

Abstract Abstract
Right ventricular (RV) function is a major determinant of the symptomatology and outcome in pulmonary hypertension. The normal RV is a thin-walled flow generator able to accommodate large changes in venous return but unable to maintain flow output in the presence of a brisk increase in pulmonary artery pressure. The RV chronically exposed to pulmonary hypertension undergoes hypertrophic changes and an increase in contractility, allowing for preserved flow output in response to peripheral demand. Failure of systolic function adaptation (homeometric adaptation, described by Anrep’s law of the heart) results in increased dimensions (heterometric adaptation; Starling’s law of the heart), with a negative effect on diastolic ventricular interactions, limitation of exercise capacity, and vascular congestion. Ventricular function is described by pressure-volume relationships. The gold standard of systolic function is maximum elastance (Emax), or the maximal value of the ratio of pressure to volume. This value is not immediately sensitive to changes in loading conditions. The gold standard of afterload is arterial elastance (Ea), defined by the ratio of pressure at Emax to stroke volume. The optimal coupling of ventricular function to the arterial circulation occurs at an Emax/Ea ratio between 1.5 and 2. Patients with severe pulmonary hypertension present with an increased Emax, a trend toward decreased Emax/Ea, and increased RV dimensions, along with progression of the pulmonary vascular disease, systemic factors, and left ventricular function. The molecular mechanisms of RV systolic failure are currently being investigated. It is important to refer biological findings to sound measurements of function. Surrogates for Emax and Ea are being developed through bedside imaging techniques.
Keywords: right ventricle, pulmonary hypertension, preload, afterload, maximum elastance, end-systolic elastance, arterial elastance
One must inquire how increasing pulmonary vascular resistance results in impaired right ventricular function.
—J. T. Reeves, 19891
In 1988, Jack Reeves and his colleagues noted that pulmonary hypertension is a common complication of cardiac and pulmonary diseases, that associated alterations in right ventricular (RV) function cause symptoms and limit survival, and that, paradoxically, research in this area had been relatively scarce.1 Accordingly, they called for more studies to improve the understanding of RV failure in health and disease. Twenty-five years later, there has been significant progress, but our present knowledge remains incomplete, and calls for more research have been repeated.2,3
The right ventricle (RV) in mammals and birds is a thin-walled flow generator, designed to accommodate the entire systemic venous return undergoing gas exchange in the pulmonary circulation, which is a separate high-flow, low-pressure system.4 The pulmonary vascular pressures at rest and at mild levels of exercise are so low that a mean systemic filling pressure in the range of 10–15 mmHg is sufficient to drive the venous return through the pulmonary circulation to the left heart without the assistance of RV pumping. In 1943, Starr and his colleagues5 showed that ablation of the RV in dogs is compatible with life with little change in pulmonary venous pressure. In 1971, Fontan and Baudet6 introduced the first cavopulmonary anastomosis bypassing the RV as a palliative intervention for cardiac malformations. Patients with the so-called Fontan circulation may enjoy a near-normal sedentary life for several decades but rapidly deteriorate in case of increased pulmonary artery pressure (Ppa), for example, with altitude exposure or when the left ventricular (LV) filling pressure increases.7
The structure of the RV is not adequate to cope with a brisk increase in pulmonary vascular resistance (PVR) produced, for example, by pulmonary arterial constriction to mimic massive pulmonary embolism.8 However, if given time, the RV is able to adapt to a progressive increase in PVR by increasing contractility and remodeling, basically much like the left ventricle (LV).9 Beat-to-beat changes in preload or afterload are accompanied by a heterometric dimension adaptation described by Starling’s law of the heart. Sustained changes in load are associated with a homeometric contractility adaptation described by Anrep’s law of the heart.
In 1912, Gleb Vassilevitch von Anrep,10 who was active at that time in Pavlov’s laboratory in St. Petersburg, reported on the rapid increase in LV contractility in response to aortic constriction. Von Anrep was sent by Pavlov to London to work, under Starling’s supervision, on the humoral control of digestion. He soon observed the rapid switch from the heterometric to the homeometric adaptation to loading in Starling’s heart-lung preparation. Thus, after an acute increase in venous return or systemic vascular resistance, the heart initially dilates, allowing for increased or maintained stroke volume (SV), respectively, but after a few minutes the cardiac dimensions are back to baseline values in spite of persistently increased loading, indicating increased contractility. Starling thought that much of this observation was related to rapid deterioration of the experimental preparation. Later studies confirmed the predominant role of the homeometric, or systolic function, adaptation within the first few minutes after an acute increase in either preload or afterload.11 This is illustrated by a 10-minute recording of RV volumes after the acute increase in venous return in Figure 1.
Figure 1.
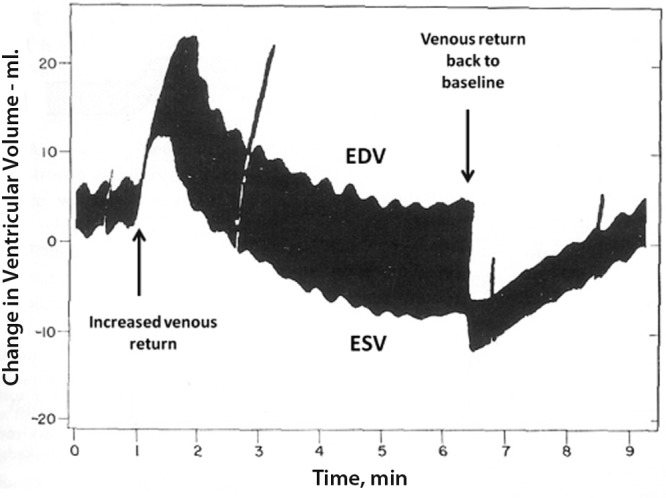
Time-course of right ventricular volume changes after a brisk increase in venous return. The initial heterometric adaptation is followed by a homeometric adaptation, allowing for a return to the initial end-diastolic volume (EDV) with decreased end-systolic volume (ESV) and increased stroke volume. Reproduced from Rosenblueth et al.11 with permission.
Whether the time course of systolic function adaptation to afterload is the same for the RV and the LV is not known. Comparisons between studies are difficult because of the differences in ventricular structure and relative changes in arterial pressure. Contractility responses to increased afterload may depend on the volume status or on systemic disease components. Ventricular hypertrophy occurs, which increases the contractile force and decreases wall tension, but the time course, pathobiology, and contribution to preserved contractility are not well understood. When loading conditions become excessive and prolonged, the homeometric adaptation eventually fails, and Starling’s heterometric adaptation comes again into play, at the price of increased dimensions and filling pressures.9
Accordingly, RV failure can be defined by a dyspnea-fatigue syndrome with eventual systemic congestion, caused by the inability of the RV to maintain flow output using Anrep’s homeometric adaptation, in response to metabolic demand, without the use of Starling’s heterometric adaptation.9 In this definition, cardiac output is not necessarily low but is low relative to oxygen uptake or aerobic exercise capacity, and vascular congestion is implicit.
Systolic function
The gold-standard measurement of cardiac contractility in an intact being is the maximal elastance (Emax), or the maximum value of the ratio of ventricular pressure to volume during the cardiac cycle.9 LV Emax coincides with end systole and is thus equal to the ratio of end-systolic pressure (ESP) to end-systolic volume (ESV). End-systolic elastance (Ees) is measured at the upper left corner of a square-shaped pressure-volume loop.12 Because of low pulmonary vascular impedance, the normal RV pressure-volume loop has a triangular shape, and Emax occurs before the end of ejection, or end systole. However, a satisfactory definition of Emax can be obtained by the generation of a family of pressure-volume loops at decreasing venous return, as illustrated in Figure 2.13 Thus, in spite of embryological and structural differences, the “laws of the heart” apply similarly to the RV and the LV.
Figure 2.
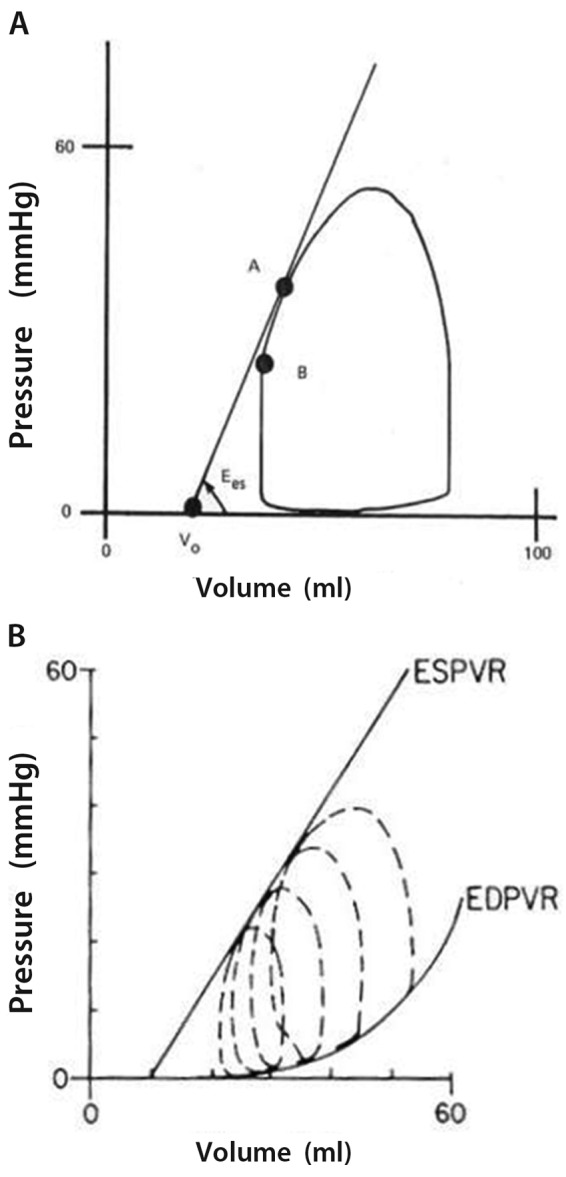
A, A normal right ventricular pressure-volume loop is of triangular shape, with maximum elastance (or Ees, point A) occurring before the end of systole (point B). B, A decrease in venous return allows for the recording of a family of right ventricular pressure-volume loops and the use of end-systolic pressure-volume relationship (ESPVR) and end-diastolic pressure-volume relationship (EDPVR) to define systolic and diastolic function. Reproduced from Maughan et al.13 with permission.
Instantaneous measurements of RV volumes are difficult at the bedside, and so are manipulations of venous return. This is why single-beat methods have been developed, initially for the LV14 and then for the RV.15 The single-beat method relies on a maximum-pressure (Pmax) calculation from a nonlinear extrapolation of the early and late portions of an RV pressure curve, an integration of pulmonary flow, and synchronization of the signals. The Emax is estimated from the slope of a tangent from Pmax to the pressure-volume curve (Figure 3). It is important to note that this graphic analysis uses relative changes in volume and thus does not require measurement of absolute volumes. This is acceptable because Emax is essentially preload, or end-diastolic volume (EDV), independent.9 On the other hand, excellent agreement between Pmax measured directly by clamping the main pulmonary artery for one beat and Pmax estimated by simple sinusoidal extrapolation of early and late portions of the RV pressure curve has been demonstrated in a large-animal experimental preparation.15
Figure 3.
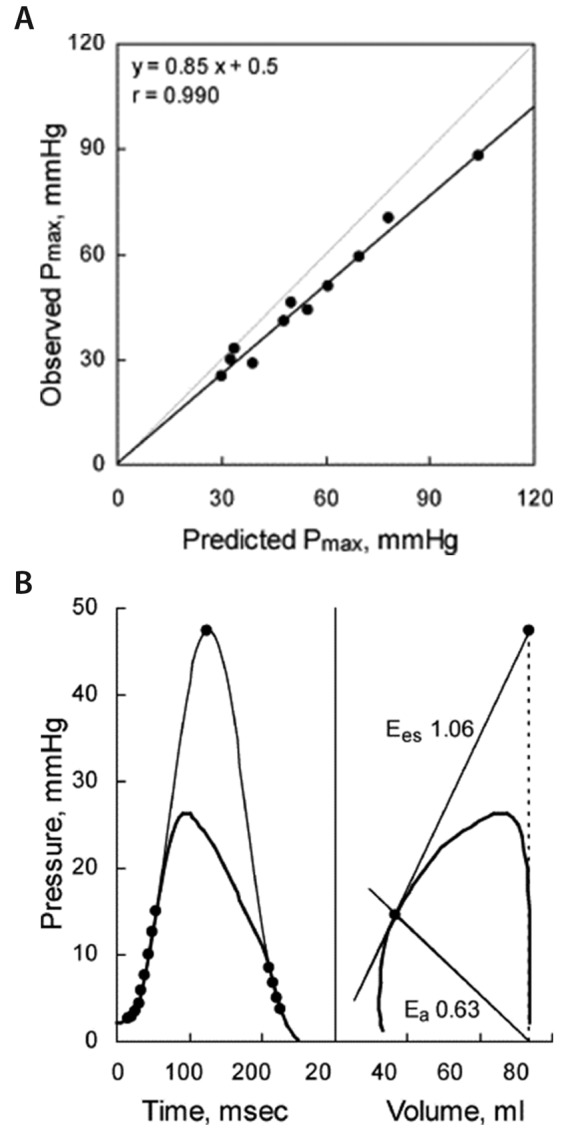
Single-beat method for measurement of right ventriculoarterial coupling in an anesthetized dog. A, Good agreement between directly measured maximum right ventricular (RV) pressure (Pmax) when the pulmonary arterial trunk is clamped during one heart beat (black line) and extrapolated Pmax (gray line). The slightly lower observed Pmax is explained by the proximal pulmonary arterial compliance. B, Pmax is calculated from early and late portions of the RV pressure curve, end-systolic elastance (Ees) or arterial elastance (Ea) graphically determined from Pmax, and relative changes in volume and pressure during systole. Reproduced from Brimioulle et al.15 with permission.
Single-beat measurements of RV Emax have been reported in patients with pulmonary arterial hypertension (PAH), using fluid-filled catheters and magnetic resonance imaging (MRI),16 and in a patient with a congenitally corrected transposition of the great arteries and a systemic RV, using high-fidelity micromanometer-tipped catheters and transonic measurements of pulmonary flow.17 Measurements of RV Emax have also been reported in idiopathic and systemic sclerosis–associated PAH, using conductance catheter methodology for instantaneous high-fidelity pressure and volume determinations.18 In that study, the Valsalva maneuver was validated against inferior vena cava occlusion in order to decrease venous return for the definition of Emax by a family of pressure-volume loops.18 These limited reports confirm the importance of systolic function adaptation to afterload previously demonstrated in various animal species19 or experimental models of acute15,20,21 or chronic22-26 pulmonary hypertension.
Coupling of systolic function to afterload
Afterload can be defined by the maximum ventricular wall stress or by arterial hydraulic load.27,28 Wall stress is approximated by the maximum value of the product of volume and pressure divided by wall thickness. This is a transposition of Laplace’s law for spheric structures and thus is problematic for the RV because of considerable regional variations in RV internal radius.27 Hydraulic load calculations require instantaneous measurements of arterial pressure and flow and spectral analysis to derive arterial impedance calculations.28 A more straightforward approach is to derive arterial elastance (Ea) as it is “seen” by the ventricle, and thus graphically determined on a pressure-volume loop, by dividing pressure at Emax by SV.9 Because contractility is homeometrically adjusted to afterload, its adequacy is best evaluated as a ratio of Emax to Ea, which defines RV-arterial coupling. The optimal mechanical coupling occurs when the ratio of Emax to Ea is equal to 1. The optimal energy transfer from the RV to the pulmonary circulation occurs at Emax/Ea ratios of 1.5–2.9
RV-arterial coupling measured with the Emax/Ea ratio has been investigated in models of pulmonary hypertension with and without RV failure and in models with persistent RV failure after transient pulmonary artery banding (to mimic a pulmonary hypertensive crisis) and pharmacological interventions. The results are summarized in Tables 1 and 2. The acute hypoxia-induced increase in PVR was associated with preserved RV-arterial coupling because of increased RV contractility.15,19,29-31 The adapted increase in RV contractility with preserved RV-arterial coupling was also reported in pulmonary hypertension following either microembolism or pulmonary arterial banding.19 Endotoxic shock–induced increase in PVR was associated with early preservation of RV-arterial coupling but deterioration as soon as 1 hour after the initial insult because of an unsustained adaptive increase in contractility.21 Chronic aortopulmonary shunting as a model of persistent ductus arteriosus in growing piglets was associated with preserved RV-arterial coupling after 3 months,24,37 but with uncoupling after 6 months because of RV systolic function failure.25 Persistent RV failure after tight transient pulmonary arterial ensnarement was characterized by a profound RV-arterial uncoupling because of a persistent decrease in contractility and a reactive increase in PVR.32-35 Monocrotaline-induced pulmonary hypertension was associated with RV-arterial uncoupling because of an insufficient increase in contractility to match increased afterload.26 Mild pulmonary hypertension in heart failure induced by several weeks of overpacing as a model of tachycardiomyopathy was associated with RV-arterial uncoupling by absence of adapted increase in RV contractility.36 Altogether, these studies support the notion of RV systolic function adaptation to increased afterload in various models of pulmonary hypertension, but with RV-arterial uncoupling in the context of inflammation (endotoxemia, monocrotaline), long-term increase in PVR, or left heart failure.
Table 1.
Right ventricular–arterial coupling in experimental models of pulmonary hypertension
| Model | Animal | Emax | Ea | Emax/Ea | Reference(s) |
|---|---|---|---|---|---|
| Hypoxia | Dog, goat, pig | Increase | Increase | No change | 15, 19, 29–31 |
| Monocrotaline | Rat | Increase | Increase | Decrease | 25 |
| Sepsis, early | Pig | Increase | Increase | No change | 21 |
| Sepsis, late | Pig | No change | Increase | Decrease | 21 |
| Embolism | Dog, goat, pig | Increase | Increase | No change | 19 |
| PA banding | Dog, goat, pig | Increase | Increase | No change | 19 |
| AP shunting, 3 months | Pig | Increase | Increase | No change | 24 |
| AP shunting, 6 months | Pig | Decrease | Increase | Decrease | 25 |
| RVF on PA banding | Dog, pig | Decrease | Increase | Decrease | 32–35 |
| Chronic heart failure | Dog | No change | Increase | Decrease | 36 |
Emax: maximum right ventricular elastance; Ea: pulmonary arterial elastance; PA: pulmonary artery; AP; aortopulmonary; RVF: right ventricular failure.
Table 2.
Effects of pharmacological intervention in experimental pulmonary hypertension
| Model | Drug | Emax | Ea | Emax/Ea | Reference(s) |
|---|---|---|---|---|---|
| Hypoxia | Dobutamine | Increase | No change | Increase | 15 |
| RHF on PA banding | Dobutamine | Increase | Decrease or no change | Increase | 32, 33 |
| RHF on PA banding | Levosimendan | Increase | Decrease | Increase | 33, 35 |
| RHF on PA banding | Norepinephrine | Increase | No change | Increase | 32 |
| Chronic heart failure | Milrinone | Increase | No change | Increase | 36 |
| Chronic heart failure | Nitroprusside | No change | No change | No change | 36 |
| Chronic heart failure | Nitric oxide | No change | No change | No change | 36 |
| Hypoxia | Propranolol | Decrease | Increase | Decrease | 15 |
| Monocrotaline | Bisoprolol | Increase | No change | Increase | 26 |
| AP shunting, 3 months | Epoprostenol | No change | Decrease | Increase | 37 |
| RHF on PA banding | Epoprostenol | No change | Decrease | Increase | 34 |
| Hypoxia | Sildenafil | No change | Decrease | Increase | 30 |
| Monocrotaline | Sildenafil | Increase | Decrease | Increase | 38 |
| Hypoxia | Isoflurane | Decrease | Increase | Decrease | 29 |
Emax: maximum right ventricular elastance; Ea: pulmonary arterial elastance; RHF: right heart failure.
Low-dose dobutamine increased RV-arterial coupling by an inotropic effect without15,33 or with32 a decreased afterload. Low-dose norepinephrine improved RV-arterial coupling through an exclusive positive inotropic effect that was, however, less pronounced than that with low-dose dobutamine.32 Acute administration of propranolol caused deterioration of RV-arterial coupling through combined negative inotropy and pulmonary vasoconstriction during an acute hypoxic exposure.15 Chronic administration of bisoprolol improved RV-arterial coupling by an improved contractility in monocrotaline-induced pulmonary hypertension.26 Acute administration of epoprostenol or inhaled nitric oxide (NO) improved RV-arterial coupling through selective pulmonary vasodilating effects in overcirculation-induced pulmonary hypertension.37 Acute epoprostenol administration partially restored RV-arterial coupling through an exclusive pulmonary vascular effect in pulmonary banding–induced persistent RV failure34 or was associated with maintained RV-arterial coupling because of decreased contractility in proportion to decreased PVR in hypoxia.31 Levosimendan improved RV-arterial coupling through combined inotropy and vasodilation in pulmonary banding–induced persistent RV failure.33,35 Sildenafil improved RV-arterial coupling in acute hypoxic pulmonary hypertension because of pulmonary vasodilating effects,30 but it improved coupling because of a positive inotropic effect in monocrotaline-induced pulmonary hypertension.38 Bosentan had no intrinsic effect on contractility in pulmonary hypertension after 3 months of aortopulmonary shunting.24 Milrinone improved RV-arterial coupling by an improved contractility in tachycardia-induced congestive heart failure with mild pulmonary hypertension, while nitroprusside or inhaled NO had no effect in this model.36 Isoflurane and enflurane caused deterioration of RV-arterial coupling because of combined decrease in contractility and increase in PVR.29
Measurements of Emax and Ea have been reported in patients with pulmonary hypertension. These results are summarized in Table 3. The reported profile was that of increased RV contractility in the face of increased PVR with or without preservation of the Emax/Ea ratio in patients with idiopathic PAH (Figure 4).16,18 Insufficient increase in contractility to preserve RV-arterial coupling was observed in a patient with a systemic RV in the context of a congenitally corrected transposition of the great arteries17 and in patients with systemic sclerosis–associated PAH.18
Table 3.
Right ventriculoarterial coupling in patients with pulmonary arterial hypertension
| Diagnosis | Emax | Ea | Emax/Ea | Reference |
|---|---|---|---|---|
| IPAH | Increase | Increase | Decrease | 16 |
| CCTGA | Increase | Increase | Decrease | 17 |
| IPAH | Increase | Increase | No change | 18 |
| SSc-PAH | Increase | Increase | Decrease | 18 |
Emax: maximum right ventricular elastance; Ea: pulmonary arterial elastance; IPAH: idiopathic pulmonary arterial hypertension; CCTGA: congenitally corrected transposition of the great arteries (systemic right ventricle); SSc-PAH: systemic sclerosis–associated pulmonary arterial hypertension.
Figure 4.
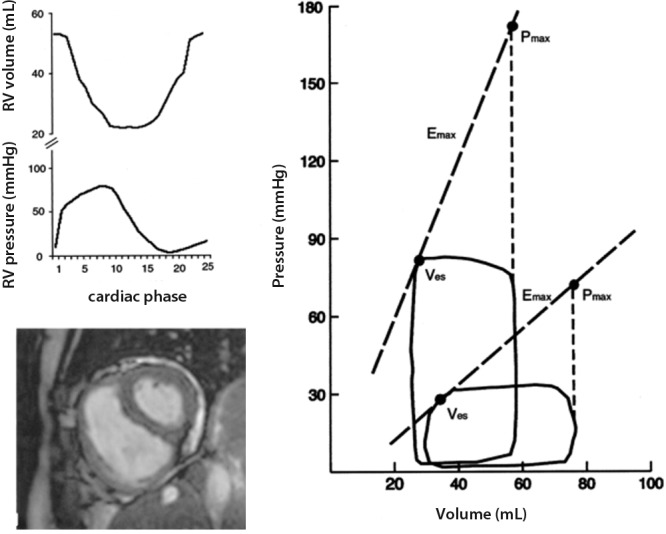
Right ventricular (RV) pressure-volume loops with calculated maximal RV pressure (Pmax), maximal elastance (Emax), and arterial elastance (Ea) in a normal subject and in a patient with pulmonary arterial hypertension (PAH). Left, source volume and pressure signals of the PAH patient (top) and a magnetic resonance image of the normal control (bottom). Right, pulmonary hypertension was associated with a marked increase in pressures and Ea, accompanied by an increase in Emax. The normal subject had an Emax/Ea ratio of 2. The Emax/Ea ratio was decreased to 1 in the PAH patient. Ves: end-systolic volume. Reproduced from Kuehne et al.16 with permission.
Thus, it appears that RV-arterial coupling tends to be maintained by an adaptive increase in Emax in models such as hypoxic exposure or a few months of aortopulmonary shunting associated with only moderate increase in Ppa. Prolonged mechanical stress, such as with 6 months of overcirculation in piglets or altered LV function after several weeks of overpacing-induced tachycardia in dogs, may cause uncoupling of the RV from the pulmonary circulation, with variable Starling’s mechanism, or increased EDV. Monocrotaline has extrapulmonary toxic effects and causes an inflammatory pulmonary vascular disease.39 This is associated with altered RV systolic function adaptation and leads to increased RV volumes. A general trend in reported studies is that increased pulmonary arterial obstruction, such as pulmonary stenosis or pulmonary artery banding, allows for better and more prolonged preservation of RV-arterial coupling when compared to conditions where the PVR is increased because of pulmonary vascular diseases.3,40 It has to be added that Starling’s mechanism may contribute to RV systolic function adaptation in any model, depending on the volume status and the contribution of preload to afterload-induced changes, with volume overload as a cause of enhanced RV hypertrophy.41
In patients with idiopathic PAH, RV-arterial coupling may be preserved with no increase in RV volume for a while, but the presence of systemic disease, such as systemic sclerosis, may be a cause of earlier RV failure.18 The determinants of long-term preservation of RV-arterial coupling in patients with severe pulmonary hypertension or a systemic RV are not known. The pathobiologic events leading to RV-arterial uncoupling and increased RV volumes remain to be identified. Knowing the signaling pathways responsible for maintained RV function in the presence of severely increased afterload may offer interesting therapeutic perspective.3
The current understanding of the pathophysiology of RV failure involves neurohumoral activation, expression of inflammatory mediators, apoptosis, capillary loss, oxidative stress, and metabolic shifts, with variable fibrosis and hypertrophy.3,42 The exact sequence of events and interactions is being explored, and each has still to be referred to sound measurements of function, as illustrated in recent studies that showed inflammation and apoptosis to be correlated with decreased Emax/Ea in acute43 as well as chronic25,44 models of RV failure as a universal relationship (Figure 5).
Figure 5.
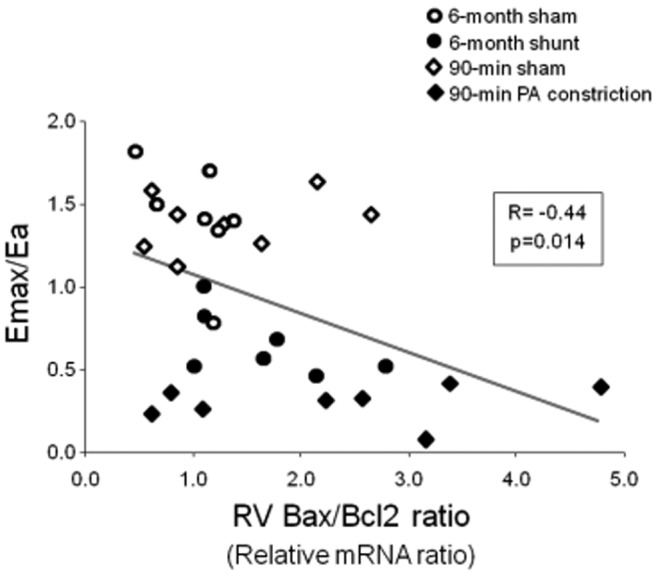
Decreased right ventricular (RV) maximum elastance/arterial elastance (Emax/Ea) ratio correlated with the Bax/Bcl2 ratio indicating activation of apoptosis as a universal mechanism in models of acute or chronic RV failure. Data from Rondelet et al.25 and Dewachter et al.43 PA: pulmonary artery; mRNA: messenger RNA.
Simplified methods for the measurement of RV-arterial coupling
Volume measurements
A ratio of elastances can be simplified to a ratio of volumes, provided that ESV is measured at the point of Emax and not at the end of RV ejection. Pressure-volume relationships obtained for the LV after the Mustard procedure connecting the LV to the pulmonary circulation are indistinguishable from the normal triangular shape of the RV, while the overall shape of the pressure-volume loop of the systemic RV resembles that of the normal LV.45 Thus, the discrepancy between Ees and Emax may increase with ventricular unloading.
Sanz and colleagues46 measured ESV and SV by MRI in a large group of patients with pulmonary hypertension and showed that the SV/ESV ratio is initially preserved and then decreases with increasing severity of pulmonary hypertension. This result calls for further evaluation of the functional and prognostic relevance of SV/ESV. It can be reasoned that the SV/ESV ratio includes information about the RV ejection fraction (EF), or SV/EDV ratio,27 but in a less preload-dependent manner. A recent study reported on the negative effect on outcome of decreased RVEF in spite of a targeted therapy-associated decrease of PVR in patients with PAH.47 Systemic vasodilating effects of targeted therapies in PAH may increase venous return and increase EDV, which decreases EF if SV remains essentially unchanged, while an increase in cardiac output may decrease PVR without any change in the functional state of the pulmonary circulation.48 The SV/ESV ratio as a measure of RV-arterial coupling could clarify these confounding effects of changes in preload.
Current progress in echocardiography makes accurate measurements of the pulmonary circulation and RV function possible,49,50 even though the precision of this approach may remain an issue for individual decision making based on cut-off values.51 Advances in 3-dimensional echocardiography now offer the perspective of easier bedside measurements of RV volumes,52 and thus of EF or SV/ESV, for the evaluation of RV-arterial coupling.
Pressure measurements
Another simplified approach for the measurement of RV-arterial coupling, introduced by Trip and colleagues,53 relies on a Pmax calculated from an RV pressure curve (which is easily obtained during right heart catheterization), mean Ppa (mPpa) taken as a surrogate for ESP, and RV volume measurements by MRI. These authors calculated Emax as (Pmax − mPpa)/(EDV − ESV) and Emax at V0 = 0 as mPpa/ESV (Figure 6); V0 is the extrapolated volume intercept of the linear best fit of a multipoint Emax pressure-volume relationship. The results showed that mPpa/ESV was lower than (Pmax − mPpa)/SV, on average about half the value, while V0 ranged from −8 to 171 mL and was correlated with EDV and ESV. From this the authors concluded that V0 is dependent on RV dilatation and thus that the estimated Emax may be more preload dependent than previously assumed. This is possible, although a more likely explanation is the uncertainties of extrapolation from linear fits of relationships that have been demonstrated to be curvilinear,54 as illustrated in Figure 7. The Ees is best determined by interpolation of pressure-volume coordinates,54 with further tightening by a correction for EDV.9,18 Further uncertainty is related to the use of the mPpa/SV ratio or the slope of (Pmax − mPpa)/SV as a surrogate for an Emax determination from single- or (better) multiple-beat pressure-volume relationships. Extrapolations amplify errors that are made by the use of surrogate pressures and volumes.
Figure 6.
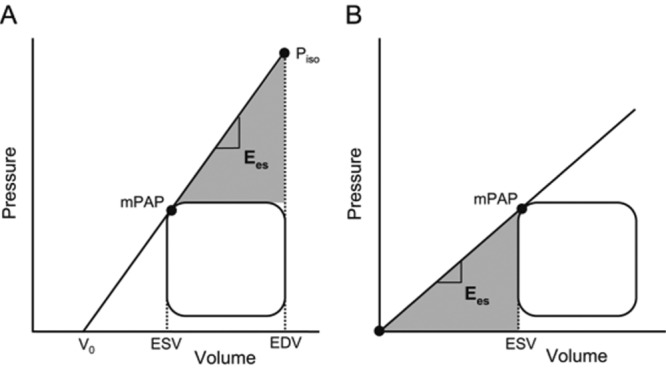
Simplified pressure method for the determination of right ventricular (RV) maximum elastance/arterial elastance (Ees/Ea) ratio. Emax is calculated as (Pmax − mPAP)/(EDV − ESV) in A and as mPAP/ESV in B. A positive V0 is associated with a higher estimated Emax. mPAP: mean pulmonary artery pressure; Pmax: maximum RV pressure; ESV: end-systolic volume; EDV: end-diastolic volume; V0: ventricular volume at zero pressure. Reproduced from Trip et al.53 with permission.
Figure 7.
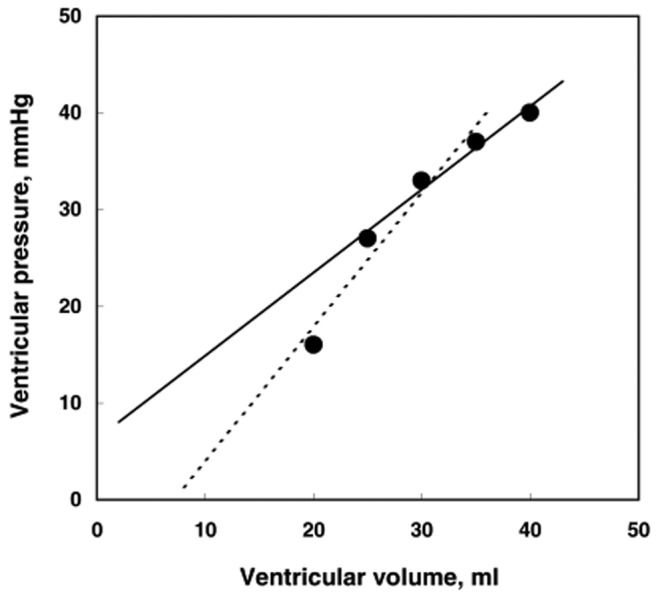
Linear extrapolation from slightly curvilinear end-systolic pressure-volume relationships may lead to markedly different pressure or volume intercepts. In this series of 5 pressure-volume coordinates, omission of the highest or the lowest pressure point changes the zero-pressure volume intercept from a negative to a positive value.
Alternative methods to evaluate RV-arterial coupling
The pump function graph
The coupling of RV function to the pulmonary circulation can also be described by pump function curves relating the mean RV pressure to SV.55 A pump function graph is built from measurements of the mean RV pressure and SV, a calculated Pmax at zero SV, and a parabolic extrapolation to a zero-pressure SV (Figure 8). In this representation, an increase in preload shifts the curve to greater SV with no change in shape, while an increased contractility leads to a higher Pmax with no change in maximum SV. This analysis helps us understand that at a high PVR a fall in pressure markedly increases SV while at a low PVR the pressure is more affected than the SV.27 The pump function graph has been used to explain more severe RV failure at any level of mPpa in systemic sclerosis.56 This was later confirmed by Emax/Ea determinations.18 The limitations of the pump function graph are in its sensitivity to changes in preload and, as mentioned above, to the use of mean RV pressure or mPpa as surrogates for the RV pressure at Emax.
Figure 8.
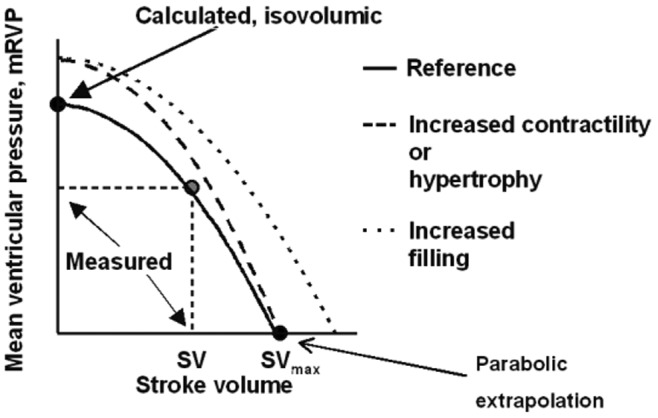
Pump function curve defined by mean right ventricular pressure as a function of stroke volume (SV). The zero-SV point is calculated from a maximum-pressure determination (see Figure 3). The zero-pressure point results from a parabolic extrapolation from measured and zero-SV points. Increased preload shifts the curve in parallel to higher SV. Increased contractility increases pressure generated at any given value of SV, but in proportion to decreased SV. Reproduced from Elzinga and Westerhof55 with permission.
The contractile reserve
Systolic function adaptation to afterload can also be tested dynamically to determine the contractile reserve, or the capacity to increase contractility at a given level of loading. Contractile or ventricular reserve determined with exercise or pharmacological stress tests (typically an infusion of dobutamine) has been shown to be a strong predictor of outcome in heart failure.57 The evaluation of the RV contractile reserve has not been reported in patients with pulmonary hypertension. In rats with pulmonary arterial banding, Emax was shown to increase to the same extent in response to 2.5 μg/kg/min of dobutamine, as in controls, suggesting preserved systolic function in this pulmonary hypertension model.23
A more straightforward, noninvasive approach was recently introduced by Grünig and his colleagues.58 These authors simply used Doppler echocardiography to measure RV systolic pressure from the maximum velocity of tricuspid regurgitation at rest and at exercise, and they showed, in 124 patients with either PAH or chronic thromboembolic pulmonary hypertension, that an exercise-induced increase by more 30 mmHg was a strong predictor of exercise capacity and survival (Figure 9). Further studies will be necessary to explore improved indices of RV contractile reserve by incorporating volume measurements and ESP determinations, because this is now becoming possible through noninvasive bedside methodology.
Figure 9.
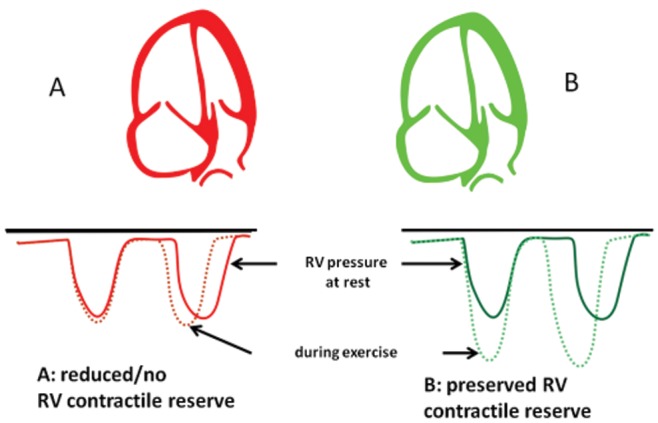
Right ventricular contractile reserve defined by exercise-induced increase in the systolic pressure. Reproduced from Grünig et al.58 with permission.
Surrogate measurements of RV-arterial coupling
RV systolic function can be estimated by a series of invasive and noninvasive measurements that are easily available in daily clinical practice. Right heart catheterization allows measurements of Ppa, right atrial pressure, and cardiac output (Fick or thermodilution) and thus calculations of RV function curves, such as cardiac output or stroke work, as functions of right atrial pressure. The limitations of these invasive measurements are the absence of RV volume measurements needed for EF calculations or estimations of preload and the insufficient definition of afterload by PVR. Imaging techniques such as MRI or 3-dimensional echocardiography allow the measurements of RV volumes, and thus EF and ESP/ESV ratios, as an estimation of RV contractility. The limitation of imaging is the absence of direct pressure measurements.
A series of imaging-derived indices of RV systolic function have been shown to be related to the functional state and prognosis of patients with severe pulmonary hypertension: MRI-determined EF, Doppler echocardiography of fractional area change (a surrogate for EF), tricuspid annual plane systolic excursion, and tissue Doppler imaging of the tricuspid annulus systolic velocity S wave and isovolumic acceleration (IVA) or maximum isovolumic velocity (IVV), strain or strain rate.49,50 Isovolumic phase indices, such as the IVA or IVV, are probably less preload dependent, and as such are the closest estimates of Emax measurements.59,60
Diastolic function
This review has focused on RV systolic function and RV-arterial coupling as the essential biomechanical mechanisms of ventricular function adaptation to increased afterload. However, a Starling mechanism may contribute at any stage of disease progression, depending on the rate of progression, a more or less inflammatory nature of pulmonary hypertension, and any systemic condition affecting cardiac function. There is thus interest in taking into account diastolic function in the RV adaptation to pulmonary hypertension.
Diastolic function is described by a diastolic elastance curve determined by a family of pressure-volume loops at variable loading. This function is curvilinear and therefore impossible to summarize as a single number. Several formulas have been proposed, but none has made it yet to clinical studies.27 A series of surrogate measurements of diastolic function are provided by Doppler echocardiography: isovolumic relaxation time and a decreased ratio of transmitral E and A waves or mitral annulus tissue Doppler imaging E′/A′ waves, increased right atrial or RV surface areas on apical 4-chamber views, altered eccentricity index on a parasternal short-axis view, estimates of right atrial pressure from RV diastolic function indices or inferior vena cava dimensions, pericardial effusion, and the so-called Tei index, which is the ratio of isovolumic time intervals to ventricular ejection time and thus integrates diastolic and systolic function.49,50
Ventricular interaction
RV function has to be understood in the light of its direct and indirect interactions with LV function.1 Direct interaction, or ventricular interdependence, is defined as the forces that are transmitted from one ventricle to the other through the myocardium and pericardium, independent of neural, humoral, or circulatory effects.61 Diastolic ventricular interaction refers to the competition for space within the indistensible pericardium when the RV dilates, which alters LV filling and may be a cause of inadequate cardiac output response to metabolic demand. Right heart catheterization and imaging studies have shown that in patients with severe pulmonary hypertension, pulmonary artery wedge pressure and LV peak filling rate are altered in proportion to decreased RVEF.62 Systolic interaction refers to positive interactions between RV and LV contractions. It can be shown experimentally that aortic constriction markedly improves RV function in animals with pulmonary arterial banding.63 This is explained by a mechanical entrainment effect but also by LV systolic function determining systemic blood pressure, which is an essential determinant of RV coronary perfusion. Increased RV filling pressures and excessive decrease in blood pressure may be causes of RV ischemia and decreased contractility.1 An additional cause of negative ventricular interaction disclosed by imaging studies is asynchrony, which has been shown to develop in parallel to increased Ppa and contributes to altered RV systolic function and LV underfilling.64
Limitations and perspectives
Measurements of systolic and diastolic function must be physiologically sound and validated against pressure-volume relationships. The validity of measurements of function is sometimes evaluated by their effect on clinical stability or survival. This is clinically useful but not helpful when it comes to a better understanding of mechanisms of disease and mode of action of pharmacological interventions.
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Reeves JT, Groves BM, Turkevich D, Morrison DA, Trapp JA. Right ventricular function in pulmonary hypertension. In: Weir EK and Reeves JT, eds. Pulmonary vascular physiology and physiopathology. New York: Dekker, 1989:325–351.
- 2.Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, Dupuis J, et al. Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation 2006;114(17):1883–1891. [DOI] [PubMed]
- 3.Voelkel NF, Gomez-Arroyo J, Abbate A, Bogaard HJ. Mechanisms of right heart failure—a work in progress and plea for further prevention. Pulm Circ 2013;3(1):137–143. [DOI] [PMC free article] [PubMed]
- 4.West JB. Role of the fragility of the pulmonary blood-gas barrier in the evolution of the pulmonary circulation. Am J Physiol Regul Integr Comp Physiol 2013;304(3):R171–R176. [DOI] [PubMed]
- 5.Starr I, Jeffers WA, Meade RH. The absence of conspicuous increments of venous pressure after severe damage to the right ventricle of the dog, with a discussion of the relation between clinical congestive heart failure and heart disease. Am Heart J 1943;26(3):291–301.
- 6.Fontan F, Baudet F. Surgical repair of tricuspid atresia. Thorax 1971;26(3):240–248. [DOI] [PMC free article] [PubMed]
- 7.Gewillig M. The Fontan circulation. Heart 2005;91(6):839–846. [DOI] [PMC free article] [PubMed]
- 8.Guyton AC, Lindsey AW, Gilluly JJ. The limits of right ventricular compensation following acute increase in pulmonary circulatory resistance. Circ Res 1954;2(4):326–332. [DOI] [PubMed]
- 9.Sagawa K, Maughan L, Suga H, Sunagawa K. Cardiac Contraction and the Pressure-Volume Relationship. New York: Oxford University Press, 1988.
- 10.von Anrep G. On the part played by the suprarenals in the normal vascular reactions of the body. J Physiol 1912;45(5):307–317. [DOI] [PMC free article] [PubMed]
- 11.Rosenblueth A, Alanís J, López E, Rubio R. The adaptation of ventricular muscle to different circulatory conditions. Arch Int Physiol Biochim 1959;67(3):358–373. [DOI] [PubMed]
- 12.Suga H, Sagawa K, Shoukas AA. Load independence of the instantaneous pressure-volume ratio of the canine left ventricle and effects of epinephrine and heart rate on the ratio. Circ Res 1973;32(3):314–322. [DOI] [PubMed]
- 13.Maughan WL, Shoukas AA, Sagawa K, Weisfeldt ML. Instantaneous pressure-volume relationship of the canine right ventricle. Circ Res 1979;44(3):309–315. [DOI] [PubMed]
- 14.Sunagawa K, Yamada A, Senda Y, Kikuchi Y, Nakamura M, Shibahara T, Nose Y. Estimation of the hydromotive source pressure from ejecting beats of the left ventricle. IEEE Trans Biomed Eng 1980;27(6):299–305. [DOI] [PubMed]
- 15.Brimioulle S, Wauthy P, Ewalenko P, Rondelet B, Vermeulen F, Kerbaul F, Naeije R. Single-beat estimation of right ventricular end-systolic pressure-volume relationship. Am J Physiol Heart Circ Physiol 2003;284(5):H1625–H1630. [DOI] [PubMed]
- 16.Kuehne T, Yilmaz S, Steendijk P, Moore P, Groenink M, Saaed M, Weber O, et al. Magnetic resonance imaging analysis of right ventricular pressure-volume loops: in vivo validation and clinical application in patients with pulmonary hypertension. Circulation 2004;110(14):2010–2016. [DOI] [PubMed]
- 17.Wauthy P, Naeije R, Brimioulle S. Left and right ventriculo-arterial coupling in a patient with congenitally corrected transposition. Cardiol Young 2005;15(6):647–649. [DOI] [PubMed]
- 18.Tedford RJ, Mudd JO, Girgis RE, Mathai SC, Zaiman AL, Housten-Harris T, Boyce D, et al. Right ventricular dysfunction in systemic sclerosis–associated pulmonary arterial hypertension. Circ Heart Fail 2013;6(5):953–963. [DOI] [PMC free article] [PubMed]
- 19.Wauthy P, Pagnamenta A, Vassali F, Naeije R, Brimioulle S. Right ventricular adaptation to pulmonary hypertension: an interspecies comparison. Am J Physiol Heart Circ Physiol 2004;286(4):H1441–H1447. [DOI] [PubMed]
- 20.de Vroomen M, Cardozo RHL, Steendijk P, van Bel F, Baan J. Improved contractile performance of right ventricle in response to increased RV afterload in newborn lamb. Am J Physiol Heart Circ Physiol 2000;278(1):H100–H105. [DOI] [PubMed]
- 21.Lambermont B, Ghuysen A, Kolh P, Tchana-Sato V, Segers P, Gérard P, Morimont P, et al. Effects of endotoxic shock on right ventricular systolic function and mechanical efficiency. Cardiovasc Res 2003;59(2):412–418. [DOI] [PubMed]
- 22.Leeuwenburgh BP, Helbing WA, Steendijk P, Schoof PH, Baan J. Biventricular systolic function in young lambs subject to chronic systemic right ventricular pressure overload. Am J Physiol Heart Circ Physiol 2001;281(6):H2697–H2704. [DOI] [PubMed]
- 23.Faber MJ, Dalinghaus M, Lankhuizen IM, Steendijk P, Hop WC, Schoemaker RG, Duncker DJ, Lamers JM, Helbing WA. Right and left ventricular function after chronic pulmonary artery banding in rats assessed with biventricular pressure-volume loops. Am J Physiol Heart Circ Physiol 2006;291(4):H1580–H1586. [DOI] [PubMed]
- 24.Rondelet B, Kerbaul F, Motte S, van Beneden R, Remmelink M, Brimioulle S, McEntee K, et al. Bosentan for the prevention of overcirculation-induced experimental pulmonary arterial hypertension. Circulation 2003;107(9):1329–1335. [DOI] [PubMed]
- 25.Rondelet B, Dewachter C, Kerbaul F, Kang X, Fesler P, Brimioulle S, Naeije R, Dewachter L. Prolonged overcirculation-induced pulmonary arterial hypertension as a cause of right ventricular failure. Eur Heart J 2012;33(8):1017–1026. [DOI] [PubMed]
- 26.de Man FS, Handoko ML, van Ballegoij JJ, Schalij I, Bogaards SJ, Postmus PE, van der Velden J, Westerhof N, Paulus WJ, Vonk-Noordegraaf A. Bisoprolol delays progression towards right heart failure in experimental pulmonary hypertension. Circ Heart Fail 2012;5(1):97–105. [DOI] [PubMed]
- 27.Vonk-Noordegraaf A, Westerhof N. Describing right ventricular function. Eur Respir J 2013;41(6):1419–1423. [DOI] [PubMed]
- 28.Chesler NC, Roldan A, Vanderpool RR, Naeije R. How to measure pulmonary vascular and right ventricular function. Conf Proc IEEE Eng Med Biol Soc 2009:177–180. doi:10.1109/IEMBS.2009.5333835. [DOI] [PMC free article] [PubMed]
- 29.Kerbaul F, Rondelet B, Motte S, Fesler P, Hubloue I, Ewalenko P, Naeije R, Brimioulle S. Isoflurane and desflurane impair right ventricular-pulmonary arterial coupling in dogs. Anesthesiology 2004;101(6):1357–1361. [DOI] [PubMed]
- 30.Fesler P, Pagnamenta A, Rondelet B, Kerbaul F, Naeije R. Effects of sildenafil on hypoxic pulmonary vascular function in dogs. J Appl Physiol 2006;101(4):1085–1090. [DOI] [PubMed]
- 31.Rex S, Missant C, Segers P, Rossaint R, Wouters PF. Epoprostenol treatment of acute pulmonary hypertension is associated with a paradoxical decrease in right ventricular contractility. Intensive Care Med 2008;34(1):179–189. [DOI] [PubMed]
- 32.Kerbaul F, Rondelet B, Motte S, Fesler P, Hubloue I, Ewalenko P, Naeije R, Brimioulle S. Effects of norepinephrine and dobutamine on pressure load-induced right ventricular failure. Crit Care Med 2004;32(4):1035–1040. [DOI] [PubMed]
- 33.Kerbaul F, Rondelet B, Demester JP, Fesler P, Huez S, Naeije R, Brimioulle S. Effects of levosimendan versus dobutamine on pressure load-induced right ventricular failure. Crit Care Med 2006;34(11):2814–2819. [DOI] [PubMed]
- 34.Kerbaul F, Brimioulle S, Rondelet B, Dewachter C, Hubloue I, Naeije R. How prostacyclin improves cardiac output in right heart failure in conjunction with pulmonary hypertension. Am J Respir Crit Care Med 2007;175(8):846–850. [DOI] [PubMed]
- 35.Missant C, Rex S, Segers P, Wouters PF. Levosimendan improves right ventriculovascular coupling in a porcine model of right ventricular dysfunction. Crit Care Med 2007;35(3):707–715. [DOI] [PubMed]
- 36.Pagnamenta A, Dewachter C, McEntee K, Fesler P, Brimioulle S, Naeije R. Early right ventriculo-arterial uncoupling in borderline pulmonary hypertension on experimental heart failure. J Appl Physiol 2010;109(4):1080–1085. [DOI] [PubMed]
- 37.Wauthy P, Kafi AS, Mooi W, Naeije R, Brimioulle S. Inhaled nitric oxide versus prostacyclin in chronic shunt-induced pulmonary hypertension. J Thorac Cardiovasc Surg 2003;126(6):1434–1441. [DOI] [PubMed]
- 38.Borgdorff MA, Bartelds B, Dickinson MG, Boersma B, Weij M, Zandvoort A, Silljé HHW, Steendijk P, de Vroomen M, Berger RMF. Sildenafil enhances systolic adaptation, but does not prevent diastolic dysfunction, in the pressure-loaded right ventricle. Eur J Heart Fail 2012;14(9):1067–1074. [DOI] [PubMed]
- 39.Gomez-Arroyo JG, Farkas L, Alhussaini AA, Farkas D, Kraskauskas D, Voelkel N, Bogaard HJ. The monocrotaline model of pulmonary hypertension in perspective. Am J Physiol Lung Cell Mol Physiol 2012;302(4):L363–L369. [DOI] [PubMed]
- 40.Bogaard HJ, Natarayan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, Ockaili R, McCord JM, Voelkel NF. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 2009;120(20):1951–1960. [DOI] [PubMed]
- 41.Borgdorff MA, Bartelds B, Dickinson MG, Steendijk P, de Vroomen M, Berger RM. Distinct loading conditions reveal various patterns of right ventricular adaptation. Am J Physiol Heart Circ Physiol 2013;305(3):H354–H364. [DOI] [PubMed]
- 42.Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right heart failure in pulmonary hypertension. Chest 2009;135(3):794–804. [DOI] [PubMed]
- 43.Dewachter C, Dewachter L, Rondelet B, Fesler P, Brimioulle S, Kerbaul F, Naeije R. Activation of apoptotic pathways in experimental acute afterload-induced right ventricular failure. Crit Care Med 2010;38(6):1405–1413. [DOI] [PubMed]
- 44.Belhaj A, Dewachter L, Kerbaul F, Brimioulle S, Dewachter C, Naeije R, Rondelet B. Heme oxygenase-1 and inflammation in experimental right ventricular failure on prolonged overcirculation-induced pulmonary hypertension. PloS ONE 2013;8(7):e69470. doi:10.1371/journal.pone.0069470. [DOI] [PMC free article] [PubMed]
- 45.Redington AN, Rigby RL, Shinebourne EA, Oldershaw PJ. Changes in pressure-volume relation of the right ventricle when its loading conditions are modified. Br Heart J 1990;63(1):45–49. [DOI] [PMC free article] [PubMed]
- 46.Sanz J, García-Alvarez A, Fernández-Friera L, Nair A, Mirelis JG, Sawit ST, Pinney S, Fuster F. Right ventriculo-arterial coupling in pulmonary hypertension: a magnetic resonance study. Heart 2012;98(3):238–243. [DOI] [PubMed]
- 47.van de Veerdonk MC, Kind T, Marcus JT, Mauritz GJ, Heymans MW, Bogaard HJ, Boonstra A, Marques KMJ, Westerhof N, Vonk-Noordegraaf A. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol 2011;58(24):2511–2519. [DOI] [PubMed]
- 48.Sniderman AD, Fitchett DH. Vasodilators and pulmonary arterial hypertension: the paradox of therapeutic success and clinical failure. Int J Cardiol 1988;20(2):173–181. [DOI] [PubMed]
- 49.Roberts JD, Forfia PR. Diagnosis and assessment of pulmonary vascular disease by Doppler echocardiography. Pulm Circ 2011;1(2):160–181. [DOI] [PMC free article] [PubMed]
- 50.Bossone E, D’Andrea A, D’Alto M, Citro R, Argiento P, Ferrara F, Cittadini A, Rubenfire M, Naeije R. Echocardiography in pulmonary arterial hypertension. J Am Soc Echocardiogr 2013;26(1):1–14. [DOI] [PubMed]
- 51.D’Alto M, Romeo E, Argiento P, D’Andrea A, Vanderpool R, Correra A, Bossone E, et al. Accuracy and precision of echocardiography versus right heart catheterization for the assessment of pulmonary hypertension. Int J Cardiol 2013;168(4):4058–4062. [DOI] [PubMed]
- 52.Zhang QB, Sun JP, Gao RF, Lee APW, Feng YL, Liu XR, Sheng W, Liu F, Yu CM. Novel single-beat full-volume capture real-time three-dimensional echocardiography and auto-contouring algorithm for quantification of left ventricular volume: validation with cardiac magnetic resonance imaging. Int J Cardiol 2013;168(3):2946–2948. [DOI] [PubMed]
- 53.Trip P, Kind T, van de Veerdonk MC, Marcus JT, de Man FS, Westerhof N, Vonk-Noordegraaf A. Accurate assessment of load-independent right ventricular systolic function in patients with pulmonary hypertension. J Heart Lung Transplant 2013;32(1):50–55. [DOI] [PubMed]
- 54.Kass DA, Beyar R, Lankford E, Heard M, Maughan WL, Sagawa K. Influence of contractile state on the curvilinearity of in situ end-systolic pressure-volume relationships. Circulation 1989;79(1):167–178. [DOI] [PubMed]
- 55.Elzinga G, Westerhof N. The effect of an increase in inotropic state and end-diastolic volume on the pumping ability of the feline left heart. Circ Res 1978;42(5):620–628. [DOI] [PubMed]
- 56.Overbeek MJ, Lankhaar JW, Westerhof N, Voskuyl AE, Boonstra A, Bronzwaer JG, Marques KMJ, Smit EF, Dijkmans BAC, Vonk-Noordegraaf A. Right ventricular contractility in systemic sclerosis–associated and idiopathic pulmonary arterial hypertension. Eur Respir J 2008;31(6):1160–1166. [DOI] [PubMed]
- 57.Haddad F, Vrtovec B, Ashley EA, Deschamps A, Haddad H, Denault AY. The concept of ventricular reserve in heart failure and pulmonary hypertension: an old metric that brings us one step closer in our quest for prediction. Curr Opin Cardiol 2011;26(2):123–131. [DOI] [PubMed]
- 58.Grünig E, Tiede H, Enyimayew EO, Ehlken N, Seyfarth HJ, Bossone E, D’Andrea A, et al. Assessment and prognostic relevance of right ventricular contractile reserve in patients with pulmonary arterial hypertension. Circulation 2013;128(18):2005–2015. [DOI] [PubMed]
- 59.Vogel M, Schmidt MR, Christiansen SB, Cheung M, White PA, Sorensen K, Redington AN. Validation of myocardial acceleration during isovolumic contraction as a novel non-invasive index of right ventricular contractility: comparison with ventricular pressure-volume relations in an animal model. Circulation 2002;105(14):1693–1699. [DOI] [PubMed]
- 60.Ernande L, Cottin V, Leroux PY, Girerd N, Huez S, Mulliez A, Bergerot C, et al. Right isovolumic contraction velocity predicts survival in pulmonary hypertension. J Am Soc Echocardiogr 2013;26(3):297–306. [DOI] [PubMed]
- 61.Santamore WP, Dell’Italia LJ. Ventricular interdependence: significant left ventricular contributions to right ventricular systolic function. Prog Cardiovasc Dis 1998;40(4):289–308. [DOI] [PubMed]
- 62.Lazar JM, Flores AR, Grandis DJ, Orie JE, Schulman DS. Effects of chronic right ventricular pressure overload on left ventricular diastolic function. Am J Cardiol 1993;72(15):1179–1182. [DOI] [PubMed]
- 63.Belenkie I, Horne SG, Dani R, Smith ER, Tyberg JV. Effects of aortic constriction during experimental acute right ventricular pressure loading: further insights into diastolic and systolic ventricular interaction. Circulation 1995;92(3):546–554. [DOI] [PubMed]
- 64.Marcus JT, Gan CT, Zwanenburg JJ, Boonstra A, Allaart CP, Götte MJ, Vonk-Noordegraaf A. Interventricular mechanical asynchrony in pulmonary arterial hypertension: left-to-right delay in peak shortening is related to right ventricular overload and left ventricular underfilling. J Am Coll Cardiol 2008;51(7):750–757. [DOI] [PubMed]