

ABSTRACT
Several recent studies have converged upon the innate immune DNA cytosine deaminase APOBEC3B (A3B) as a significant source of genomic uracil lesions and mutagenesis in multiple human cancers, including those of the breast, head/neck, cervix, bladder, lung, ovary, and other tissues. A3B is upregulated in these tumor types relative to normal tissues, but the mechanism is unclear. Because A3B also has antiviral activity in multiple systems and is a member of the broader innate immune response, we tested the hypothesis that human papillomavirus (HPV) infection causes A3B upregulation. We found that A3B mRNA expression and enzymatic activity were upregulated following transfection of a high-risk HPV genome and that this effect was abrogated by inactivation of E6. Transduction experiments showed that the E6 oncoprotein alone was sufficient to cause A3B upregulation, and a panel of high-risk E6 proteins triggered higher A3B levels than did a panel of low-risk or noncancer E6 proteins. Knockdown experiments in HPV-positive cell lines showed that endogenous E6 is required for A3B upregulation. Analyses of publicly available head/neck cancer data further support this relationship, as A3B levels are higher in HPV-positive cancers than in HPV-negative cancers. Taken together with the established role for high-risk E6 in functional inactivation of TP53 and published positive correlations in breast cancer between A3B upregulation and genetic inactivation of TP53, our studies suggest a model in which high-risk HPV E6, possibly through functional inactivation of TP53, causes derepression of A3B gene transcription. This would lead to a mutator phenotype that explains the observed cytosine mutation biases in HPV-positive head/neck and cervical cancers.
IMPORTANCE
The innate immune DNA cytosine deaminase APOBEC3B (A3B) accounts for a large proportion of somatic mutations in cervical and head/neck cancers, but nothing is known about the mechanism responsible for its upregulation in these tumor types. Almost all cervical carcinomas and large proportions of head/neck tumors are caused by human papillomavirus (HPV) infection. Here, we establish a mechanistic link between HPV infection and A3B upregulation. The E6 oncoprotein of high-risk, but not low-risk, HPV types triggers A3B upregulation, supporting a model in which TP53 inactivation causes a derepression of A3B gene transcription and elevated A3B enzyme levels. This virus-induced mutator phenotype provides a mechanistic explanation for A3B signature mutations observed in HPV-positive head/neck and cervical carcinomas and may also help to account for the preferential cancer predisposition caused by high-risk HPV isolates.
INTRODUCTION
Seven APOBEC3 (A3) enzymes have broad and overlapping functions in innate immunity by restricting viruses, transposons, and other foreign DNA elements (reviewed in references 1 to 3). These proteins are part of a larger family that includes the DNA deaminase AID and the RNA editing protein APOBEC1, which also have specialized functions in antibody diversification and APOB mRNA editing, respectively, (reviewed in references 4 and 5). The defining activity of all members of this protein family is single-stranded DNA cytosine-to-uracil (C-to-U) deamination, as even APOBEC1 prefers DNA over RNA cytosines (6–8). The current working model for HIV-1 restriction illustrates the overlapping antiviral activities of these enzymes (reviewed in references 1 to 3). In a Trojan horse-like mechanism, A3D, A3F, A3G, and A3H (not A3A, A3B, or A3C) specifically package into budding viral particles, breach the viral core during maturation, and deaminate viral cDNA cytosines during reverse transcription. The resulting uracils template the addition of genomic-strand adenines and account for the well-known phenomenon of viral G-to-A hypermutation often observed in clinical isolates. A3D, A3F, and A3H are responsible for mutations in GA-to-AA dinucleotide contexts, whereas A3G accounts for mutations in GG-to-AG contexts (illustrating the distinct local specificity of these enzymes for 5′ TC and 5′ CC substrates). Remarkably, a single HIV-1 protein, Vif, is able to physically bind and counteract all four of these restriction factors by recruiting an E3 ubiquitin ligase complex to target them for proteasomal degradation. Based on evidence for restriction and/or G-to-A mutation, other potentially A3-susceptible human viruses include adeno-associated virus (AAV), Epstein-Barr virus (EBV), hepatitis B virus (HBV), herpes simplex virus 1 (HSV-1), human T-cell lymphotropic virus (HTLV), and, most relevant to this study, human papillomavirus (HPV) (9–13; reviewed in reference 14).
HPV is an ~8-kb double-stranded DNA virus that replicates in the nucleus of mucosal or cutaneous keratinocytes (reviewed in reference 15). Over 170 HPV types have been identified thus far, and these can be classified into high- and low-risk groups based on carcinogenic risk (16–18). HPV infection is necessary but not sufficient for the development of cervical cancer, and it is also strongly associated with other anogenital and a growing subset of head/neck squamous cancers (19, 20). The viral oncoproteins E6 and E7 are invariably expressed in HPV-positive cancers (21, 22), and the expression of these proteins from high-risk isolates is sufficient to immortalize human keratinocytes (23, 24). The most critical functions of E6 and E7, respectively, are thought to be functional inactivation of tumor suppressors TP53 and RB (25–28).
Recently, the enzymatic activity of A3B has been implicated as a major source of mutagenesis in multiple human cancers (29–37). A3B is a nuclear enzyme and the only detectable source of single-stranded DNA cytosine deaminase activity in multiple cancer cell lines (29, 32). A3B mRNA levels are upregulated in many cancer types, including those of the breast, bladder, cervix, lung, head/neck, and ovary (29–32). The trinucleotide preference of A3B (5′ TCA and 5′ TCG) is highly enriched in the mutation spectrum of these cancer types (29–32). Moreover, positive correlations are evident between A3B mRNA levels and somatic mutation loads (30–32). Interestingly, head/neck and cervical cancers are among the tumor types displaying the highest A3B expression levels and cytosine mutational loads in A3B-preferred trinucleotide contexts (30, 31). Overall, a compelling case has been made for A3B mutagenesis in multiple human cancers.
Given the fact that A3B is expressed at low levels or not at all in most normal tissues (29, 30, 38), a major unresolved question is how it becomes upregulated in cancer. Since A3B is a member of the A3 family of innate immune effector proteins with demonstrated antiviral activities (though not against HIV-1 in T lymphocytes [39, 40]) and given the tendency of HPV-associated cancers of the head/neck and cervix to be among the highest A3B-impacted tumor types, here we test the hypothesis that HPV directly causes A3B mRNA upregulation. Moreover, because E6 is invariably expressed in HPV-positive tumors (21, 22) and A3B upregulation is associated with genetic inactivation of TP53 (29), we tested the specific hypothesis that E6 is the primary trigger of A3B upregulation in virus-positive tumor types.
RESULTS
HPV genomic DNA causes A3B upregulation.
We first tested whether high-risk HPV genomes could trigger A3B upregulation. Normal immortalized keratinocytes (NIKS) were transfected with full-length HPV16 or HPV18 genomes. Pools of transfectants were selected and expanded to allow for establishment of the viral genomes as nuclear plasmids and viral gene expression, and then reverse transcription-quantitative PCR (RT-qPCR) was used to quantify A3B mRNA levels. In comparison to a control vector-transfected pool of NIKS established in parallel, A3B mRNA levels were induced significantly by transfection of either HPV16 or HPV18 genomes (Fig. 1A). HPV18 genomic DNA consistently caused higher levels of A3B induction, routinely 5- to 10-fold above the negative control.
FIG 1 .

APOBEC3B upregulation by transfection of full-length HPV genomes. (A) Histograms reporting APOBEC family member mRNA levels in NIKS transfected with a full-length HPV16 or HPV18 or a control plasmid (Cont.). Each histogram bar shows the mean expression level of each APOBEC family member normalized to TBP (error bars report standard deviations from triplicate assays). (B) Image of the results of a representative DNA cytosine deaminase assay performed with cell extracts from the same cells as in panel A. The single-stranded DNA substrate was treated with reaction buffer as a negative control (−) and recombinant APOBEC3A as a positive control (+).
To ask whether the effect of HPV genomic DNA is specific to A3B or to a more general antiviral response, RT-qPCR assays were used to quantify expression of all A3 family members (Fig. 1A). Most of these genes, including A3A, A3D, A3H, AID, A1, A2, and A4, were expressed at very low or undetectable levels and not affected by HPV genomic DNA transfection. Two family members, A3F and A3G, were expressed at similar levels in both control and HPV-transfected NIKS. The only exception was A3C, which showed an inverse relationship with higher levels in control-transfected cells and lower levels in HPV-transfected cells, especially with HPV18 genomic DNA. Thus, A3B is the only DNA deaminase family member upregulated at the level of transcription in NIKS harboring HPV genomes. Since there are no commercial antibodies for A3B, the current gold standard for detecting A3B protein levels is by quantifying its functional activity (29, 32). Therefore, we performed single-stranded DNA deaminase assays using protein extracts from the same cells as used for mRNA quantification (Fig. 1B). As expected, protein extracts produced from cells transfected with HPV18 had deaminase activity more than five times higher than that of the transfection control (68% versus 12% substrate deamination, respectively). These results show that transfection of HPV18 genome results in an increase in DNA deaminase activity that is proportional to the increase observed for A3B mRNA.
HPV E6 is sufficient for A3B upregulation.
Considering that the viral oncoproteins are invariably expressed in HPV-positive tumors, we next tested for a possible role of E6 in A3B upregulation. NIKS were transfected either with the full-length HPV18 genome or with a full-length HPV18 genome containing a stop codon within the E6 open reading frame (HPV18 E6-STOP) (41). As shown above, transfection with the wild-type HPV18 genome resulted in a significant upregulation of A3B mRNA levels. However, most of this effect was lost upon transfection with the HPV18 genome containing an E6-STOP mutation, indicating that E6 is required for induction of A3B (Fig. 2A). E6 mRNA levels were also reduced likely due to nonsense-mediated decay (Fig. 2B). To reconfirm the correlation between upregulation of A3B mRNA levels and enzymatic activity, DNA deaminase assays were performed using cell extracts. As expected, the DNA cytosine deaminase activity induced by transfection with wild-type HPV18 genome was ablated by inactivation of E6 (Fig. 2C).
FIG 2 .
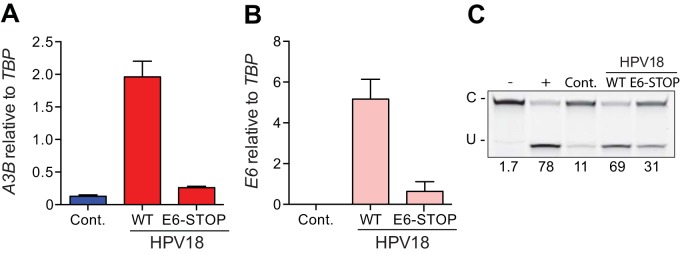
HPV18 E6 is necessary for APOBEC3B upregulation. (A) A3B and (B) E6 mRNA levels in NIKS transfected with full-length HPV18 (WT), with HPV18 with a stop codon truncating the E6 open reading frame (E6-STOP), or with a control plasmid (Cont.). Each histogram bar shows the mean mRNA expression level normalized to TBP (error bars report standard deviations from triplicate assays). (C) Image of the results of a representative DNA cytosine deaminase assay performed with cell extracts from the same cells as in panels A and B. The single-stranded DNA substrate was treated with reaction buffer as a negative control (−) and recombinant APOBEC3A as a positive control (+).
To test if expression of E6 is sufficient to induce A3B upregulation, we used a panel of transduced cell lines based on the hTERT-immortalized keratinocyte cell line N/TERT-1. Each line expressed a different E6 protein from a genus alpha high-risk type (HPV16, -18, -33, -45, and -52), a low-risk type (HPV6b and -11), or a type with no known cancer association (HPV2a and -57) (42, 43). Interestingly, only cells expressing high-risk E6 proteins showed significant increases in A3B mRNA levels in comparison to an empty vector control and E6 from low-risk and non-cancer-associated HPV types (P < 0.01; Fig. 3).
FIG 3 .
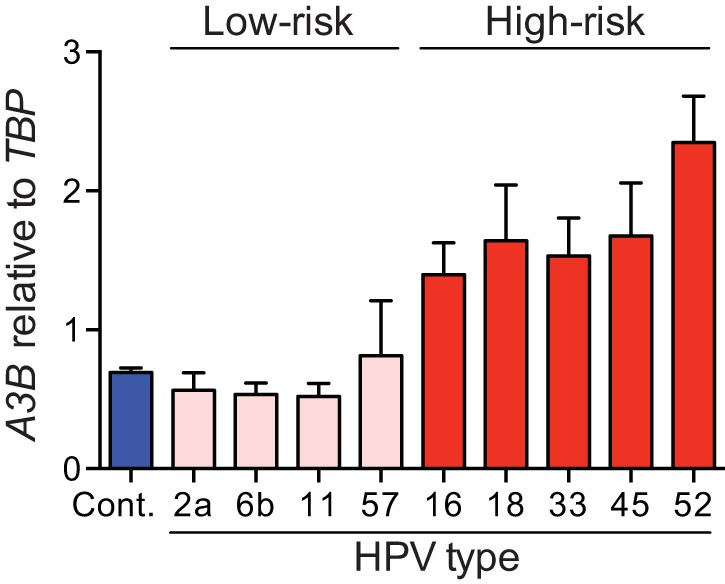
Upregulation of APOBEC3B by expression of HPV E6. A3B mRNA levels in N/TERT-1 cells transduced with HPV E6 from different high-risk types (HPV16, -18, -33, -45, and -52), low-risk types (HPV6b and -11), or noncancer types (HPV2a and -57) or with an empty vector (Cont.). Each histogram bar shows the mean A3B expression level normalized to TBP (error bars report standard deviations from triplicate assays). Low-risk/noncancer E6 proteins did not cause significant A3B upregulation compared to high-risk E6 proteins (P < 0.01; two-tailed Student’s t test).
We next asked if these results extended to primary keratinocytes. Early-passage human keratinocyte G5-Ep cells were transduced with the same panel of retroviruses expressing E6 from different HPV types. As above, A3B upregulation was induced and high-risk E6 proteins caused higher levels of induction (P < 0.05) (see Fig. S1 in the supplemental material). Together, the data with NIKS, N/TERT-1, and early-passage keratinocytes demonstrate that E6 alone, especially from high-risk HPV types, is sufficient to induce A3B upregulation.
E6 is required for endogenous A3B expression in HPV-positive cancer cell lines.
To test if endogenous E6 could contribute to upregulation of endogenous A3B, we depleted the HPV early transcript from the HPV16-positive CaSki cell line. Two different small interfering RNAs (siRNAs) were used to interfere with E6 expression. In each instance, the level of E6 depletion was proportional to the decrease in endogenous A3B mRNA levels with an approximately 3-fold reduction in E6 mRNA levels and a corresponding 3-fold reduction in A3B mRNA levels (Fig. 4). These results indicate that endogenous E6 contributes to upregulation of endogenous A3B.
FIG 4 .

HPV E6 knockdown reduces endogenous A3B expression. E6 mRNA levels (A) and A3B mRNA levels (B) in CaSki cells transfected with siRNA targeting the HPV16 early transcript (#1 or #2) or with a nontargeting control siRNA (Cont.). Each histogram bar shows the mean E6 or A3B expression level relative to TBP (error bars report standard deviations from triplicate assays).
Increased levels of A3B in HPV-positive head and neck tumors.
Finally, we asked if the observed relationship between HPV status and A3B levels occurs in vivo. TCGA RNA sequencing (RNA-seq) data from head and neck cancers with reported HPV status were analyzed for A3B mRNA levels relative to the housekeeping gene TBP. Using TCGA clinical data, we were able to acquire RNA-seq counts for 23 of the HPV-positive patients and 69 of the HPV-negative patients. A3B mRNA levels were significantly increased in HPV-positive compared to HPV-negative cancers (P = 0.0006) (Fig. 5A). Notably, a significant increase in A3B expression was evident for the subset of HPV-positive patients with no smoking history (n = 6) compared to corresponding HPV-negative nonsmokers (n = 10; P = 0.0013).
FIG 5 .
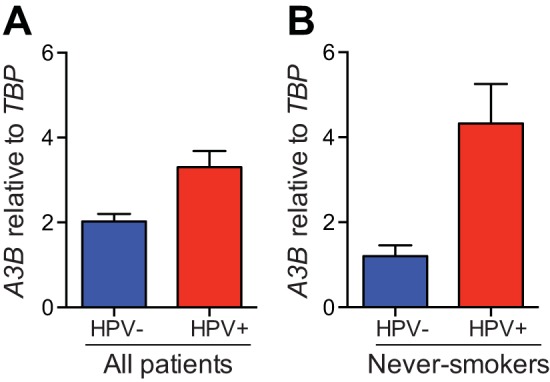
APOBEC3B overexpression in HPV-positive head/neck tumors. (A) A3B mRNA levels in HPV-positive and HPV-negative head/neck cancers (HPV positive, n = 69; HPV negative, n = 23; P = 0.0006). (B) A3B mRNA levels in the subset of patients in panel A reported as never-smokers (HPV positive, n = 6; HPV negative, n = 10; P = 0.0013). Each histogram bar shows the average A3B expression level normalized to TBP, with values derived from TCGA RNA-seq data sets (error bars report the standard deviations).
DISCUSSION
This is the first study to demonstrate a mechanistic link between HPV infection and upregulation of the DNA cytosine deaminase A3B. Here we show that transfection of the HPV genome triggers A3B upregulation and that E6 expression is required. A3B upregulation is apparent at both mRNA and activity levels. We demonstrate that high-risk E6 alone is sufficient for the induction of A3B in keratinocytes and that continuous expression of E6 is required to maintain higher A3B levels in HPV-positive cancer cell lines. Finally, analyses of available TCGA data show that A3B levels are higher in head/neck HPV-positive cancers than in HPV-negative cancers. Taken together, these results suggest a model in which high-risk HPV E6 induces A3B gene expression, leading to a mutator phenotype and the observed cytosine mutation biases in HPV-positive head/neck and cervical cancers.
A3B has been strongly implicated in mutagenesis in a wide variety of human cancers (29–37). Given its relatively low expression level in almost all normal tissues (29, 30, 38), a major question is how A3B upregulation occurs in cancer cells. As described here, the E6 oncoprotein provides the first mechanistic link between viral infection and A3B-mediated cancer mutagenesis. Although E6 has numerous functions, we propose a direct model in which high-risk E6 proteins inactivate TP53 and cause derepression of A3B gene transcription. This mechanism has the potential to explain A3B upregulation in HPV-positive cervical and head/neck cancers (and possibly other cancers such as some bladder carcinomas where HPV may also contribute [19, 20, 44]). This connection between TP53 function and A3B upregulation is supported by our previous observation that genetic inactivation of TP53 correlates positively with elevated A3B levels in breast cancer cell lines and primary tumors (29). Therefore, a model involving TP53 inactivation may apply more generally and contribute to tumorigenesis on at least two distinct levels, by elevating levels of DNA damage and mutation through A3B and by preventing the DNA damage response and apoptosis. Additional studies are necessary to distinguish between this model and other, less-direct possibilities such as an association of E6 with cellular PDZ domain proteins, a characteristic that is also shared by genus alpha high-risk E6 proteins (45, 46). These E6 proteins have a PDZ-binding domain that interacts with a number of PDZ targets with a wide array of functions, including cell signaling, polarity determination, and cell proliferation (reviewed in references 47 and 48).
Although E6 and E7 are sufficient to immortalize primary keratinocytes (23, 24), complete cellular transformation also requires the introduction of additional activated oncogenes or extensive periods of cell culture (49–51). These observations strongly suggest that additional somatic mutations are required for transformation (reviewed in reference 52). We hypothesize that E6 expression leads to elevated A3B levels and an increased, but still stochastic, mutational process that leads eventually to transformation. The same deamination process may also explain genomic instability phenotypes previously shown to be inducible by high-risk E6 oncoproteins (reviewed in reference 52).
Our data provide evidence that HPV infection causes upregulation of A3B, a phenomenon generally regarded as an innate antiviral response and previously observed for HIV-1 infection of primary T lymphocytes (1, 39, 40). This relationship prompts the additional question of how HPV avoids restriction in the presence of increased A3B activity and constitutive levels of other A3 proteins. Despite the fact that overexpression studies have shown that HPV can be mutated by APOBEC3 (A/C/H) (11, 53), clinical isolates rarely show evidence for hypermutation (11), consistent with an effective APOBEC3 counteraction or avoidance strategy operating in vivo. Based on precedents with other viruses (notably lentiviruses and foamy viruses [reviewed in reference 1]), the answer to this question may provide fundamental mechanistic insights into the HPV replication and transmission cycle. Finally, the robust cellular response to HPV infection characterized by A3B upregulation strongly suggests that other viruses may also be able to provoke similar responses. A mechanistic linkage to the innate antiviral response may also help to explain A3B upregulation and genomic mutagenesis observed in other cancers such as those of the lung, bladder, and breast tissues.
MATERIALS AND METHODS
Cell lines.
Normal immortal keratinocytes (NIKS; provided by Lynn Allen-Hoffman [54]) were cultured in E medium supplemented with 24 µg/ml adenine, 8.4 µg/ml cholera toxin, 10 ng/ml epidermal growth factor (EGF), 400 ng/ml hydrocortisone, 5 µg/ml insulin, 1% penicillin-streptomycin, and 5% fetal bovine serum and grown in the presence of mitomycin C-treated J2-3T3 feeder cells (55).
Human hTERT-immortalized keratinocytes (N/TERT-1) and G5-Ep primary human foreskin keratinocytes (provided by James Rheinwald [56–58]) were cultured in keratinocyte serum-free medium (K-SFM) supplemented with 0.3 mM CaCl2, 0.2 ng/ml EGF, 25 µg/ml bovine pituitary extract, and 1% penicillin-streptomycin.
HPV genome transfections.
NIKS were transfected with the HPV genome as described previously (41). Full-length HPV genomes from wild-type HPV16 and HPV18 and from HPV18 containing a stop codon at E6 (E6-STOP) were excised from their bacterial vectors with either BamHI or NcoI and recircularized with T4 DNA ligase (15 U/µl) at a concentration of 8 ng/µl DNA. One day prior to transfection, 3 × 105 NIKS were plated in low-Ca2+ incomplete E medium in the absence of J2-3T3 feeders. Cells were transfected with 3 µg of religated HPV and 1.2 µg of a plasmid conferring neomycin resistance (pEGFP-N1) using Effectene (Qiagen). HPV-negative controls were transfected with 1.2 µg of pEGFP-N1 alone. The next day, cells were transferred to a 10-cm dish containing J2-3T3 feeders. Cells were selected for 4 days in the presence of G418 (125 µg/ml for 2 days followed by 250 µg/ml for 2 days). Two to 3 weeks after transfection, colonies were pooled and expanded. Cells were passaged on a weekly basis and were grown until approximately 90% confluent prior to harvesting of total RNA.
Retroviral transductions.
N/TERT-1 and G5-Ep primary keratinocytes stably expressing HPV16 E6 have been described elsewhere (42, 43). Cells were transduced with a panel of retroviruses (pMSCV-N-HA-IRES-PURO) expressing E6 from genus alpha high-risk types (HPV16, -18, -33, -45, and -52), from low-risk types (HPV6b and -11), or from types with no known cancer association (HPV2a and -57) (42). As a negative control, cells were transduced with the empty vector (pMSCV-N-HA-IRES-PURO empty). Cells were selected with puromycin and grown to approximately 30% confluence prior to harvesting of total RNA. Expression differences were assessed using a two-tailed Student t test.
siRNA transfections.
CaSki cells were transfected using DharmaFECT 1 (Dharmacon/GE Life Sciences) as described elsewhere (59). siRNA duplexes used were as follows: nontargeting siRNA (Dharmacon/GE Life Sciences D-001810-01) and two custom-designed siRNAs targeting the HPV16 early transcript. SiGLO red transfection indicator (Dharmacon/GE Life Sciences D-0011630-02) was used to visualize efficient transfection in a control well. Sequences of the custom siRNAs are as follows: HPV16 #1, CAACAUUAGAACAGCAAUAUU, and HPV16 #2, GGACAGAGCCCAUUACAAUUU. siRNAs were used at a final concentration of 40 nM. Cells were harvested at 72 h posttransfection.
Quantification of cellular and viral RNA.
Reverse transcription-quantitative PCR (RT-qPCR) was used to measure APOBEC and E6 mRNA levels as described previously (38). Total RNA was isolated using the NucleoSpin RNA kit (Clontech). One microgram from each sample was reverse transcribed with the cDNA Transcriptor reverse transcriptase kit (Roche; catalog no. 03531287001). qPCR was performed using 2× master mix (Roche; catalog no. 04887301001). Primer and probe sets (Universal Probe Library; Roche) for HPV E6 were as follows: 16E6-F, 5′-GCACCAAAAGAGAACTGCAA; 16E6-R, 5′-TGTTTGCAGCTCTGTGCATAA; UPL#115; 18E6-F, 5′-ACATTGGAAAAACTAACTAACACTGG; 18E6-R, 5′-TCGTTTTTCATTAAGGTGTCTAAGTTT; UPL#120. For each condition, qPCRs were performed in triplicate, mRNA expression levels were normalized to those of the housekeeping gene TBP mRNA, and the mean and standard deviation were reported.
DNA deaminase activity assays.
Deamination reactions were performed at 37°C for 2 h using 16.5 µl of cell extract, 4 pmol of oligonucleotide (5′-ATTATTATTATTCAAATGGATTTATTTATTTATTTATTTATTT-fluorescein), 0.025 U uracil DNA glycosylase (UDG), 2 µl 10× UDG buffer (NEB), and 1.75 U RNase A. Reaction mixtures were treated with 100 mM NaOH at 95°C for 10 min to achieve complete backbone breakage. Reaction mixtures were separated on 15% Tris-borate-EDTA (TBE)–urea gels to separate substrate from product. Gels were scanned using a Fujifilm FLA-7000 image reader, and densitometry was performed using ImageQuant TL (GE Healthcare Life Science).
Head and neck cancer data retrieval and analyses.
Head/neck cancer data were acquired from The Cancer Genome Atlas (TCGA) (60), and individuals were selected for analysis if HPV status was clear. RNA-seq counts were used to quantify A3B mRNA expression levels and calculate abundance relative to TBP. This metric facilitates cross-comparisons with RT-qPCR data similarly normalized. Expression differences were assessed using a two-tailed Student t test. Results were considered significant if the calculated P value was under 0.05.
SUPPLEMENTAL MATERIAL
Upregulation of APOBEC3B in primary keratinocytes by expression of HPV E6. A3B mRNA levels in G5-Ep primary keratinocytes transduced with HPV E6 from different high-risk (HPV16, -18, -33, -45, and -52), low-risk (HPV6b and -11), or noncancer (HPV2a and -57) types or with an empty vector (Cont.). Each histogram bar shows the mean A3B expression level normalized to TBP (error bars report standard deviations from triplicate assays). Low-risk/noncancer E6 proteins did not cause significant A3B upregulation compared to high-risk E6 proteins (P < 0.05; two-tailed Student’s t test). Download
ACKNOWLEDGMENTS
This work was supported by NIH grants R01 AI064046 (R.S.H.), P01 CA050661 (P.M.H.), and P01 CA022443 (P.F.L.). V.C.V. was supported by a scholarship from CNPq-Brazil. This study is part of the requirements for V.C.V. to obtain her Ph.D. degree from the Graduate Program in Oncology at the Brazilian Cancer Institute (Rio de Janeiro, Brazil).
Footnotes
Citation Vieira VC, Leonard B, White EA, Starrett GJ, Temiz NA, Lorenz LD, Lee D, Soares MA, Lambert PF, Howley PM, Harris RS. 2014. Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B. mBio 5(6):e02234-14. doi:10.1128/mBio.02234-14.
REFERENCES
- 1. Harris RS, Hultquist JF, Evans DT. 2012. The restriction factors of human immunodeficiency virus. J. Biol. Chem. 287:40875–40883. 10.1074/jbc.R112.416925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Malim MH, Bieniasz PD. 2012. HIV restriction factors and mechanisms of evasion. Cold Spring Harb. Perspect. Med. 2:a006940. 10.1101/cshperspect.a006940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Refsland EW, Harris RS. 2013. The APOBEC3 family of retroelement restriction factors. Curr. Top. Microbiol. Immunol. 371:1–25. 10.1007/978-3-642-37765-5_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Di Noia JM, Neuberger MS. 2007. Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 76:1–22. 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- 5. Conticello SG. 2008. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 9:229. 10.1186/gb-2008-9-7-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Harris RS, Petersen-Mahrt SK, Neuberger MS. 2002. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell 10:1247–1253. 10.1016/S1097-2765(02)00742-6. [DOI] [PubMed] [Google Scholar]
- 7. Petersen-Mahrt SK, Neuberger MS. 2003. In vitro deamination of cytosine to uracil in single-stranded DNA by apolipoprotein B editing complex catalytic subunit 1 (APOBEC1). J. Biol. Chem. 278:19583–19586. 10.1074/jbc.C300114200. [DOI] [PubMed] [Google Scholar]
- 8. Severi F, Chicca A, Conticello SG. 2011. Analysis of reptilian APOBEC1 suggests that RNA editing may not be its ancestral function. Mol. Biol. Evol. 28:1125–1129. 10.1093/molbev/msq338. [DOI] [PubMed] [Google Scholar]
- 9. Turelli P, Mangeat B, Jost S, Vianin S, Trono D. 2004. Inhibition of hepatitis B virus replication by APOBEC3G. Science 303:1829. 10.1126/science.1092066. [DOI] [PubMed] [Google Scholar]
- 10. Chen H, Lilley CE, Yu Q, Lee DV, Chou J. 2006. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr. Biol. 16:480–485. 10.1016/j.cub.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 11. Vartanian JP, Guétard D, Henry M, Wain-Hobson S. 2008. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science 320:230–233. 10.1126/science.1153201. [DOI] [PubMed] [Google Scholar]
- 12. Suspène R, Aynaud MM, Koch S, Pasdeloup D, Labetoulle M, Gaertner B, Vartanian JP, Meyerhans A, Wain-Hobson S. 2011. Genetic editing of herpes simplex virus 1 and Epstein-Barr herpesvirus genomes by human APOBEC3 cytidine deaminases in culture and in vivo. J. Virol. 85:7594–7602. 10.1128/JVI.00290-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ooms M, Krikoni A, Kress AK, Simon V, Münk C. 2012. APOBEC3A, APOBEC3B, and APOBEC3H haplotype 2 restrict human T-lymphotropic virus type 1. J. Virol. 86:6097–6108. 10.1128/JVI.06570-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vieira VC, Soares MA. 2013. The role of cytidine deaminases on innate immune responses against human viral infections. Biomed. Res. Int. 2013:1–18. 10.1155/2013/683095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Doorbar J, Quint W, Banks L, Bravo IG, Stoler M, Broker TR, Stanley MA. 2012. The biology and life-cycle of human papillomaviruses. Vaccine 30:F55–F70. 10.1016/j.vaccine.2012.06.083. [DOI] [PubMed] [Google Scholar]
- 16. Bernard HU, Burk RD, Chen Z, van Doorslaer K, zur Hausen H, de Villiers EM. 2010. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 401:70–79. 10.1016/j.virol.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Villiers EM. 2013. Cross-roads in the classification of papillomaviruses. Virology 445:2–10. 10.1016/j.virol.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 18. Muñoz N, Bosch FX, de Sanjosé S, Herrero R, Castellsagué X, Shah KV, Snijders PJ, Meijer CJ, International Agency for Research on Cancer Multicenter Cervical Cancer Study Group 2003. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 348:518–527. 10.1056/NEJMoa021641. [DOI] [PubMed] [Google Scholar]
- 19. Zur Hausen H. 2009. Papillomaviruses in the causation of human cancers—a brief historical account. Virology 384:260–265. 10.1016/j.virol.2008.11.046. [DOI] [PubMed] [Google Scholar]
- 20. D’Souza G, Dempsey A. 2011. The role of HPV in head and neck cancer and review of the HPV vaccine. Prev. Med. 53:S5–S11. 10.1016/j.ypmed.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zur Hausen H. 2002. Papillomaviruses and cancer: from basic studies to clinical application. Nat. Rev. Cancer 2:342–350. 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 22. Schwarz E, Freese UK, Gissmann L, Mayer W, Roggenbuck B, Stremlau A, zur Hausen H. 1985. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature 314:111–114. 10.1038/314111a0. [DOI] [PubMed] [Google Scholar]
- 23. Münger K, Phelps WC, Bubb V, Howley PM, Schlegel R. 1989. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J. Virol. 63:4417–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hawley-Nelson P, Vousden KH, Hubbert NL, Lowy DR, Schiller JT. 1989. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. EMBO J. 8:3905–3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. 1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63:1129–1136. 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 26. Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. 1993. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75:495–505. 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 27. Münger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM. 1989. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 8:4099–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dyson N, Howley PM, Münger K, Harlow E. 1989. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243:934–937. 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 29. Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, Yee D, Temiz NA, Donohue DE, McDougle RM, Brown WL, Law EK, Harris RS. 2013. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494:366–370. 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burns MB, Temiz NA, Harris RS. 2013. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 45:1–8. 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, Harris S, Shah RR, Resnick MA, Getz G, Gordenin DA. 2013. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 45:1–8. 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leonard B, Hart SN, Burns MB, Carpenter MA, Temiz NA, Rathore A, Vogel RI, Nikas JB, Law EK, Brown WL, Li Y, Zhang Y, Maurer MJ, Oberg AL, Cunningham JM, Shridhar V, Bell DA, April C, Bentley D, Bibikova M, Cheetham RK, Fan JB, Grocock R, Humphray S, Kingsbury Z, Peden J, Chien J, Swisher EM, Hartmann LC, Kalli KR, Goode EL, Sicotte H, Kaufmann SH, Harris RS. 2013. APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 73:7222–7231. 10.1158/0008-5472.CAN-13-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Taylor BJ, Nik-Zainal S, Wu YL, Stebbings LA, Raine K, Campbell PJ, Rada C, Stratton MR, Neuberger MS. 2013. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. eLife 2:e00534. 10.7554/eLife.00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Henderson S, Chakravarthy A, Su X, Boshoff C, Fenton TR. 2014. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep. 7:1833–1841. 10.1016/j.celrep.2014.05.012. [DOI] [PubMed] [Google Scholar]
- 35. Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, Menzies A, Martin S, Leung K, Chen L, Leroy C, Ramakrishna M, Rance R, Lau KW, Mudie LJ, Varela I, McBride DJ, Bignell GR, Cooke SL, Shlien A, Gamble J, Whitmore I, Maddison M, Tarpey PS, Davies HR, Papaemmanuil E, Stephens PJ, McLaren S, Butler AP, Teague JW, Jönsson G, Garber JE, Silver D, Miron P, Fatima A, Boyault S, Langerød A, Tutt A, Martens JWM, Aparicio SA, Borg Å, Salomon AV, Thomas G, Børresen-Dale AL, Richardson AL, Neuberger MS, Futreal PA, Campbell PJ, Stratton MR, Cancer Breast, Working Group of the International Cancer Genome Consortium 2012. Mutational processes molding the genomes of 21 breast cancers. Cell 149:979–993. 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jäger N, Jones DT, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt ANJ, Valdés-Mas R, van Buuren MM, van ’t Veer L, Vincent-Salomon A, Waddell N, Yates LR, Australian Pancreatic Cancer Genome Initiative. ICGC Breast Cancer Consortium. ICGC MMML-Seq Consortium. ICGC PedBrain. Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR. 2013. Signatures of mutational processes in human cancer. Nature 500:415–421. 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shinohara M, Io K, Shindo K, Matsui M, Sakamoto T, Tada K, Kobayashi M, Kadowaki N, Takaori-Kondo A. 2012. APOBEC3B can impair genomic stability by inducing base substitutions in genomic DNA in human cells. Sci. Rep. 2:806. 10.1038/srep00806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. 2010. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 38:4274–4284. 10.1093/nar/gkq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, Harris RS. 2011. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 85:11220–11234. 10.1128/JVI.05238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Refsland EW, Hultquist JF, Harris RS. 2012. Endogenous origins of HIV-1 G-to-A hypermutation and restriction in the nonpermissive T cell line CEM2n. PLoS Pathog. 8:e1002800. 10.1371/journal.ppat.1002800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lorenz LD, Rivera Cardona J, Lambert PF. 2013. Inactivation of p53 rescues the maintenance of high risk HPV DNA genomes deficient in expression of E6. PLoS Pathog. 9:e1003717. 10.1371/journal.ppat.1003717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. White EA, Kramer RE, Tan MJ, Hayes SD, Harper JW, Howley PM. 2012. Comprehensive analysis of host cellular interactions with human papillomavirus E6 proteins identifies new E6 binding partners and reflects viral diversity. J. Virol. 86:13174–13186. 10.1128/JVI.02172-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. White EA, Walther J, Javanbakht H, Howley PM. 2014. Genus beta human papillomavirus E6 proteins vary in their effects on the transactivation of p53 target genes. J. Virol. 88:8201–8212. 10.1128/JVI.01197-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chrisofos M, Skolarikos A, Lazaris A, Bogris S, Deliveliotis CH. 2004. HPV 16/18-associated condyloma acuminatum of the urinary bladder: first international report and review of literature. Int. J. STD AIDS 15:836–838. 10.1258/0956462042563783. [DOI] [PubMed] [Google Scholar]
- 45. Lee SS, Weiss RS, Javier RT. 1997. Binding of human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. U. S. A. 94:6670–6675. 10.1073/pnas.94.13.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kiyono T, Hiraiwa A, Fujita M, Hayashi Y, Akiyama T, Ishibashi M. 1997. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. U. S. A. 94:11612–11616. 10.1073/pnas.94.21.11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Javier RT. 2008. Cell polarity proteins: common targets for tumorigenic human viruses. Oncogene 27:7031–7046. 10.1038/onc.2008.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nagasaka K, Kawana K, Osuga Y, Fujii T. 2013. PDZ domains and viral infection: versatile potentials of HPV-PDZ interactions in relation to malignancy. Biomed. Res. Int. 2013:369712. 10.1155/2013/369712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dürst M, Gallahan D, Jay G, Rhim JS. 1989. Glucocorticoid-enhanced neoplastic transformation of human keratinocytes by human papillomavirus type 16 and an activated ras oncogene. Virology 173:767–771. 10.1016/0042-6822(89)90595-3. [DOI] [PubMed] [Google Scholar]
- 50. Pei XF, Meck JM, Greenhalgh D, Schlegel R. 1993. Cotransfection of HPV-18 and v-fos DNA induces tumorigenicity of primary human keratinocytes. Virology 196:855–860. 10.1006/viro.1993.1546. [DOI] [PubMed] [Google Scholar]
- 51. Crook T, Storey A, Almond N, Osborn K, Crawford L. 1988. Human papillomavirus type 16 cooperates with activated ras and fos oncogenes in the hormone-dependent transformation of primary mouse cells. Proc. Natl. Acad. Sci. U. S. A. 85:8820–8824. 10.1073/pnas.85.23.8820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Münger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K. 2004. Mechanisms of human papillomavirus-induced oncogenesis. J. Virol. 78:11451–11460. 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang Z, Wakae K, Kitamura K, Aoyama S, Liu G, Koura M, Monjurul AM, Kukimoto I, Muramatsu M. 2014. APOBEC3 deaminases induce hypermutation in human papillomavirus 16 DNA upon beta interferon stimulation. J. Virol. 88:1308–1317. 10.1128/JVI.03091-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Allen-Hoffmann BL, Schlosser SJ, Ivarie CA, Sattler CA, Meisner LF, Connor SL. 2000. Normal growth and differentiation in a spontaneously immortalized near-diploid human keratinocyte cell line, NIKS. J. Invest. Dermatol. 114:444–455. 10.1046/j.1523-1747.2000.00869.x. [DOI] [PubMed] [Google Scholar]
- 55. Todaro GJ, Green H. 1963. Quantitative studies of the growth of mouse embryo cells in culture and their development into established cell lines. J. Cell Biol. 17:299–313. 10.1083/jcb.17.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dickson MA, Hahn WC, Ino Y, Ronfard V, Wu JY, Weinberg RA, Louis DN, Li FP, Rheinwald JG. 2000. Human keratinocytes that express hTERT and also bypass a p16(INK4α)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol. Cell. Biol. 20:1436–1447. 10.1128/MCB.20.4.1436-1447.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wei W, Barron PD, Rheinwald JG. 2010. Modulation of TGF-β-inducible hypermotility by EGF and other factors in human prostate epithelial cells and keratinocytes. In Vitro Cell. Dev. Biol. Anim. 46:841–855. 10.1007/s11626-010-9353-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Degen M, Natarajan E, Barron P, Widlund HR, Rheinwald JG. 2012. MAPK/ERK-dependent translation factor hyperactivation and dysregulated laminin γ2 expression in oral dysplasia and squamous cell carcinoma. Am. J. Pathol. 180:2462–2478. 10.1016/j.ajpath.2012.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Smith JA, White EA, Sowa ME, Powell ML, Ottinger M, Harper JW, Howley PM. 2010. Genome-wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. Proc. Natl. Acad. Sci. U. S. A. 107:3752–3757. 10.1073/pnas.0914818107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Broad Institute. TCGA Genome Data Analysis Center 2013. Mutation analysis (MutSigCV v0.9). Broad Institute of MIT and Harvard, Cambridge, MA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Upregulation of APOBEC3B in primary keratinocytes by expression of HPV E6. A3B mRNA levels in G5-Ep primary keratinocytes transduced with HPV E6 from different high-risk (HPV16, -18, -33, -45, and -52), low-risk (HPV6b and -11), or noncancer (HPV2a and -57) types or with an empty vector (Cont.). Each histogram bar shows the mean A3B expression level normalized to TBP (error bars report standard deviations from triplicate assays). Low-risk/noncancer E6 proteins did not cause significant A3B upregulation compared to high-risk E6 proteins (P < 0.05; two-tailed Student’s t test). Download