

Abstract Abstract
There is unequivocal evidence that exercise results in considerable health benefits. These are the result of positive hormonal, metabolic, neuronal, and structural changes brought about by the intermittent physiological challenge of exercise. However, there is evolving evidence that intense exercise may place disproportionate physiological stress on the right ventricle (RV) and the pulmonary circulation. Both echocardiographic and invasive studies are consistent in demonstrating that pulmonary arterial pressures increase progressively with exercise intensity, such that the harder one exercises, the greater the load on the RV. This disproportionate load can result in fatigue or damage of the RV if the intensity and duration of exercise is sufficiently prolonged. This is distinctly different from the load imposed by exercise on the left ventricle (LV), which is moderated by a greater capacity for reductions in systemic afterload. Finally, given the increasing RV demand during exercise, it may be hypothesized that chronic exercise–induced cardiac remodeling (the so-called athlete’s heart) may also disproportionately affect the RV. Indeed, there is evidence, although somewhat inconsistent, that RV volume increases may be relatively greater than those for the LV. Perhaps more importantly, there is a suggestion that chronic endurance exercise may cause electrical remodeling, predisposing some athletes to serious arrhythmias originating from the RV. Thus, a relatively consistent picture is emerging of acute stress, prolonged fatigue, and long-term remodeling, which all disproportionately affect the RV. Thus, we contend that the RV should be considered a potential Achilles’ heel of the exercising heart.
Keywords: right ventricle, pulmonary circulation, exercise, athlete, athlete’s heart, exercise induced, fibrosis
Introduction
The structural, functional, and electrical remodeling of all four cardiac chambers that results from regular endurance exercise practice comprises a syndrome often termed the “athlete’s heart.”1,2 This cardiac remodeling is believed to represent an adaptive response, providing the means for enhanced cardiovascular performance during exercise. Changes in the left ventricle (LV) have been relatively well characterized, but there is still much to be learned about the right ventricle (RV) of athletes. Recent research has suggested that structural and functional changes in the RV of endurance athletes may be profound and may not always mirror those seen in the LV. Moreover, a syndrome has been reported whereby the RV in some endurance athletes undergoes structural and electrical remodeling that may create a substrate for life-threatening arrhythmias—although it is extremely important to place this in the context that sudden cardiac death remains a rare event in young athletes (approximate incidence of 0.6–3.6 per 100,000 athletes).3 Thus, study of the RV in endurance athletes is justified both by the need for a more complete understanding of athletic physiology and by the clinical need for better risk stratification of athletes presenting with arrhythmias.
Hundreds of studies performed over more than a century have led to a relatively thorough description of what constitutes the normal limits of cardiac structure and function in athletes.1 The volume and pressure load imposed by repeated bouts of exercise result in an increase in the size and thickness of all four cardiac chambers, as illustrated in Figure 1. The cardiac remodeling reflects the intermittent increases in wall stress in each of the cardiac chambers during exercise. The majority of research has focused on the LV, and it has been well detailed that LV mass increases with exercise and that the extent of LV remodeling correlates reasonably well with the athlete’s fitness.4,5 The orthodox view on LV remodeling is summarized by the Morganroth hypothesis, which states that endurance exercise results in an increase in LV cavity dimensions (eccentric remodeling), whereas strength-based sports promote an increase in wall thickness more than an increase in cavity dimensions (concentric remodeling).6,7 This has been challenged recently, with some prospective data suggesting that aerobic exercise promotes functional and structural changes in the LV, whereas strength training contributes little.8 However, there has also been an expanding appreciation that the LV may not be the most important chamber with respect to exercise-induced remodeling. Evolving evidence suggests that the load during short and prolonged bouts of exercise is disproportionately greater for the RV and that this, in turn, causes specific structural, functional, and electrical remodeling of the RV.
Figure 1.

Morphological changes in an athlete’s heart compared with a nonathlete’s heart. The normal heart of a 24-year-old librarian (left) is compared with that of a 23-year-old professional cyclist (right). Note the difference in scale such that the nonathlete’s ventricle is ∼10 cm in length (circled in red), whereas the 10-cm mark reaches only halfway along the ventricle of the athlete. Note that all cardiac chambers are enlarged and that increases in wall thickness are relative to dilation.
In this review, we will detail how intense exercise places a disproportionate physiological stress on the RV. We will discuss how pulmonary arterial pressures increase progressively with exercise intensity in a near-linear manner, such that the harder one exercises, the greater the load on the RV. This disproportionate load can result in fatigue or damage of the RV if the intensity and duration of exercise is sufficiently prolonged. This is distinctly different from the load imposed by exercise on the LV, which is moderated by a greater capacity for reductions in systemic afterload. Finally, we will discuss the nature and consequences of specific RV remodeling in athletes. Thus, we will contend that during short intense exercise, prolonged endurance exercise, and chronic exercise–induced remodeling, the RV represents the Achilles’ heel of the exercising heart.
Normal pulmonary vascular physiology
Although the pulmonary circulation shares much in common with the systemic circulation, there are also some important differences. The pulmonary circulation receives the entire cardiac output but has a pressure approximately one-fifth that of the systemic circulation, a figure that is remarkably consistent across mammalian species.9 The relationship between pressure (P), flow (F), and resistance (R) can be simplified as F ∝ P/R, which is a simile of Ohm’s law for electric circuits and is often referred to as the simplified Poiseuille’s law in vascular circuits. Thus, the lower-pressure pulmonary system equates to a lower-resistance system. This lower resistance is a product of a number of unique features: first, there is rapid and prolific branching of vessels in the pulmonary circulation. Total vascular resistance may be quantified as the sum of the reciprocal of the resistance of each branch (i.e., 1/Rtotal = 1/R1 + 1/R2 + 1/R3 + …), such that the greater the number of parallel branches, the lower the total resistance. In addition, the pulmonary arteries and arterioles are thinner walled and have much lower basal tone than their systemic counterparts. This has a very important implication in that the thinner-walled vessels are more compliant and this greater compliance causes a further reduction in resistance and pressure. This concept is summarized in the Windkessel model of vascular flow.10,11 This model predicts that compliance is inversely proportional to resistance, and thus the “stretching” of compliant vessels with pulsatile flow serves to decrease the resistance and blunt pressure rises. Finally, hypoxia-induced vasoconstriction may occur in the pulmonary circulation during exercise but is not a feature of the systemic circulation. Therefore, the differences between the pulmonary and the systemic circulation are that the former forms a more extensive and earlier parallel circuit, is more compliant, and has lower basal tone, serving to make it a lower-pressure and lower-resistance circuit.
Another potential difference is that there is relative independence between the arteriole and the venule pressures in the systemic system. The high systemic arteriolar pressures mean that systemic venous pressures would need to be very high to contribute significantly to afterload. In contrast, the venous pressures in the pulmonary vasculature are considerable relative to the lower arteriolar pressures, thereby contributing far more significantly to total afterload. It has been suggested that left atrial pressures explain ∼80% of the variance in pulmonary arterial pressures in the absence of pulmonary vascular disease.12
RV and pulmonary vascular coupling in the healthy circulation
At rest, RV measures of mass and contractility are one-third to one-fifth those of the LV, and this appropriately matches the pressure requirements of each.13,14 This lesser myocardial mass of the RV suggests that it may have a diminished contractile reserve and may be less able to accommodate marked changes in loading. This is supported by studies that demonstrate that afterload increases result in a marked reduction in RV stroke volume but only a slight decrease in LV stroke volume.15,16 MacNee16 quantified the relative reductions in stroke volume in both ventricles at rest with increasing afterload. The ∼30% reduction in RV stroke volume compared with the ∼10% fall in LV stroke volume should be viewed as a combination of the effect of insufficient contractile reserve to overcome increases in afterload. To our knowledge, there are no studies that compare the contractile response of each ventricle during exercise in normal subjects. However, it may be reasonable to assume that sensitivity to afterload excess will be similar to those demonstrated at rest and that significant exercise-induced increases in pulmonary pressures may promote decreases in RV output, thereby limiting exercise cardiac capacity.
Strenuous exercise imposes a greater hemodynamic load on the RV
There are differences between the RV and the LV in the arterial load imposed by exercise and in the ventricular capacity to counter that load (i.e., maintain ventricular arterial coupling). As illustrated in Table 1, at rest the LV contracts against a systemic circulation with moderate resistance and compliance compared with the low-resistance and high-compliance pulmonary circulation. As a result, the RV has to perform little work in the healthy circulation at rest. During exercise, CO increases many fold (outputs as high as 35–40 L/min have been measured in well-trained athletes17,18), which would be expected to result in an increase in vascular pressures unless sufficiently counterbalanced by decreases in resistance and increases in compliance. However, the pulmonary vasculature has very low resistance at rest, and it has somewhat limited capacity for further decreases.19 Recruitment of upper lobe vessels in combination with flow-mediated and neurohormonal vasodilation effect a reduction in pulmonary vascular resistance of ∼20%–50%.20 Such changes are limited compared with the profound reductions in systemic vascular resistance, which is enabled by the greater capacity for redistribution to vascular territories of low resistance. The moderating effect of vascular compliance is also less than may be anticipated. During exercise, the vasculature is distended by the high flow rates, meaning that compliance (the ability to further distend the vessels with any given change in pressure) is reduced.21,22 Therefore, vascular pressures increase, and these increases are greater for the pulmonary circulation than for the systemic circulation.
Table 1.
Left and right ventricular arterial load at rest and during exercise
| Left ventricle and systemic circulation | Right ventricle and pulmonary circulation | |
|---|---|---|
| At rest | ||
| Cardiac output, L/min | 5 | 5 |
| Vascular resistance, dyn s cm5 | ∼1,100 | ∼70 |
| Vascular compliance | ++ | +++ |
| Afterload pressure, mmHg | 130/75 (85) | 25/9 (15) |
| During exercise | ||
| Cardiac output, L/min | 25 | 25 |
| Vascular resistance, dyn s cm5 | ↓↓↓ | ↓ |
| Vascular compliance | + | ++ |
| Afterload pressure, mmHg | ↑ | ↑↑↑ |
A number of studies have reported substantial increases in pulmonary arterial pressures during exercise using echocardiographic estimates23-26 and direct invasive measures.27,28 Figure 2 summarizes data from three recent studies that demonstrate remarkable consistency in the linear regression derived for the relationship between pulmonary arterial pressures and CO. For example, Lewis et al.27 observed an increase of 1.5 mmHg in mean pulmonary arterial pressure for each liter increase in CO. Thus, an increase in CO of 30 L/min would equate to a mean pulmonary arterial pressure exceeding 50 mmHg, representing an increase of threefold or more from rest. We sought to assess this seemingly disproportionate load using a combination of magnetic resonance and echocardiographic imaging at rest and during exercise to quantify RV systolic wall stress compared with that of the LV.25 Using the simple construct of the Laplace relationship, we found that during exercise increases in both pressures and volumes were greater for the RV, while increases in wall thickness were relatively less than those for the LV. As a result, RV wall stress estimates increased 125% during exercise, compared with a modest 14% increase in LV wall stress.25 Thus, it may be contended that the stress, work, and metabolic demands placed on the RV during exercise are relatively far greater than those placed on the LV.
Figure 2.
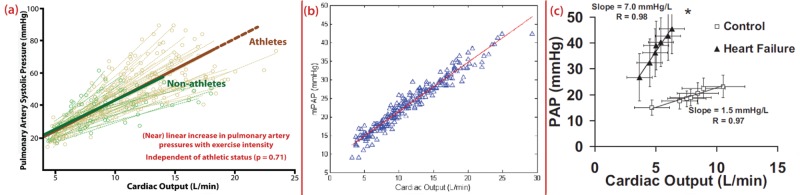
Agreement in multiple studies describing the near-linear relationship between increases in cardiac output and pulmonary arterial pressures (PAPs). Data are from two echocardiographic studies24,26 and one invasive study27 demonstrating a consistent relationship between increases in PAPs and cardiac output. Although there were methodological differences between the studies, all three converge on an approximately linear increase of 1.5 mmHg in mean PAP (mPAP) for every 1-L increase in cardiac output. As demonstrated by La Gerche et al.,26 this relationship is similar in the healthy pulmonary circulation of nonathletes and athletes, such that the greater increases in PAPs are determined solely by cardiac output. Lewis et al.27 observed a steeper relationship in heart failure patients than in healthy controls. Figures are reproduced with permission.
Such substantive afterload would seem a significant burden for the contractile reserve of the RV and raises the possibility that in the extremes of exercise the RV/pulmonary vascular unit may limit output, just as it does in some disease states. This hypothesis has been raised previously,29-31 but it has not been actively pursued over the last three decades. This is in part because it is difficult to assess RV function during exercise. We sought to assess the RV contractile response during exercise by means of two echocardiographic measures—the RV pressure/area relationship and the systolic strain rate. Both measures are relatively less influenced by changes in load and therefore may represent a reasonable approximation of RV contractility. In a cohort of athletes and healthy nonathletes, we demonstrated a consistent increase in both measures during strenuous exercise, suggesting that the RV had sufficient reserve to counter the considerable increases in afterload during brief strenuous exercise.
Assessment of RV function during exercise is difficult. The complex geometry, heterogeneous function, and substernal location of the RV make assessment by echocardiography challenging. With this challenge in mind, we have invested considerable effort into developing an exercise imaging modality capable of accurately defining RV volumes and function during exercise. Cardiac magnetic resonance (CMR) imaging represents a noninvasive gold standard for RV assessment at rest, and we have extended this technique to enable real-time images to be acquired during strenuous exercise and free breathing (see Video 1). We have validated this technique against gold standards and have demonstrated that both RV and LV volumes can be accurately measured with exercise requiring cardiac outputs in excess of 35 L/min.17 This represents a promising methodology for the evaluation of pulmonary vascular function, which has already provided some important insights into pathophysiology.32,33 Furthermore, our current studies combine these gold standard volumetric measures with invasive catheter measures of pulmonary and systemic vascular pressures. This provides a comprehensive approach to understanding the role of the “forgotten ventricle” in defining exercise capacity in health and disease.
Video 1.

Intense endurance exercise causes RV fatigue
While the healthy RV seems capable of meeting the work requirements of short bouts of intense exercise, the aforementioned disparity in ventricular load would suggest that if prolonged exercise can induce cardiac “fatigue,” then the RV would be most susceptible. Numerous studies have demonstrated evidence of cardiac fatigue or injury following intense exercise sustained over many hours, such as marathon running and ultraendurance triathlons. Biochemical evidence of cardiac injury is common, usually minor, and mostly associated with little if any LV dysfunction.34 In contrast, virtually all studies in which the RV has been assessed following intense endurance exercise have reported RV dysfunction.35-42
We studied 40 highly trained athletes before and after an endurance sporting event using the best available echocardiographic measures of biventricular function and observed three key findings.39 First, every measure of RV function was reduced in the postrace setting, while LV function was unchanged. 3D echo was used to demonstrate an increase in RV volumes with a corresponding reduction in LV volumes. RV end-systolic volumes increased 16%, reflecting a combination of ventricular dilation and reduced systolic contraction, while LV end-systolic volumes decreased 8%. As illustrated in Figure 3, this ventricular interaction resulted in the interventricular septum pushing toward the LV in early diastole in a manner similar to that described by Marcus et al.43 in pulmonary hypertension and attributed to the delayed deformation of the dysfunctional RV. Second, we identified a dose-dependent effect on RV function, such that the duration of endurance exercise was associated with greater postrace reductions in RV ejection fraction. This supports the hypothesis that exercise exerts a disproportionate load on the RV that can be accommodated by the RV in the short term but results in progressive fatigue or injury as time progresses. Finally, we noted a moderate correlation between the reduction in RV ejection fraction and the increase in cardiac troponin and B-type natriuretic peptide (Fig. 4). Consistent with previous studies, we found no relationship between the expression of these biomarkers and changes in LV function but, rather, that these markers of myocardial injury and strain were associated with the part of the myocardium that is most greatly stressed during exercise—the RV.
Figure 3.
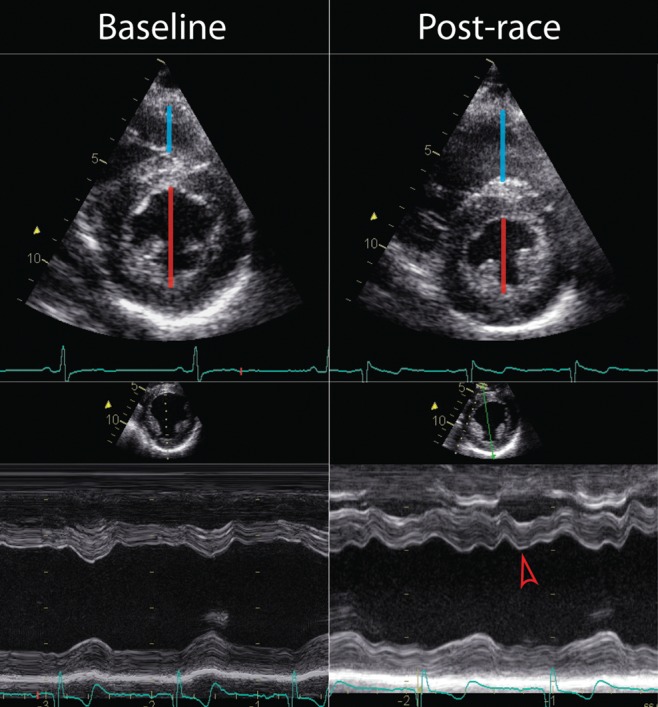
Acute right ventricular dilation and dysfunction following ultraendurance exercise causing early diastolic septal shift toward the left ventricle. Compared with baseline, right ventricular (RV) dimensions (blue line) are increased and left ventricular (LV) dimensions (red line) reduced immediately following an ultraendurance triathlon. The dilation and delayed contraction of the RV results in septal shift toward the LV in early diastole (red arrow) such that RV dysfunction impairs early LV filling. This would suggest that overall cardiac output may be predominantly limited by reduced RV function in the endurance exercise setting.
Figure 4.
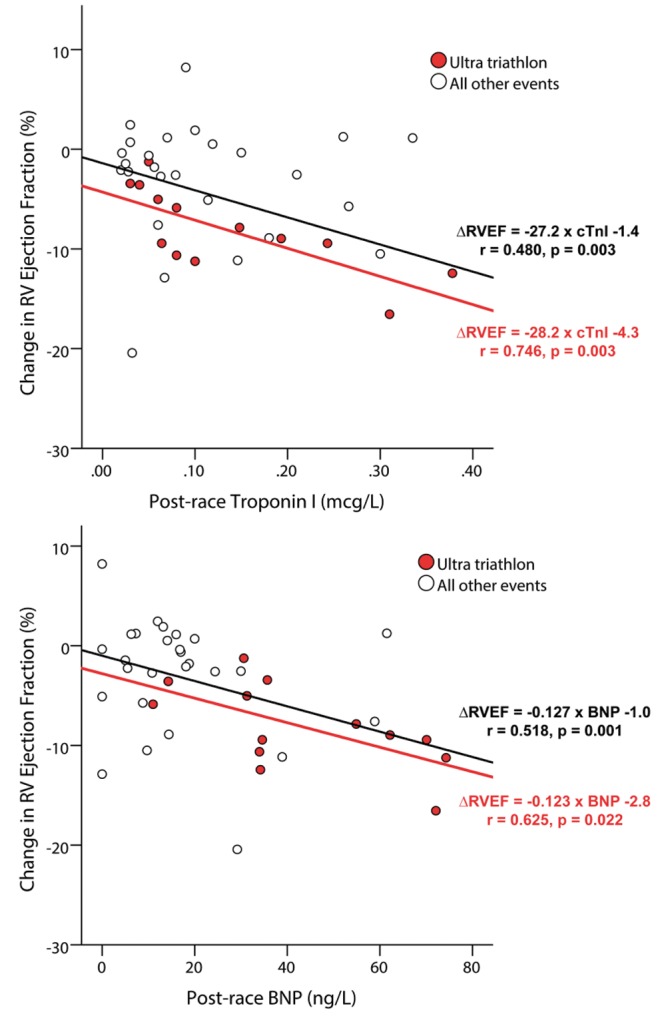
Moderate correlation between expression of cardiac biomarkers and decreases in right ventricular (RV) ejection fraction (RVEF). The release of cardiac troponin (cTnl) and B-type natriuretic peptide (BNP) has been repeatedly observed following ultraendurance exercise but has not correlated with changes in left ventricular function. We hypothesized that, given that the RV was the predominant source of exercise injury, changes in RV function might explain the increases in markers of myocardial injury and strain. Indeed, we demonstrated a moderate correlation between change in RVEF and increases in biomarkers taken within 1 hour after the race. We also observed that this relationship was strongest in those who performed exercise of greatest duration.39
Chronic RV remodeling in athletes
Given the increasing RV demand during exercise, it could be hypothesized that with increasing amounts of habitual exercise the RV may remodel in an attempt to approximate the morphology and function of the LV. However, evidence of disproportionate RV remodeling in endurance athletes is conflicting. Scharhag et al.44 described symmetrical ventricular enlargement in 21 young endurance athletes (aged 27 ± 5 years), whereas in a slightly larger cohort we found larger relative increases in the RV∶LV ratio for end-systolic volumes when comparing endurance athletes to healthy controls.25 This could relate to the fact that our cohort was older (aged 36 ± 8 years) and had greater exposure to exercise-induced load excess. However, it is difficult to draw firm conclusions from these disparate results, especially when one considers that the degree of ventricular asymmetry resulting from exercise is likely to be slight. It could be argued that all prior studies in athletic cohorts have been underpowered to assess subtle ventricular differences. This proposition is supported by the results of Aaron et al.,45 who recruited a large cohort of 1,867 nonathletes and demonstrated that the amount of strenuous activity predicted RV mass and volumes independent of LV size.
Some parallel insights may be gained from animal experiments on the basis of an induced aortacaval fistula in pigs.46 This intervention leads to a chronic high-output cardiac state with an increase in volume and pressure load somewhat akin to exercise. After 3 months, there was a disproportionate increase in RV stroke work index relative to that of the LV (+216% vs. +70%), RV fibrosis, and the development of RV dysfunction. Compared with the more “physiological” LV hypertrophy characterized by myocyte length increase and increased local production of insulin-like growth factor (IGF), the RV showed more pronounced hypertrophy, an increase in both myocyte length and diameter, and an associated increased collagen deposition. Apart from IGF-1 increase, there also was an increase in endothelin 1 and angiotensin 2 production. The authors postulated that the differential loads led to different gene expression and different structural changes.46 More recently, Benito et al.47 established an animal model of endurance activity in which rats exercised vigorously for 16 weeks (which was estimated to be equivalent to 10 years of endurance exercise in humans). At the end of the intervention, the running rats showed increased interstitial fibrosis in both atria and the RV but none in the LV. This was associated with inducibility of ventricular arrhythmias in 42% of the exercise rats compared with only 6% in controls (P = 0.05). Importantly, cessation of exercise reversed the inflammatory and fibrotic changes.
These animal studies share some striking resemblance to findings in some endurance athletes in whom many years of intense endurance training are associated with exaggerated RV remodeling and a propensity to arrhythmias. Heidbuchel et al.48 coined the term “exercise-induced arrhythmogenic RV cardiomyopathy” after observing that among endurance athletes, ventricular arrhythmias were frequently associated with electrical, structural, and functional changes of the RV but rarely of the LV. A comprehensive evaluation was undertaken in 46 high-level endurance athletes (performing ≥3 × 2 hours of sports per week for more than 5 years; 80% competitive; 80% cyclists) who had been diagnosed with sustained or nonsustained ventricular tachycardia or frequent ventricular ectopy (37%, 52%, and 11% of cases, respectively) in the context of nonspecific symptoms, such as palpitations and dizziness. The great majority of ventricular arrhythmias were of RV origin (86% of cases), which was surprising given that inherited cardiomyopathies, ischemic heart disease (due to congenital coronary anomalies or atherosclerosis), and myocarditis are most frequently associated with ventricular arrhythmias and sudden cardiac death in young athletic cohorts.49-51 Most of these pathologies would imply a LV or biventricular substrate contrasting with the high prevalence of RV pathology, which could only be explained by a major selection bias resulting in athletes with familial arrhythmogenic RV cardiomyopathy (ARVC) being disproportionately represented. However, such a bias did not seem likely and was not supported by the fact that only one athlete (2%) had a family history suggestive of ARVC. Rather, Heidbuchel and colleagues proposed that the association between RV abnormalities and endurance sport practice could be better explained if it were endurance sport itself that was contributing to the creation of an arrhythmic substrate in the RV. In other words, the unifying feature of the cohort (the very high volume of sport practice) was the likely explanation for the unexpectedly high prevalence of RV abnormalities.
Two recent studies have reported small patches of delayed gadolinium enhancement (DGE) on magnetic resonance studies in 13% and 50% of athletes with a long-standing history of endurance competition.52,53 These findings could be consistent with the hypothesis that sustained exercise excess results in chronic fibrous remodeling and arrhythmogenicity. However, it is important to note that a number of studies have failed to identify DGE in endurance athletes,35,37 and there are no studies assessing the relationship between DGE and clinical events (such as arrhythmias) in athletes. Thus, it would be premature to suggest that patches of DGE represent a substrate for arrhythmias, although some investigators are drawing this link in cases of unexplained cardiac arrest in highly trained athletes.54
The mechanism by which endurance exercise may promote myocardial fibrosis has not been established. As already discussed in this review, our contention is that the hemodynamic load of exercise plays an integral part in defining the metabolic and mechanical stresses on the heart. Of great interest, therefore, is the fact that the location of the DGE in athletes is most commonly localized to the interventricular septum in a pattern reminiscent of that associated with pulmonary hypertension and other conditions in which right heart strain is increased. Exercise CMR provides an intriguing insight into this process. Video 2 illustrates septal DGE in an endurance athlete and corresponding cine imaging through the same short-axis slice during strenuous exercise. It is notable that the DGE is at the very site where there is rapid excursion of the septum toward the LV in early diastole.
Video 2.

Summary and clinical significance
There is evolving evidence that the pulmonary circulation and RV play an important role in the augmentation of blood flow during exercise. The extent to which cardiac output and oxygen delivery to the working muscles is determined by factors upstream of the LV has been somewhat overlooked. This is due in large part to the fact that it is far more challenging to study pulmonary vascular and RV function, particularly during exercise. However, noninvasive imaging advances such as exercise CMR and novel echocardiographic techniques are providing invaluable insights in combination with a number of recent studies reappraising RV physiology using direct invasive measures. These findings are starting to build a clear picture; the pulmonary circulation and RV are placed under disproportionate stress during intense exercise, and when that exercise load is sustained the RV can suffer fatigue or injury. In well-trained athletes, the complete picture can be observed with increased acute hemodynamic stress causing transient RV dysfunction. According to normal physiological principals of growth through damage and compensatory repair, the heart muscle would gradually increase its size and strength to adapt to the intermittent exercise load. However, as illustrated in Figure 5, we hypothesize that if training is too intense or frequent to enable sufficient compensation, then a maladaptive process may result in proarrhythmic RV remodeling termed “exercise-induced RV cardiomyopathy.” The exact determinants and individual predispositions (genetic or otherwise) are still to be established.
Figure 5.
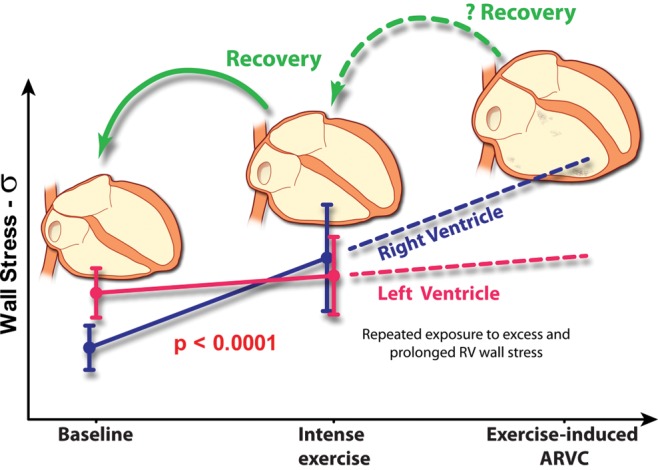
Healthy training versus overtraining of the heart. Healthy training with balanced exercise and recovery results in physiological remodeling in which enhanced cardiac structure and function enable greater cardiac performance during exercise. On the other hand, we propose that excessive exercise (training that is too intense and/or recovery that is too short) may cause cardiac injury and proarrhythmic remodeling, which predominantly affects the right ventricle (RV; adapted from the doctoral thesis of La Gerche55). ARVC: arrhythmogenic RV cardiomyopathy.
In the normal healthy circulation, extreme exercise is required to place sufficient strain on the RV to result in functional impairment. However, in patients with pulmonary vascular or RV pathology, the normal exercise-induced augmentation of cardiac output may be attenuated by these presystemic factors. Thus, exercise measures of the RV and pulmonary circulation may prove important in the diagnosis of exertional intolerance, the guidance of physiologically targeted therapies and prediction of patient outcomes. As we progress from “one size fits all” for the management of heart failure to a more patient-specific approach, assessment of the RV can no longer continue to be ignored.
Source of Support: Nil.
Conflict of Interest: None declared.
Supplements
References
- 1.Prior DL, La Gerche A. The athlete’s heart. Heart 2012;98:947–955. [DOI] [PubMed]
- 2.La Gerche A, Taylor AJ, Prior DL. Athlete’s heart: the potential for multimodality imaging to address the critical remaining questions. JACC Cardiovasc Imaging 2009;2:350–363. [DOI] [PubMed]
- 3.La Gerche A, Baggish AL, Knuuti J, et al. Cardiac imaging and stress testing asymptomatic athletes to identify those at risk of sudden cardiac death. JACC Cardiovasc Imaging 2013;6:993–1007. [DOI] [PubMed]
- 4.La Gerche A, Burns AT, Taylor AJ, MacIsaac AI, Heidbuchel H, Prior DL. Maximal oxygen consumption is best predicted by measures of cardiac size rather than function in healthy adults. Eur J Appl Physiol 2012;112:2139–2147. [DOI] [PubMed]
- 5.Steding K, Engblom H, Buhre T, et al. Relation between cardiac dimensions and peak oxygen uptake. J Cardiovasc Magn Reson 2010;12:8. [DOI] [PMC free article] [PubMed]
- 6.Morganroth J, Maron BJ, Henry WL, Epstein SE. Comparative left ventricular dimensions in trained athletes. Ann Intern Med 1975;82:521–524. [DOI] [PubMed]
- 7.Pluim BM, Zwinderman AH, van der Laarse A, van der Wall EE. The athlete’s heart: a meta-analysis of cardiac structure and function. Circulation 2000;101:336–344. [DOI] [PubMed]
- 8.Spence AL, Naylor LH, Carter HH, et al. A prospective randomised longitudinal MRI study of left ventricular adaptation to endurance and resistance exercise training in humans. J Physiol 2011;589:5443–5452. [DOI] [PMC free article] [PubMed]
- 9.West JB, Watson RR, Fu Z. Major differences in the pulmonary circulation between birds and mammals. Respir Physiol Neurobiol 2007;157:382–390. [DOI] [PMC free article] [PubMed]
- 10.Lankhaar JW, Westerhof N, Faes TJ, et al. Pulmonary vascular resistance and compliance stay inversely related during treatment of pulmonary hypertension. Eur Heart J 2008;29:1688–1695. [DOI] [PubMed]
- 11.Slife DM, Latham RD, Sipkema P, Westerhof N. Pulmonary arterial compliance at rest and exercise in normal humans. Am J Physiol 1990;258:H1823–H1828. [DOI] [PubMed]
- 12.Reeves JT, Groves BM, Cymerman A, et al. Operation Everest II: cardiac filling pressures during cycle exercise at sea level. Respir Physiol 1990;80:147–154. [DOI] [PubMed]
- 13.Buechel EV, Kaiser T, Jackson C, Schmitz A, Kellenberger CJ. Normal right- and left ventricular volumes and myocardial mass in children measured by steady state free precession cardiovascular magnetic resonance. J Cardiovasc Magn Reson 2009;11:19. [DOI] [PMC free article] [PubMed]
- 14.Faber MJ, Dalinghaus M, Lankhuizen IM, et al. Right and left ventricular function after chronic pulmonary artery banding in rats assessed with biventricular pressure-volume loops. Am J Physiol Heart Circ Physiol 2006;291:H1580–H1586. [DOI] [PubMed]
- 15.Chin KM, Kim NH, Rubin LJ. The right ventricle in pulmonary hypertension. Coron Artery Dis 2005;16:13–18. [DOI] [PubMed]
- 16.MacNee W. Pathophysiology of cor pulmonale in chronic obstructive pulmonary disease: part one. Am J Respir Crit Care Med 1994;150:833–852. [DOI] [PubMed]
- 17.La Gerche A, Claessen G, Van de Bruaene A, et al. Cardiac MRI: a new gold standard for ventricular volume quantification during high-intensity exercise. Circ Cardiovasc Imaging 2013;6:329–338. [DOI] [PubMed]
-
18.Levine BD. /article/back/ref-list/ref/mixed-citation/inline-formula
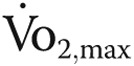 : what do we know, and what do we still need to know? J Physiol 2008;586:25–34. [DOI] [PMC free article] [PubMed]
: what do we know, and what do we still need to know? J Physiol 2008;586:25–34. [DOI] [PMC free article] [PubMed] - 19.Argiento P, Chesler N, Mulè M, et al. Exercise stress echocardiography for the study of the pulmonary circulation. Eur Respir J 2010;35:1273–1278. [DOI] [PMC free article] [PubMed]
- 20.Dawson CA. Role of pulmonary vasomotion in physiology of the lung. Physiol Rev 1984;64:544–616. [DOI] [PubMed]
- 21.McLaughlin VV, Langer A, Tan M, et al. Contemporary trends in the diagnosis and management of pulmonary arterial hypertension: an initiative to close the care gap. Chest 2013;143:324–332. [DOI] [PubMed]
- 22.Jacobs KA, Kressler J, Stoutenberg M, Roos BA, Friedlander AL. Sildenafil has little influence on cardiovascular hemodynamics or 6-km time trial performance in trained men and women at simulated high altitude. High Alt Med Biol 2011;12:215–222. [DOI] [PubMed]
- 23.Bidart CM, Abbas AE, Parish JM, Chaliki HP, Moreno CA, Lester SJ. The noninvasive evaluation of exercise-induced changes in pulmonary artery pressure and pulmonary vascular resistance. J Am Soc Echocardiogr 2007;20:270–275. [DOI] [PubMed]
- 24.Argiento P, Chesler N, Mulè M, et al. Exercise stress echocardiography for the study of the pulmonary circulation. Eur Respir J 2010;35:1273–1278. [DOI] [PMC free article] [PubMed]
- 25.La Gerche A, Heidbuchel H, Burns AT, et al. Disproportionate exercise load and remodeling of the athlete’s right ventricle. Med Sci Sports Exerc 2011;43:974–981. [DOI] [PubMed]
- 26.La Gerche A, MacIsaac AI, Burns AT, et al. Pulmonary transit of agitated contrast is associated with enhanced pulmonary vascular reserve and right ventricular function during exercise. J Appl Physiol 2010;109:1307–1317. [DOI] [PubMed]
- 27.Lewis GD, Murphy RM, Shah RV, et al. Pulmonary vascular response patterns during exercise in left ventricular systolic dysfunction predict exercise capacity and outcomes. Circ Heart Fail 2011;4:276–285. [DOI] [PMC free article] [PubMed]
- 28.Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J 2009;34:888–894. [DOI] [PubMed]
- 29.Stanek V, Jebavy P, Hurych J, Widimsky J. Central haemodynamics during supine exercise and pulmonary artery occlusion in normal subjects. Bull Physiopathol Respir (Nancy) 1973;9:1203–1217. [PubMed]
- 30.Gurtner HP, Walser P, Fassler B. Normal values for pulmonary haemodynamics at rest and during exercise in man. Prog Resp Res 1975;9:1203–1217.
- 31.Stanek V, Widimsky J, Degre S, Denolin H. The lesser circulation during exercise in healthy subjects. Prog Resp Res 1975;9:295–315.
- 32.Holverda S, Rietema H, Westerhof N, et al. Stroke volume increase to exercise in chronic obstructive pulmonary disease is limited by increased pulmonary artery pressure. Heart 2009;95:137–141. [DOI] [PubMed]
- 33.Holverda S, Gan CT, Marcus JT, Postmus PE, Boonstra A, Vonk-Noordegraaf A. Impaired stroke volume response to exercise in pulmonary arterial hypertension. J Am Coll Cardiol 2006;47:1732–1733. [DOI] [PubMed]
- 34.Shave R, George K, Whyte G, Hart E, Middleton N. Postexercise changes in left ventricular function: the evidence so far. Med Sci Sports Exerc 2008;40:1393–1399. [DOI] [PubMed]
- 35.Trivax JE, Franklin BA, Goldstein JA, et al. Acute cardiac effects of marathon running. J Appl Physiol 2010;108:1148–1153. [DOI] [PubMed]
- 36.Neilan TG, Yoerger DM, Douglas PS, et al. Persistent and reversible cardiac dysfunction among amateur marathon runners. Eur Heart J 2006;27:1079–1084. [DOI] [PubMed]
- 37.Mousavi N, Czarnecki A, Kumar K, et al. Relation of biomarkers and cardiac magnetic resonance imaging after marathon running. Am J Cardiol 2009;103:1467–1472. [DOI] [PubMed]
- 38.Douglas PS, O’Toole ML, Hiller WD, Reichek N. Different effects of prolonged exercise on the right and left ventricles. J Am Coll Cardiol 1990;15:64–69. [DOI] [PubMed]
- 39.La Gerche A, Burns AT, Mooney DJ, et al. Exercise-induced right ventricular dysfunction and structural remodelling in endurance athletes. Eur Heart J 2012;33:998–1006. [DOI] [PubMed]
- 40.La Gerche A, Connelly KA, Mooney DJ, MacIsaac AI, Prior DL. Biochemical and functional abnormalities of left and right ventricular function after ultra-endurance exercise. Heart 2008;94:860–866. [DOI] [PubMed]
- 41.Oxborough D, Shave R, Warburton D, et al. Dilatation and dysfunction of the right ventricle immediately after ultraendurance exercise: exploratory insights from conventional two-dimensional and speckle tracking echocardiography. Circ Cardiovasc Imaging 2011;4:253–263. [DOI] [PubMed]
- 42.Neilan TG, Januzzi JL, Lee-Lewandrowski E, et al. Myocardial injury and ventricular dysfunction related to training levels among nonelite participants in the Boston Marathon. Circulation 2006;114:2325–2333. [DOI] [PubMed]
- 43.Marcus JT, Gan CT, Zwanenburg JJ, et al. Interventricular mechanical asynchrony in pulmonary arterial hypertension: left-to-right delay in peak shortening is related to right ventricular overload and left ventricular underfilling. J Am Coll Cardiol 2008;51:750–757. [DOI] [PubMed]
- 44.Scharhag J, Schneider G, Urhausen A, Rochette V, Kramann B, Kindermann W. Athlete’s heart: right and left ventricular mass and function in male endurance athletes and untrained individuals determined by magnetic resonance imaging. J Am Coll Cardiol 2002;40:1856–1863. [DOI] [PubMed]
- 45.Aaron CP, Tandri H, Barr RG, et al. Physical activity and right ventricular structure and function: the MESA–Right Ventricle Study. Am J Respir Crit Care Med 2011;183:396–404. [DOI] [PMC free article] [PubMed]
- 46.Modesti PA, Vanni S, Bertolozzi I, et al. Different growth factor activation in the right and left ventricles in experimental volume overload. Hypertension 2004;43:101–108. [DOI] [PubMed]
- 47.Benito B, Gay-Jordi G, Serrano-Mollar A, et al. Cardiac arrhythmogenic remodeling in a rat model of long-term intensive exercise training. Circulation 2011;123:13–22. [DOI] [PubMed]
- 48.Heidbuchel H, Hoogsteen J, Fagard R, et al. High prevalence of right ventricular involvement in endurance athletes with ventricular arrhythmias: role of an electrophysiologic study in risk stratification. Eur Heart J 2003;24:1473–1480. [DOI] [PubMed]
- 49.Corrado D, Basso C, Rizzoli G, Schiavon M, Thiene G. Does sports activity enhance the risk of sudden death in adolescents and young adults? J Am Coll Cardiol 2003;42:1959–1963. [DOI] [PubMed]
- 50.Maron BJ, Doerer JJ, Haas TS, Tierney DM, Mueller FO. Sudden deaths in young competitive athletes: analysis of 1866 deaths in the United States, 1980–2006. Circulation 2009;119:1085–1092. [DOI] [PubMed]
- 51.de Noronha SV, Sharma S, Papadakis M, Desai S, Whyte G, Sheppard MN. Aetiology of sudden cardiac death in athletes in the United Kingdom: a pathological study. Heart 2009;95:1409–1414. [DOI] [PubMed]
- 52.Wilson M, O’Hanlon R, Prasad S, et al. Diverse patterns of myocardial fibrosis in lifelong, veteran endurance athletes. J Appl Physiol 2011;110:1622–1626. [DOI] [PMC free article] [PubMed]
- 53.La Gerche A, Burns AT, Mooney DJ, et al. Exercise-induced right ventricular dysfunction and structural remodelling in endurance athletes. Eur Heart J 2012;33:1006. [DOI] [PubMed]
- 54.Trivax JE, McCullough PA. Phidippides cardiomyopathy: a review and case illustration. Clin Cardiol 2012;35:69–73. [DOI] [PMC free article] [PubMed]
- 55.La Gerche A. Aetiology of structural and functional remodeling of the right ventricle in endurance athletes [doctoral thesis]. University of Melbourne, 2010.