

Abstract
In the early 1900s, the abnormal toxicity test (ATT) was developed as an auxiliary means to ensure safe and consistent antiserum production. Today, the ATT is utilized as a quality control (QC) release test according to pharmacopoeial or other regulatory requirements. The study design has not been changed since around 1940. The evidence of abnormal toxicity testing as a prediction for harmful batches is highly questionable and lacks a scientific rationale. Numerous reviews of historical ATT results have revealed that no reliable conclusions can be drawn from this QC measure. Modern pharmaceutical manufacturers have thorough control of the manufacturing process and comply with good manufacturing practice rules. Contaminants are appropriately controlled by complying with the validated manufacturing processes and strict QC batch release confirming batch-to-batch consistency. Recognizing that product safety, efficacy, and stability can be ensured with strict QC measures, nowadays most regulatory authorities do not require the ATT for most product classes. In line with the replacement, reduction, and refinement (3Rs) initiative, the test requirement has been deleted from approximately 80 monographs of the European Pharmacopoeia and for the majority of product classes in the United States. For these reasons, it is recommended that the ATT should be consistently omitted world-wide and be removed from pharmacopoeias and other regulatory requirements.
Keywords: abnormal toxicity test, analysis, biotechnology, general safety, innocuity test, pharmacopoeia, quality control, regulatory science, toxicity, vaccines
INTRODUCTION
The abnormal toxicity test (ATT) [European Pharmacopoeia (EP) nomenclature]1 is also referred to as the general safety (US reference)2 or innocuity test (WHO nomenclature).3 The principle of this animal test consists of a single injection of a specified volume of a product batch into guinea pigs and/or mice followed by an observation period. Typically, a batch passes the test if the findings seen in animals follow the below criteria:
animals survive the test period
animals do not exhibit any response, which is not specific for or expected from the product and may indicate a difference in its quality; and
animals weigh not less at the end of the test period than that at the time of injection.2
The ATT was developed in the early 1900s, when production processes and quality control (QC) for biological products were poorly established and licensing procedures did not yet exist. At this time, the test was intended to ensure the safe and consistent production of serum products, for example, to titrate the preservative phenol level of diphtheria antiserum. Analytical techniques were not available to appropriately detect phenol in serum products. Therefore, mice—as a susceptible species—were used for the detection of potentially toxic phenol levels. The test with guinea pigs was introduced around 1900 as a biological indicator for the presence of tetanus toxin in antiserum preparations.4,5
The test was later expanded to a general safety test to detect extraneous contaminants (other than, for example, bacterial endotoxins) in biological products. In spite of significant evolution of analytical techniques as well as advanced process understanding and validation approaches, this biological test remains and changed from being an analytical test to an additional safety test intended to detect product/process contaminants to avoid batch-to-batch differences in quality.
The test has not significantly changed since around 1940. National implementation leads to today's variations between the different pharmacopoeias/international requirements. Table1 presents requirements from the pharmacopoeias of Europe,1 the United States,2 Russia,6 and China7 as well as WHO requirements.3
Table 1.
Comparison of Test Conditions According to Different Pharmacopoeias/Requirements (Examples)
| European Pharmacopoeia1 | United Statesa | WHO3 | Russian Pharmacopoeia6 | Chinese Pharmacopoeia7 | ||||
|---|---|---|---|---|---|---|---|---|
| Scope | General Test | Immunosera/ Vaccines | Biological Products (with exemptionsb) | Vaccines | General Test | Vaccines/ Sera | Biologics/ Vaccines | Chemicals, Traditional Medicines |
| Blank control | No | No | No | No | No | No | Blank control | No |
| Animal quantity | 5 mice | 5 mice | ≥5 mice | 5 mice | 5 mice | 5 mice | 5 mice | 5 mice |
| 2 guinea pigs | ≥2 guinea pigs | 2 guinea pigs | 2 guinea pigs | 2 guinea pigs | ||||
| Body weight (g) | 17–24 | 17–24 (m) | <22 (m) | 17–22 (m) | 19–21 | 17–20 (m) | 18–22 (m) | 17–20 |
| 250–400 (gp) | <400 (gp) | 250–350 (gp) | 250–300 (gp) | 250–350 (gp) | ||||
| Dose/ administration volume | One human dose | One human dose | ≤0.5 mL (m) | One human dose | 0.5 mL | One human dose | 0.5 mL (m) | 0.5 mL (m) |
| ≤1.0 mL | ≤1.0 mL (m) | ≤5.0 mL (gp) | ≤ 1.0 mL (m) | ≤ 1.0 mL (m) | 5.0 mL (gp) | |||
| ≤5.0 mL (gp) | ≤ 1.0 mL (gp) | ≤ 5.0 mL (gp) | ||||||
| Injection route | i.v. | i.p. | i.p.or following the approved route of product administration | i.p. | i.v. | i.p. | i.p. | Following the approved route of product administration |
| Observation time | 24 h | 7 days | 48 h | 48 h | 48 h | 7 days | 7 days | 2 days |
| Acceptance criteria | No animal dies within 24 h or within such time as specified in the individual monograph | No animal shows signs of ill health | No animal dies or exhibits any response, which is not specific for or expected from the product and may indicate a difference in its quality. No loss of body weight | No animal dies within at least 7 days or shows significant signs of toxicity | No animal dies within the specified follow-up period | No animal dies within at least 7 days, shows significant signs of toxicity, or a decrease in body weight | All animals remain healthy and survive the observation period, without any abnormal reaction, and with an increase in body weight by the end of observation period | All animals survive the observation period |
| Retest(s) number/ description | One | One | Two | No | One | One | One | One |
| If one animal dies, repeat the test | If one animal dies or shows signs of ill health, repeat the test | If the initial test/first repeat test fails, a repeat test may be conducted | If an animal dies, repeat the experiment with five mice (20 ± 0.5 g) | If an animal dies, shows clinical signs of intoxication or a decrease in body weight, repeat experiment under the same conditions | If the test fails, it may be repeated once with 10 mice/4 guinea pigs | If the test fails, it may be repeated once with 10 mice (18–19 g) | ||
The United States Pharmacopoeia (USP 36) refers to US Code of Federal Regulations (21 CFR, Part 610).2
Exemptions: therapeutic DNA plasmid products, therapeutic synthetic peptide products of 40 or fewer amino acids, monoclonal antibody products for in vivo use, or therapeutic recombinant DNA-derived products.
i.p., intraperitoneal; i.v., intravenous; m, mice; gp, guinea pigs.
Of note, the administration volumes in the case of intravenous (i.v.) dosing do not comply with today's best practices and animal welfare considerations. According to Diehl et al.,8 a maximum of 5 mL/kg should be administered to mice, which would result in a maximum volume of 0.1 mL for mice with a body weight of around 20 g. Following the pharmacopoeias/requirements (0.5–1.0 mL administration volume), a mouse receives 5–10-fold of the volume considered good practice.
HISTORICAL DATA ANALYSES
Publications by the German Paul Ehrlich Institute provide evidence that the test does not serve its purpose and does not add any further information to that already obtained from QC release testing under good manufacturing practice (GMP).9 A retrospective analysis of several thousand test results conducted for vaccines revealed that there were no true positive results10,11 and no batch rejection was obtained by the authority.12 In conclusion, there is no evidence that the ATT is useful as a predictor or control for harmful batches. Table2 presents details on the analysis of historical data from Kraemer et al.12
Table 2.
Retrospective Analysis of Abnormal Toxicity Tests of Vaccine Products, Test Results, and Number of Animals12
| Number of Tests | Analyzed Preparations | Used Mice | Used Guinea Pigs | Batch Rejections |
|---|---|---|---|---|
| 5896 | 416 | 30193 | 12420 | 0 |
As a result of the historical experience, the US Food and Drug Administration (FDA) amended the respective regulations regarding general biological products standards by adding an administrative procedure for obtaining exemptions from the test requirements.13 A similar conclusion was drawn by the European agency, and the test was removed from over 80 product monographs.11,14 For details, see the section given below.
TEST PERFORMANCE
Specificity (False Positive Results)
The ATT is nonspecific, as many factors other than contaminants can influence the result (e.g., body weight, species and strain differences, stress levels of the animals). Accordingly, misinterpretation of responses caused by the active ingredients themselves, or formulation components, may lead to false positive results, for example, as administered concentrations may be unrealistically high compared with what is administered to humans.15 The formulations are optimized for safe use in humans but not in mice or guinea pigs at such high concentrations.
Depending on the test design, a fixed volume is administered, irrespective of the dose used in humans. Thus, the full human dose may be administered to guinea pigs of 250–400 g body weight.1 In this case, assuming a human body weight of 60 kg, a guinea pig would receive 150-fold of the human dose. A mouse of 20 g would receive 3000-fold of the human dose.
As already mentioned, formulation components (e.g., preservative and vaccine adjuvants) may cause false positive responses. There are some examples of drug products that have yielded false positive test results:
A response following intraperitoneal injection of high concentrations of benzyl alcohol, which is used as a formulation component for a recombinant protein.
A reaction caused by the high sugar content in an oral pediatric vaccine, when administered according to a national pharmacopoeia by i.v. injection.
Reproducibility
Kraemer et al.12 investigated the test reproducibility if the ATT is performed with strict adherence to the same study design/protocol (German Pharmacopoeia DAB 10). Identical batches tested in different laboratories have produced significantly different test results. Positive results never showed a correlation with product quality or contamination, and the same batches passed the ATT in subsequent test repetitions.
In addition, the ATT design is not harmonized and varies between national pharmacopoeias/international requirements (Table1).
We conclude that test responses of the same batch may be unpredictable and may give contradictory results if performed:
repeatedly in the same laboratory,
in different laboratories according to the same protocol, and
in different countries according to different protocols.
Reliability
Reliability is a measure of consistency/reproducibility. Considering the lack of reproducibility, the ATT must be classified as unreliable.
Suitability
For common analytical procedures (e.g., quantitative tests for impurities’ content), it is mandatory to demonstrate suitability for the intended purpose. Typical parameters of such validation are precision (repeatability) and specificity.16 The ATT is neither reproducible nor specific. Accordingly, the ATT would not fulfill international validation criteria of analytical methods.
No adequate positive control is possible as the test seeks for unknown contamination and nonspecific toxicity. Furthermore, the ATT lacks explicit acceptance criteria, as no definite endpoint is defined (“signs of ill health,”1 “significant signs of toxicity,”3 and “abnormal reaction”7).
In conclusion, this animal test is considered as not suitable for its intended purpose.
Would Modification Lead to an Improved Test Performance?
Mizukami et al.17 made an effort to improve the ATT design for vaccines (Fig. 1). It is based on the observation that body weight changes of animals are characteristic for each vaccine, and such standardized changes can be used as references for evaluating vaccines. In addition to body weight analysis, histopathological and hematological analyses are proposed in case of required retests.
Figure 1.
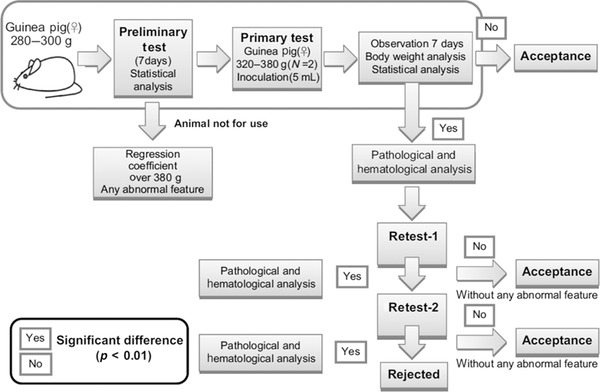
Proposed test scheme according to Mizukami et al.17
In our opinion, this modified scheme does not overcome the principal deficiencies of the test as discussed above, or the inherent variations of the animal model. This variability is also evident in the fact that still up to two retests are foreseen in the scheme. Furthermore, the test design is not considered appropriate to be used as a routine QC release test due to the high experimental efforts (statistical and pathological/hematological analysis) and the long test duration (several weeks including retests). From an ethical perspective, such studies are not in line with animal welfare considerations as a significant number of animals are used for the initial statistical analysis (in order to standardize body weight changes after inoculation of various vaccines) and the potential retests.
CONTROL OF CONTAMINANTS
Scientifically, there is no rationale as to why an animal test for batch release would be more appropriate than other measures to detect and control contamination. For such a test to be adequate and useful, a clear understanding of the mechanism of how contaminations lead to the study read-out, i.e. positive result, should be in place.
Furthermore, it should be known based on the understanding of the manufacturing process what potential contaminants might be and of what nature they could be. The endpoints and acceptance criteria should be deduced from this understanding so that any QC test can be appropriately validated.
Modern methodologies, such as those presented in Table3, are far better suited and have a clear scientific rationale because the relationship between the measured endpoint and the causing contamination (e.g., bacterial endotoxin) is well understood and established.
Table 3.
Measures to Verify the Absence of Different Types of Contaminants (Examples)
| Type of Contaminant | Measure to Verify the Absence of Contaminants in a Product Batch |
|---|---|
| Microbiological | – Bioburden test (in-process control) |
| – Sterility test | |
| Pyrogena | – Validation of depyrogenization (as part of the process validation) |
| Endotoxin | – Bacterial endotoxins (limulus amebocyte lysate) test |
| Residual contaminantsb | – Extended product characterization |
| – Process validation | |
| – Manufacture under GMP | |
| – QC during batch release to confirm batch-to-batch consistency |
A set of measures are nowadays available to detect and control different types of contaminants. These include:
extended product characterization during process development and process validation,
manufacture according to GMPs, and
routine QC release testing, which verifies batch-to-batch consistency and ensures that a specific batch has been manufactured according to the previously validated process.
Table 3 lists the current control measures used to verify the absence of certain contaminants in a product batch, exemplified by parenteral preparations.11,14
MODERN PRODUCT DEVELOPMENT ENSURES COMPREHENSIVE PROCESS UNDERSTANDING AND WELL-CHARACTERIZED PRODUCTS
Nowadays, pharmaceutical manufacturing is highly regulated and controlled. The modern pharmaceutical industry together with regulatory authorities have established appropriate control of the manufacturing process through substantially advanced process understanding, in-process controls, validation of the manufacturing process, and release testing complying with international GMP standards.
During formulation and process development, many studies are conducted with different formulation components (including preservatives) to investigate degradation profiles, product compatibility with various materials/surfaces, and leachables, which may be sources for contaminants.18–21 Pharmaceutical compounds are tested extensively with regard to their safety/toxicity profile in in vitro assays and animals models as well as in clinical trials in accordance with international (e.g., International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) and national guidelines. A marketing authorization is granted by the relevant health authorities only when a positive benefit/risk assessment can be demonstrated.
Today, pharmaceutical manufacturers produce highly developed medicines with well-defined purity and safety characteristics. Risk of contamination is extremely low if a manufacturer complies with GMP rules (e.g., globally recognized regulations22–25) and if consistency in production is guaranteed.26,27 Abnormal product contamination is extremely unlikely if the validated manufacturing process is followed.
Appropriate analytical methods (e.g., mass spectrometry applications) are capable of detecting contamination and ensure batch-to-batch consistency. Advanced product testing is applied for the extended product characterization and release testing.
RELEASE SPECIFICATIONS ARE SET ACCORDING TO INTERNATIONAL REQUIREMENTS AND ENSURE PRODUCT SAFETY, EFFICACY, AND STABILITY
Many multinational manufacturers supply innovative medicines globally. Thus, a batch is usually released for use in the global market. Accordingly, all countries get the same high quality drug. In line with international regulations, in most instances abnormal toxicity testing is not part of the release specifications for these globally marketed products. For example, European Medicines Agency (EMA)- and FDA-approved specifications for commercial drug products do not require abnormal toxicity testing as part of the QC release analysis for the majority of product classes. However, a batch already released for EU and/or the United States would have to be tested for abnormal toxicity in other countries, for example, the Russian Federation6 and China,7 to be released for the local market. To the best of our knowledge, no batch which met the EMA- or FDA-approved specifications delivered a positive ATT result in both of these countries (apart from false positive test results, as aforementioned).10–12
INCREASING INTERNATIONAL CONVERGENCE
Several health authorities have conducted an evaluation of the ATT and came to the same conclusion.
European Pharmacopoeia generally does not require abnormal toxicity testing in the monographs for “parenteral preparations,”28 “monoclonal antibodies for human use,”29 or “products of recombinant DNA technology.”30 Aforementioned reviews of test results revealed that no additional value could be concluded from abnormal toxicity testing. As a consequence, and in accordance with the European Convention on the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes, the test has been deleted from approximately 80 monographs for biotechnological products, blood products, antibiotics, and vaccines based on the review of historical data.11,14 In addition, further replacement alternatives for abnormal toxicity testing for batch release of existing vaccines have been currently evaluated and strongly recommended.10,31–35
The US Code of Federal Regulations Title 21 requires general safety testing be done for biological products (21 CFR, Part 610.112). However, FDA realized that “after more than a decade of experience with these products, we found that we could evaluate many aspects of a biological product's safety, purity, or potency with tests other than those prescribed in part 610.”13 Thus, the FDA amended the biologics regulations regarding general biological products standards by adding an administrative procedure for obtaining exemptions from the general safety test requirements13: 21 CFR, Part 601.2 (Ref. 36) specifies that the test is exempted as a requirement for license applications for therapeutic DNA plasmid products, therapeutic synthetic peptide products of 40 or fewer amino acids, monoclonal antibody products for in vivo use, or therapeutic recombinant DNA-derived products.
In 2002, the WHO Expert Committee on Biological Harmonization “noted that, in one region of the world, the abnormal toxicity test had been deleted for most products. This was linked to the implementation of, and compliance with, good manufacturing practices and, where this occurred, there was abundant evidence that the abnormal toxicity test did not provide additional assurances of the quality of the product.”37
ANIMAL WELFARE
The EU adopted a new directive on the protection of animals used for scientific purposes (2010/63/EU38). The directive plays a significant role in minimizing the number of animals used in experiments and the European Directorate for the Quality of Medicines & HealthCare continues to push forward the implementation of replacement, reduction, and refinement (3Rs) alternatives.39
The substantial number of laboratory animals used for the ATT cannot be justified in view of its unproven and questionable suitability to detect contaminants and increase the product safety.15 As a consequence, the European Convention on the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes reviewed the test for 3Rs. As aforementioned, the test has been consequently deleted from numerous EP monographs.11,14
CONCLUSIONS
The ATT was implemented in the early 1900s as auxiliary means for production, i.e., to titrate the level of the preservative phenol in antiserum preparations, and has persisted for the detection of contaminants, without clear rationale and reasoning. The test lacks scientific merit and is neither specific, reproducible, reliable, nor suitable for the intended use. Considering its performance, the ATT is regarded as not appropriate as a QC release test, which should be capable of enabling batch release decisions based on explicit and reproducible results.
A retrospective analysis of several thousand test results revealed that no true positive result and no batch rejection were obtained. Positive results never showed a correlation to the product quality or contamination. Accordingly, the test does not provide added value for QC. This conclusion was also drawn by the FDA considering that safety, purity, and potency of products are ensured without the ATT.
The ATT has therefore been deleted from numerous product monographs, and test exemptions are granted for many product classes, respectively. However, the test is still required as a safety test according to a number of pharmacopoeias and other regulatory requirements for certain product classes.
Based on the rationale provided in this review and in line with the scientific knowledge and regulatory trends outlined herein, it is considered fully justified to completely eliminate abnormal toxicity testing from pharmacopoeias and other regulatory requirements as already recommended by various scientific experts.12,15,35,40,41 This would also be in agreement with animal welfare concerns (e.g., 3Rs initiatives) and contemporary directives on the protection of animals.
Glossary
Abbreviations used
- 3Rs
replacement, reduction, and refinement
- ATT
abnormal toxicity test
- EMA
European Medicines Agency
- EP
European Pharmacopoeia
- FDA
US Food and Drug Administration
- GMP
good manufacturing practice
- QC
quality control.
REFERENCES
- 1.European Pharmacopoeia (8.0). 2013. Chapter 2.6.9: Abnormal toxicity.
- 2.US Food and Drug Administration. U.S. Food and Drug Administration's Code of Federal Regulations (CFR), 2013. title 21, part 610: General biological products standards.
- 3.World Health Organization. Geneva, Switzerland: WHO: 1990. Expert Committee on Biological Standardization. Fortieth report. Technical report series. Vol. 800 (Annex. 2) [PubMed] [Google Scholar]
- 4.Otto R. The state control of immunosera. In: Ehrlich P, editor. Work from the Royal Institute for Experimental Therapy in Frankfurt a.M. Vol. 2. Jena, Germany: Gustav Fischer; 1906. [Google Scholar]
- 5.Marxer A. Technology of vaccines and immunosera. Braunschweig, Germany: Friedr. Vieweg & Sohn; 1915. [Google Scholar]
- 6.Russian State Pharmacopoeia (XII). Monograph on biological methods of control; 25: Abnormal toxicity. 2007.
- 7.Appendix XII F: Test for abnormal toxicity. Pharmacopoeia of The People's Republic of China. 2010.
- 8.Diehl KH, Hull R, Morton D, Pfister R, Rabemampianina Y, Smith D, Vidal JM, van deVorstenboschC. A good practice guide to the administration of substances and removal of blood, including routes and volumes. J Appl Toxicol. 2001;21(1):15–23. doi: 10.1002/jat.727. [DOI] [PubMed] [Google Scholar]
- 9.Sponer G. Animal experiments in the context of quality control of pharmaceuticals. ALTEX. 2004;21(2):73–80. [PubMed] [Google Scholar]
- 10.Schwanig M, Nagel M, Duchow K, Kraemer B. Elimination of abnormal toxicity test for sera and certain vaccines in the European Pharmacopoeia. Vaccine. 1997;15(10):1047–1048. doi: 10.1016/s0264-410x(97)00074-1. [DOI] [PubMed] [Google Scholar]
- 11.Castle P. Replacement, reduction, refinement (3Rs): Animal welfare progress in European Pharmacopoeia monographs. Pharmeuropa. 1997;19(3):430–441. [PubMed] [Google Scholar]
- 12.Kraemer B, Nagel M, Duchow K, Schwanig M, Cussler K. Is the abnormal toxicity test still relevant for the safety of vaccines, sera and immunoglobulins? ALTEX. 1996;13(1):7–16. [PubMed] [Google Scholar]
- 13.U.S. Food and Drug Administration. Revision to the general safety requirements for biological products. Fed Reg. 2003;68(42):10157–10160. [Google Scholar]
- 14.Artiges A. Alternatives to animals in development and control of biological products for human and veterinary use. The role of the European Pharmacopoeia. Dev Biol Stand. 1999;101:29–35. [PubMed] [Google Scholar]
- 15.Duchow K, Kramer B. Abnormal toxicity—A relevant safety test under GLP- and GMP-conditions in the production of vaccines? ALTEX. 1994;11(5):11–18. [PubMed] [Google Scholar]
- 16.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Guideline Q2(R1) Validation of analytical procedures: Text and methodology. Fed Reg. 1997;62(96):27463–27467. [Google Scholar]
- 17.Mizukami T, Masumi A, Momose H, Kuramitsu M, Takizawa K, Naito S, Maeyama J, Furuhata K, Tsuruhara M, Hamaguchi I, Yamaguchi K. An improved abnormal toxicity test by using reference vaccine-specific body weight curves and histopathological data for monitoring vaccine quality and safety in Japan. Biologicals. 2009;37(1):8–17. doi: 10.1016/j.biologicals.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 18.Hawe A, Wiggenhorn M, De Weert M Van, Garbe JHO, Mahler HC, Jiskoot W. Forced degradation of therapeutic proteins. J Pharm Sci. 2012;101:895–913. doi: 10.1002/jps.22812. [DOI] [PubMed] [Google Scholar]
- 19.Wakankar AA, Wang YJ, E Canova-Davis, Ma S, Schmalzing D, Grieco J, Milby T, Reynolds T, Mazzarella K, Hoff E, Gomez S, S Martin-Moe. On developing a process for conducting extractable–leachable assessment of components used for storage of biopharmaceuticals. J Pharm Sci. 2010;99(5):2209–2218. doi: 10.1002/jps.22012. [DOI] [PubMed] [Google Scholar]
- 20.Kamerzell TJ, Esfandiary R, Joshi SB, Middaugh CR, Volkin DB. Protein–excipient interactions: Mechanisms and biophysical characterization applied to protein formulation development. Adv Drug Deliver Rev. 2011;63(13):1118–1159. doi: 10.1016/j.addr.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 21.Wu Y, Levons J, Narang AS, Raghavan K, Rao VM. Reactive impurities in excipients: Profiling, identification and mitigation of drug–excipient incompatibility. AAPS Pharm Sci Tech. 2011;12(4):1248–63. doi: 10.1208/s12249-011-9677-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.World Health Organization. WHO good manufacturing practices for pharmaceutical products: Main principles. 2011. Technical report series, No. 961 (Annex 3)
- 23.EU guidelines to good manufacturing practice medicinal products for human and veterinary use. 2003. EudraLex. Vol. 4.
- 24.US Food and Drug Administration. U.S. Food and Drug Administration's code of federal regulations (CFR), title 21, part 211: Current good manufacturing practice for finished pharmaceuticals. 2013.
- 25.PIC/S. Guide to good manufacturing practice for medical products. PE 009–11. 2014.
- 26.Cussler K. A 4R concept for the safety testing of immunobiologicals. Dev Biol Stand. 1999;101:121–126. [PubMed] [Google Scholar]
- 27.Milstien J, Grachev V, Padilla A, Griffiths E. WHO activities towards the three Rs in the development and control of biological products. Dev Biol Stand. 1996;86:31–9. [PubMed] [Google Scholar]
- 28.European Pharmacopoeia (8.0). Monograph on parenteral preparations. 2013.
- 29.European Pharmacopoeia (8.0). Monograph on monoclonal antibodies for human use. 2013.
- 30.European Pharmacopoeia (8.0). Monograph on products of recombinant DNA technology. 2013.
- 31.Metz B, Hendriksen CF, Jiskoot W, Kersten GF. Reduction of animal use in human vaccine quality control: Opportunities and problems. Vaccine. 2002;20(19–20):2411–2430. doi: 10.1016/s0264-410x(02)00192-5. [DOI] [PubMed] [Google Scholar]
- 32.Castle P. Policy and progress of the European Pharmacopoeia in the use of alternatives to animal testing in vaccine production and quality control. In: van Iersel AAJ HendriksenCFM., editor. Alternatives to animal testing in the production and control of vaccines: Present practice and perspectives. The Netherlands: National Institute for Public Health and the Environment: Bilthoven; 1993. pp. 30–36. [Google Scholar]
- 33.Cussler K, Kulpa J, Calver J. The international symposium on regulatory testing and animal welfare: Recommendations on best scientific practices for biologicals: Safety and potency evaluations. ILAR J. 2002;43(Suppl):S126–S128. doi: 10.1093/ilar.43.suppl_1.s126. [DOI] [PubMed] [Google Scholar]
- 34.Hendriksen C. Three Rs achievements in vaccinology. AATEX. 2007;14(Special Issue):575–579. [Google Scholar]
- 35.Gupta RK. Is the test for abnormal toxicity, general safety or innocuity necessary for vaccines? Vaccine. 1996;14(17/18):1716. doi: 10.1016/s0264-410x(96)00132-6. [DOI] [PubMed] [Google Scholar]
- 36.US Food and Drug Administration. U.S. Food and Drug Administration's Code of Federal Regulations (CFR), title 21, part 601.2. Applications for biologics licenses, procedures for filing. 2013.
- 37.World Health Organization. Geneva, Switzerland: WHO: 2002. Expert Committee on Biological Standardization. Fortieth Report. Technical Report Series. Vol. 904. [PubMed] [Google Scholar]
- 38.European Parliament and the Council. Directive 2010/63/EU of 22 September 2010 on the protection of animals used for scientific purposes. 2010.
- 39.Milne C, Buchheit KH. EDQM's 3R activities in the field of quality control of vaccines. ALTEX Proc. 2012;1/12:65–69. [Google Scholar]
- 40.Hendriksen CFM, Garthoff B, Aggerbeck H, Bruckner L, Castle P, Cussler K, Dobbelaer R, van deDonkH, van derGunJ, Lefrancois S, Milstien J, Minor PD, Mougeot H, Rombaut B, Ronneberger HD, Spieser JM, Stolp R, Straughann DW, Tollis M, Zigtermans G. Alternatives to animal testing in the quality control of immunobiologicals: Current status and future prospects. ATLA. 1994;22:420–434. [Google Scholar]
- 41.Associate Parliamentary Group for Animal Welfare (represented by the British Union for the Abolition of Vivisection, BUAV; the British Veterinary Association, BVA; the Fund for the Replacement of Animals in Medical Experiments, FRAME; the Royal Society for the Prevention of Cruelty to Animals, RSPCA; and the Association of the British Pharmaceutical Industry, ABPI) The use of animals in vaccine testing for humans. 2005. Working Group Report.