Wu CY, Schoolnik GK. 2005. Chitin induces natural competence in Vibrio cholerae. Science 310:1824–1827. 10.1126/science.1120096. [DOI] [PubMed] [Google Scholar]
- 19. Centers for Disease Control and Prevention 2012. Notes from the field: identification of Vibrio cholerae serogroup O1, serotype Inaba, biotype el Tor strain—Haiti, March 2012. MMWR Morb. Mortal. Wkly. Rep. 61:309. [PubMed] [Google Scholar]
- 20. Alam M, Nusrin S, Islam A, Bhuiyan NA, Rahim N, Delgado G, Morales R, Mendez JL, Navarro A, Gil AI, Watanabe H, Morita M, Nair GB, Cravioto A. 2010. Cholera between 1991 and 1997 in Mexico was associated with infection by classical, El Tor, and El Tor variants of Vibrio cholerae. J. Clin. Microbiol. 48:3666–3674. 10.1128/JCM.00866-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Katz LS, Turnsek M, Kahler A, Hill VR, Boyd EF, Tarr CL. 2014. Draft genome sequence of environmental Vibrio cholerae 2012EL-1759 with similarities to the V. cholerae O1 classical biotype. Genome Announc. 2(4):e00617-14. 10.1128/genomeA.00617-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bruen TC, Philippe H, Bryant D. 2006. A simple and robust statistical test for detecting the presence of recombination. Genetics 172:2665–2681. 10.1534/genetics.105.048975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Strimmer K, Von Haeseler A. 1997. Likelihood-mapping: a simple method to visualize phylogenetic content of a sequence alignment. Proc. Natl. Acad. Sci. U. S. A. 94:6815–6819. 10.1073/pnas.94.13.6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chin CS, Sorenson J, Harris JB, Robins WP, Charles RC, Jean-Charles RR, Bullard J, Webster DR, Kasarskis A, Peluso P, Paxinos EE, Yamaichi Y, Calderwood SB, Mekalanos JJ, Schadt EE, Waldor MK. 2011. The origin of the Haitian cholera outbreak strain. N. Engl. J. Med. 364:33–42. 10.1056/NEJMvcm0904262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Drummond AJ, Rambaut A, Shapiro B, Pybus OG. 2005. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22:1185–1192. 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
- 26. Haitian Ministry of Public Health and Population May 2013. Daily reports of cholera cases by commune May 2013. (In French.) http://mspp.gov.ht/newsite/documentation.php.
- 27. Kass R. 1995. Bayes factors. J. Am. Stat. Assoc. 90:773–795. [Google Scholar]
- 28. Baele G, Lemey P, Vansteelandt S. 2013. Make the most of your samples: Bayes factor estimators for high-dimensional models of sequence evolution. BMC Bioinformatics 14:85. 10.1186/1471-2105-14-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baele G, Lemey P, Bedford T, Rambaut A, Suchard MA, Alekseyenko AV. 2012. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 29:2157–2167. 10.1093/molbev/mss084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang Z. 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13:555–556. [DOI] [PubMed] [Google Scholar]
- 31. Yang Z, Wong WS, Nielsen R. 2005. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 22:1107–1118. 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- 32. Kryazhimskiy S, Plotkin JB. 2008. The population genetics of dN/dS. PLoS Genet. 4:e1000304. 10.1371/journal.pgen.1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lemey P, Kosakovsky Pond SL, Drummond AJ, Pybus OG, Shapiro B, Barroso H, Taveira N, Rambaut A. 2007. Synonymous substitution rates predict HIV disease progression as a result of underlying replication dynamics. PLoS Comput. Biol. 3:e29. 10.1371/journal.pcbi.0030029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sharp PM. 1997. In search of molecular Darwinism. Nature 385:111–112. 10.1038/385111a0. [DOI] [PubMed] [Google Scholar]
- 35. Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24:1586–1591. 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 36. Hu SH, Peek JA, Rattigan E, Taylor RK, Martin JL. 1997. Structure of TcpG, the DsbA protein folding catalyst from Vibrio cholerae. J. Mol. Biol. 268:137–146. 10.1006/jmbi.1997.0940. [DOI] [PubMed] [Google Scholar]
- 37. Peek JA, Taylor RK. 1992. Characterization of a periplasmic thiol:disulfide interchange protein required for the functional maturation of secreted virulence factors of Vibrio cholerae. Proc. Natl. Acad. Sci. USA 89:6210–6214. 10.1073/pnas.89.13.6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Alam MT, Weppelmann TA, Weber CD, Johnson JA, Rashid MH, Birch CS, Brumback BA, Beau de Rochars VE, Morris JG, Ali A. 2014. Monitoring water sources for environmental reservoirs of toxigenic Vibrio cholerae O1, Haiti. Emerg. Infect. Dis. 20:356–363. 10.3201/eid2003.131293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huq A, Colwell RR, Rahman R, Ali A, Chowdhury MA, Parveen S, Sack DA, Russek-Cohen E. 1990. Detection of Vibrio cholerae O1 in the aquatic environment by fluorescent-monoclonal antibody and culture methods. Appl. Environ. Microbiol. 56:2370–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stine OC, Alam M, Tang L, Nair GB, Siddique AK, Faruque SM, Huq A, Colwell R, Sack RB, Morris JG. 2008. Seasonal cholera from multiple small outbreaks, rural Bangladesh. Emerg. Infect. Dis. 14:831–833. 10.3201/eid1405.071116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bertuzzo E, Finger F, Mari L, Gatto M, Rinaldo A. 7 June 2014. On the probability of extinction of the Haiti cholera epidemic. Stoch. Environ. Res. Risk Assess. 10.1007/s00477-014-0906-3. [DOI] [Google Scholar]
- 42. Haley BJ, Choi SY, Hasan NA, Abdullah AS, Cebula TA, Huq A, Colwell RR. 2013. Genome sequences of clinical Vibrio cholerae Isolates from an oyster-borne cholera outbreak in Florida. Genome Announc. 1(6):e00966-13. 10.1128/genomeA.00966-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Folster JP, Katz L, McCullough A, Parsons MB, Knipe K, Sammons SA, Boncy J, Tarr CL, Whichard JM. 2014. Multidrug-resistant IncA/C plasmid in Vibrio cholerae from Haiti. Emerg. Infect. Dis. 20:1951–1953. 10.3201/eid2011.140889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Seo TK, Kishino H, Thorne JL. 2004. Estimating absolute rates of synonymous and nonsynonymous nucleotide substitution in order to characterize natural selection and date species divergences. Mol. Biol. Evol. 21:1201–1213. 10.1093/molbev/msh088. [DOI] [PubMed] [Google Scholar]
- 45. Kamp HD, Patimalla-Dipali B, Lazinski DW, Wallace-Gadsden F, Camilli A. 2013. Gene fitness landscapes of Vibrio cholerae at important stages of its life cycle. PLoS Pathog. 9:e1003800. 10.1371/journal.ppat.1003800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Croucher N. 2013. Bacterial genomes in epidemiology—present and future. Philos. Trans. R. Soc. B Biol. Sci. 368:20120202. 10.1098/rstb.2012.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Goecks J, Nekrutenko A, Taylor J, Team TG 2010. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11:R86. 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20:1297–1303. 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Garrison E, Marth G. 2012. Haplotype-based variant detection from short-read sequencing. http://arxiv.org/pdf/1207.3907.pdf.
- 51. Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6:80–92. 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zerbino DR. 2010. Using the Velvet de novo assembler for short-read sequencing technologies. Curr. Protoc. Bioinformatics Chapter 11:Unit 11.5. 10.1002/0471250953.bi1105s31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rissman AI, Mau B, Biehl BS, Darling AE, Glasner JD, Perna NT. 2009. Reordering contigs of draft genomes using the Mauve aligner. Bioinformatics 25:2071–2073. 10.1093/bioinformatics/btp356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schmidt HA, Strimmer K, Vingron M, von Haeseler A. 2002. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18:502–504. 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- 56. Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23:254–267. 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 57. Lemey P, Salemi M, Vandamme AM. (ed). 2009. The phylogenetic handbook: a practical approach to phylogenetic analysis and hypothesis testing, 2nd ed. Cambridge University Press, New York, NY. [Google Scholar]
- 58. Swofford DL, Sullivan J. 2003. Phylogeny inference based on parsimony and other methods using PAUP*, p 1600-206. In Salemi M, Vandamme A-M. (ed), The phylogenetic handbook: a practical approach to DNA and protein phylogeny. Cambridge University Press, New York, NY. [Google Scholar]
- 59. Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Minin VN, Bloomquist EW, Suchard MA. 2008. Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics. Mol. Biol. Evol. 25:1459–1471. 10.1093/molbev/msn090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gill MS, Lemey P, Faria NR, Rambaut A, Shapiro B, Suchard MA. 2013. Improving Bayesian population dynamics inference: a coalescent-based model for multiple loci. Mol. Biol. Evol. 30:713–724. 10.1093/molbev/mss265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Baele G, Li WL, Drummond AJ, Suchard MA, Lemey P. 2013. Accurate model selection of relaxed molecular clocks in Bayesian phylogenetics. Mol. Biol. Evol. 30:239–243. 10.1093/molbev/mss243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Baele G, Lemey P. 2013. Bayesian evolutionary model testing in the phylogenomics era: matching model complexity with computational efficiency. Bioinformatics 29:1970–1979. 10.1093/bioinformatics/btt340. [DOI] [PubMed] [Google Scholar]
- 65. Newton MA, Raftery AE. 1994. Approximate Bayesian inference with the weighted likelihood bootstrap. J. R. Stat. Soc. B Stat. Methodol. 56:3–48. [Google Scholar]
- 66. Suchard MA, Weiss RE, Sinsheimer JS. 2001. Bayesian selection of continuous-time Markov chain evolutionary models. Mol. Biol. Evol. 18:1001–1013. 10.1093/oxfordjournals.molbev.a003872. [DOI] [PubMed] [Google Scholar]
- 67. Bouckaert RR. 2010. DensiTree: making sense of sets of phylogenetic trees. Bioinformatics 26:1372–1373. 10.1093/bioinformatics/btq110. [DOI] [PubMed] [Google Scholar]
- 68. Delano WL. 2002. The PyMOL Molecular Graphics System, Version 1.7.2 Schrödinger, LLC. http://www.pymol.org/contact.
- 69. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Kosakovsky Pond SL, Scheffler K. 2013. FUBAR: a fast, unconstrained Bayesian approximation for inferring selection. Mol. Biol. Evol. 30:1196–1205. 10.1093/molbev/mst030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R, 1000 Genomes Project Analysis Group 2011. The variant call format and VCFtools. Bioinformatics 27:2156–2158. 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Study sites. Blue dots designate the locations where clinical and environmental samples were collected by the University of Florida Emerging Pathogens Institute. Samples collected during 2010 were from the St. Marc Hospital in the Artibonite Department. Samples obtained during 2012 were from the Ouest Department. Clinical samples were collected from the Gressier Cholera Treatment Center, and environmental strains were isolated from samples collected in the surrounding Gressier region. Samples included in study from Katz et al. (9) are designated by black dots. The inset shows the locations of the departments on a Haiti country map. Download
Maximum likelihood (ML) tree of V. cholerae O1 strains. The ML tree was inferred from 68 genome-wide SNPs among the 60 V. cholerae O1 strains using the GTR nucleotide substitution model and ascertainment bias correction implement in RAxML. Tree was rooted using closely related strains from the Nepalese epidemic as an outgroup. Strain labels were colored to represent the sampling time according to the table in the figure. Branch lengths are scaled in nucleotide substitutions per SNP site according to the bar at the bottom. Values along the branches represent statistical support according to bootstrapping (1,000 replicates). Download
Mean nonsynonymous (dN) and synonymous (dS) divergence over the course of the V. cholerae epidemic in Haiti for internal branches. Blue and green lines represent, respectively, mean nonsynonymous (dN) and synonymous (dS) divergence along internal branches over the course of the V. cholerae epidemic in Haiti. Estimates 1 standard deviation above and below the mean dN or dS are represented by broken lines. The y axis represents the number of nucleotide (dN or dS) changes per (synonymous or nonsynonymous) SNP site. Estimates were obtained by reconstructing and comparing ancestral sequences along V. cholerae trees sampled from the posterior distribution obtained with the Bayesian analysis. Download
Three-dimensional structure of the thiol:disulfide interchange protein. The three-dimensional protein structure of the thiol:disulfide interchange protein DbsA (TcpG) was visualized in the PyMOL molecular graphics system obtained from the Protein Data Bank (accession no. 4DVC). The white circle represents the site of the alanine-to-threonine amino acid substitution (A169T) occurring near a β-sheet in the hydrophobic binding site region. This protein plays an important role in the secretion of virulence factors and represents one example of how point mutations could potentially affect pathogen success. Download
Bioinformatics analysis pipeline of V. cholerae genome-wide SNPs. Raw paired-end Illumina reads in the FASTQ format were filtered by Phred quality score using Sickle (https://github.com/najoshi/sickle). Filtered FASTQ files were then mapped to the V. cholerae 2010EL-1786 closed reference genome (accession no. NC_016445.1 and NC_016446.1) with Bow tie 2 short read alignment software using default settings to produce BAM files (48). Base quality scores were recalibrated, and realignment around insertions and deletions was performed using the Genome Analysis Toolkit, a framework for analyzing next-generation sequence data (49). We called SNPs using the FreeBayes Bayesian genetic variant detector for haploid organisms requiring a minimum base quality of 20 and a minimum alternate fraction of 0.75 (50). Putative SNPs were initially filtered by quality and likelihood using VCFtools, and SNP locations not represented among all samples were removed (71). The remaining high-quality SNPs were then assessed and manually curated based on mapping quality and distance from neighboring SNPs to remove clusters. A FASTA alignment of SNPs was generated for phylogenetic analysis. Finally, SnpEFF was used to annotate SNPs, and codons of synonymous and nonsynonymous mutations were abstracted (51). Selection analysis was conducted by tracing the annotated codons on the V. cholerae tree using the site-specific and branch/site-specific models implemented in PAML (30). The bioinformatics pipeline was constructed using the University of Florida High Performance Computing Center’s local instance of Galaxy (47). Download
List of sequenced cholera strains. Listed are the sample name, site of isolation, source (clinical versus environmental), date of sample, and serogroup. Accession numbers for genomes in the present study are to be determined (TBD).
List of single nucleotide polymorphisms (SNP) identified among the 56 clinical and 4 environmental toxigenic V. cholerae O1 isolates. Listed are the SNP sites, effect (noncoding, coding synonymous, and coding nonsynonymous), and gene location (if coding).
Pairwise SNP differences between the 56 clinical and 4 environmental toxigenic V. cholerae O1 isolates.
Bayesian coalescent estimates inferred from the SNPs of the Haiti V. cholerae O1 strains collected from 2010 to 2012. Shown are comparisons of molecular clock models, demographic models, the times of the most recent common ancestor (TMRCA), and the evolutionary rates for each model.