

Abstract Abstract
Pulmonary arterial hypertension (PAH) contributes to poor outcomes in diverse diseases in newborns, infants, and children. Many aspects of pediatric PAH parallel the pathophysiology and disease courses observed in adult patients; however, critical maturational differences exist that contribute to distinct outcomes and therapeutic responses in children. In comparison with adult PAH, disruption of lung vascular growth and development, or angiogenesis, plays an especially prominent role in the pathobiology of pediatric PAH. In children, abnormalities of lung vascular development have consequences well beyond the adverse hemodynamic effects of PAH alone. The developing endothelium also plays critical roles in development of the distal airspace, establishing lung surface area for gas exchange and maintenance of lung structure throughout postnatal life through angiocrine signaling. Impaired functional and structural adaptations of the pulmonary circulation during the transition from fetal to postnatal life contribute significantly to poor outcomes in such disorders as persistent pulmonary hypertension of the newborn, congenital diaphragmatic hernia, bronchopulmonary dysplasia, Down syndrome, and forms of congenital heart disease. In addition, several studies support the hypothesis that early perinatal events that alter lung vascular growth or function may set the stage for increased susceptibility to PAH in adult patients (“fetal programming”). Thus, insights into basic mechanisms underlying unique features of the developing pulmonary circulation, especially as related to preservation of endothelial survival and function, may provide unique therapeutic windows and distinct strategies to improve short- and long-term outcomes of children with PAH.
Keywords: pulmonary vascular development, angiogenesis, alveolarization, persistent pulmonary hypertension of the newborn, congenital diaphragmatic hernia, bronchopulmonary dysplasia, Down syndrome, pediatric pulmonary hypertension
Pulmonary arterial hypertension (PAH) is a devastating and progressive disease that is characterized by abnormalities of vascular tone and reactivity, vessel wall structure, growth, and thrombosis.1 Mechanisms contributing to PAH and its progression are complex and remain incompletely understood. Insights into vascular biology over the past few decades have led to remarkable improvements in the clinical course, outcomes, and survival in adults with PAH, yet relatively few studies have been performed in children. As in adults, PAH contributes to poor outcomes in diverse cardiac, pulmonary, and systemic diseases in newborns, infants, and children (Table 1).2 There is a clear need to define the natural history of pediatric PAH in diverse settings; to develop new strategies to identify at-risk patients early in their course; and to establish novel approaches to better diagnose, evaluate, and treat children with PAH.3,4
Table 1.
A new pediatric pulmonary hypertensive vascular disease classification system (Panama)
| Category | Description |
|---|---|
| 1 | Prenatal or developmental pulmonary hypertensive vascular disease |
| 2 | Perinatal pulmonary vascular maladaptation |
| 3 | Pediatric cardiovascular disease |
| 4 | Bronchopulmonary dysplasia |
| 5 | Isolated pediatric pulmonary hypertensive vascular disease (isolated pediatric pulmonary arterial hypertension) |
| 6 | Multifactorial pulmonary hypertensive vascular disease in congenital malformation syndromes |
| 7 | Pediatric lung disease |
| 8 | Pediatric thromboembolic disease |
| 9 | Pediatric hypobaric hypoxic exposure |
| 10 | Pediatric pulmonary vascular diseases associated with other system disorders |
From del Cerro et al.2
Despite many similarities between pediatric and adult PAH, critical differences exist that currently limit our clinical approaches to children with PAH. Pediatric PAH is intrinsically linked with lung vascular growth and is modulated by prenatal and early postnatal factors (as reviewed by Abman and Raj3 and by Barst et al.;5 Table 2). Development of PAH in the neonate and young infant is often related to impaired functional and structural adaptation of the pulmonary circulation during the transition from fetal to postnatal life. Since the vascular endothelium plays critical roles in lung growth and the maintenance of lung structure, abnormalities of the lung circulation have consequences far beyond the adverse hemodynamic effects of PAH alone (Figure 1).6-8
Table 2.
Unique aspects of pediatric pulmonary vascular disease
| Developmental biology of the cardiopulmonary system |
| Timing of vascular injury during susceptible periods of growth and adaptation |
| Impact of lung vascular disease beyond pulmonary hypertension alone |
| Differences in genetics, maturational changes in vascular function and growth, and responsiveness to therapeutic strategies |
| Developmental differences in pharmacokinetics and pharmacodynamics, potential adverse effects |
| Additional importance of “preventive” strategies as well as approaches that target “reverse remodeling” |
| Maturational changes in right and left ventricular function |
Figure 1.

Pediatric vascular diseases associated with decreased alveolarization.
Disruption of lung vascular growth impairs alveolarization during development and contributes to the pathobiology of diverse cardiorespiratory diseases in children.6-8 In addition, differences exist between adult and pediatric PAH regarding vascular function and structure, genetics, natural history, response of the right ventricle to PH, and perhaps responsiveness to PAH-specific therapies. Therapeutic strategies for adult PAH have not been sufficiently studied in children, especially regarding potential toxicities or optimal dosing, and age-appropriate endpoints for clinical care and research are lacking. There is a critical need to better characterize unique aspects of the developing lung circulation and its response to injury as well as the need to establish biomarkers that can predict disease risk, severity, and progression, and outcome measures that are applicable to young children with PAH.
In this article, we briefly review several key examples of pediatric disorders that illustrate unique features of pulmonary vascular disease (PVD) and PAH in children. In particular, we highlight the primary theme that impaired angiogenic mechanisms underlie abnormalities of lung vascular growth, which uniquely contribute to the pathogenesis of pediatric PAH, as observed in neonatal PH (persistent pulmonary hypertension of the newborn [PPHN]), congenital diaphragmatic hernia (CDH), chronic lung disease in preterm infants (bronchopulmonary dysplasia [BPD]), and cardiopulmonary disease in Down syndrome (Figure 1). Finally, we briefly discuss several key issues regarding clinical strategies needed to enhance the care and treatment of children with PVD.
Impaired lung vascular development in pediatric PAH
General principles
Normal lung growth and structure are dependent on closely orchestrated signaling between the developing epithelium and its vasculature.9 Although much emphasis has been placed on the role of airway epithelium in guiding vascular growth, a growing body of evidence supports the hypothesis that lung vessels actively promote alveolar growth during development and contribute to the maintenance of alveolar structures throughout postnatal life.10 Angiocrine signaling mechanisms are particularly important during late gestation and early postnatal life. Disruption of endothelial cell survival and function during this critical window of rapid alveolar and capillary growth and development has been implicated in the pathogenesis of neonatal PAH, as discussed below.6-8,11,12 In addition, recent studies suggest that sustained “cross talk” between endothelial and epithelial cells within the distal airspace remains essential during lung regeneration after pneumonectomy13 and as part of a key “maintenance program” for maintaining alveolar health in response to injury throughout adulthood.10,14,15
The lung vasculature largely grows through two basic but overlapping processes: vasculogenesis and angiogenesis. Vasculogenesis is defined as the de novo formation of blood vessels from angioblasts or endothelial progenitor cells (EPCs) that migrate, differentiate, and grow into tubes in response to signals from neighboring cells. Angiogenesis is most simply defined as growth by direct extension from preexisting vessels.
Controversies exist regarding the relative roles of vasculogenesis and angiogenesis during early lung development, generating at least 3 alternate hypotheses.16-18 DeMello16 originally proposed concurrent roles for both vasculogenesis and angiogenesis. In this model, proximal pulmonary arteries and veins expand by endothelial proliferation from existing vessels while distal vessels develop from blood islands within the mesenchyme through differentiation of endothelial cells from immature precursors. In contrast, a primary role for vasculogenesis in the distal lung has also been suggested, in which intrapulmonary vessels derive from continuous expansion of the primary capillary plexus within the developing mesenchyme.17 Finally, Parera et al.18 argue that distal angiogenesis plays a more prominent role, as capillary networks surrounding the terminal buds expand by formation of new capillaries from preexisting vessels, along with the expanding airway.
Much excitement has been generated regarding potential roles for EPCs during lung vascular development; however, experimental data that clearly demonstrate EPC-related mechanisms in this process are lacking.19-21 Demonstrations of the critical role of the “secretome” in mediating proangiogenic effects of EPCs and other progenitor cells, independent of cell engraftment, further illustrate the close interplay between vasculogenic and angiogenic mechanisms.22-24
During formation of secondary septae in the alveolar stage of lung development, the double capillary network in the immature gas-exchange region fuses to form a single capillary layer.25,26 Between birth and adulthood, the alveolar and capillary surface areas expand nearly 20-fold and the capillary volume by 35-fold, primarily by sprouting angiogenesis from preexisting vessels and through energy-efficient intussusceptive growth.27 Thus, early disruption of these mechanisms during this critical window of development has long-lived implications for both short- and long-term outcomes of pulmonary vascular health throughout the “life course.”28
Multiple molecular mechanisms play important autocrine and paracrine roles during vascular development, but perhaps the most potent and best-studied angiogenic factor is vascular endothelial growth factor (VEGF).29-39 VEGF is a highly specific mitogen and survival factor for endothelium, including the lung vasculature. VEGF binds transmembrane tyrosine kinase receptors, including VEGFR-1 (or flt-1) and VEGFR-2 (flk-1/KDR), which are strongly expressed in vascular endothelium. VEGF messenger RNA (mRNA) and protein are strongly expressed in distal airway epithelial cells. Gene expression of VEGFR-1 and VEGFR-2 is prominent in the pulmonary endothelial cells closely apposed to the developing epithelium and increases during development.35,36 VEGF is absolutely required for embryonic vascular growth and survival in mice, because gene inactivation of VEGF or its receptors is embryonic lethal and is characterized by deficient organization of endothelial cells.31-33
During late gestation and the early postnatal period, pharmacologic and genetic strategies to inhibit VEGF signaling arrest alveolar as well as vascular development. Targeted exon deletion of the VEGF gene reveals that mice that lack the heparin-binding isoforms VEGF164 and VEGF188 have a significant reduction in the formation of airspaces and capillaries, resulting in enlarged and underdeveloped alveoli.39 Likewise, pharmacological and genetic VEGF inhibition during alveolar development decreases alveolarization and pulmonary arterial density, which are features observed in clinical BPD.11,12,34,40 Conversely, overexpression of VEGF during normal development disrupts distal lung architecture, suggesting a tight regulation of VEGF-driven angiogenesis to insure proper lung development.41,42 Work from Thiennu Vu’s laboratory37,43 has shown that genetic VEGF inactivation in respiratory epithelium dramatically reduces vascular growth, clearly demonstrating the dependence of pulmonary capillary development on epithelium-derived VEGF-A during development. VEGF-A-deficient lungs were further characterized by a striking defect in primary septae formation, suppression of epithelial cell proliferation, and decreased expression of hepatocyte growth factor (HGF) and its receptor, cMet.43 As HGF is an endothelium-derived factor that is required for normal septation, these studies illustrate the vital importance of angiocrine signaling mechanisms during lung airspace development.
The role of the endothelium-derived relaxing factor nitric oxide (NO) in the regulation of pulmonary vascular tone in the perinatal period is well established.44 NO also modulates lung vascular growth during development. The effects of VEGF during lung development are partly mediated through enhanced NO production, and disruption of endothelial NO synthase (eNOS) expression impairs lung vascular function and growth.45,46
Lungs of late-fetal and neonatal eNOS-deficient mice have a paucity of distal arteries and reduced alveolarization45 and are more susceptible to failed vascular and alveolar growth after exposure to mild hypoxia and hyperoxia.47 Prolonged infusions of SU5416, a VEGF receptor antagonist, cause endothelial dysfunction, with decreased eNOS expression, pulmonary vascular remodeling, and reduced arterial and alveolar growth in late-fetal sheep.48 VEGF inhibition after birth inhibited alveolar and vascular growth in newborn rats and was also associated with decreased lung eNOS protein expression and NO production; treatment with inhaled NO improved vascular and alveolar growth in this model.49
As noted above, enthusiasm has been generated regarding potential roles for EPCs during lung vascular development; however, data that clearly implicate EPC-related mechanisms in this process remain lacking.19-21 Classic studies of the adult systemic circulation first revealed the existence of highly proliferative endothelial cells within the aortic intima that were enriched with replication-competent cells required to repopulate injured or senescent cells.50-52 More recently, David Ingram, Merv Yoder, and colleagues19 demonstrated that a “complete hierarchy” exists within the vasculature, including a population of self-renewing, highly proliferative endothelial cells. In addition, lung microvascular endothelial cells in rats have been shown to proliferate twice as fast as macrovascular endothelial cells.53-55 Thus, the pulmonary macro- and microvasculature are enriched with EPC populations that display vasculogenic and angiogenesis-promoting capabilities while maintaining functional endothelial specificity that includes the ability to form vascular networks in vitro. Surprisingly, the existence and roles of these cells in the developing lung, as well as their therapeutic potential, remain largely unexplored, and more work is needed to define the role of angiogenic progenitors and endothelium-related mechanisms during lung vascular development.
Impaired angiogenesis in experimental PPHN and CDH
PPHN represents failure to achieve or sustain the normal drop in pulmonary vascular resistance (PVR) after birth.56,57 PPHN is associated with diverse cardiac and pulmonary disorders or can occur as an idiopathic disorder and is characterized by abnormal pulmonary vascular tone and reactivity, hypertensive vascular remodeling, and, in severe cases, decreased vascular growth.58-60 Autopsy studies of the lungs of newborns with fatal PPHN have revealed severe vascular remodeling even in newborns who die shortly after birth, suggesting that severe disease is associated with chronic intrauterine stress.61-66 The exact intrauterine events that alter pulmonary vascular reactivity and growth in PPHN are poorly understood, but perinatal stresses, including chorioamnionitis, placental vascular lesions, and intrauterine growth restriction, are associated with PPHN. Maternal smoking, use of nonsteroidal anti-inflammatory medications, obesity, and diabetes have been linked with increased risk as well.61 Recent epidemiologic studies suggest that maternal use of some antidepressants might increase the risk for PPHN, as seen in some animal studies.66
Several experimental models have been used to explore the pathogenesis and pathophysiology of PPHN.63,67-70 PH induced experimentally by early closure of the ductus arteriosus in fetal sheep alters lung vascular reactivity and structure, causing the failure of postnatal adaptation at delivery and providing a useful model of PPHN. Studies with this model show that chronic hypertension without high blood flow impairs fetal lung vascular structure and function.63 This model is characterized by endothelial cell dysfunction, with decreased NO production and activity as a result of downregulation of lung eNOS mRNA and protein expression.71,72 In addition to striking changes in vasoreactivity and wall structure, chronic fetal PH also decreases vascular density and causes lung hypoplasia, with reduced alveolarization and lung weight (Figure 2).48 These anatomic, histologic, and physiologic findings of severe PPHN are associated with marked decreases in lung VEGF and VEGFR2 protein expression and can be mimicked in normal sheep by prolonged infusions of VEGF antagonists.48 Chronic recombinant human VEGF (rhVEGF) treatment increases eNOS expression, restores pulmonary vasoreactivity, attenuates hypertensive arterial remodeling, and improves vascular growth in this fetal model of PPHN.48 Overall, these findings demonstrate a critical role for VEGF in the pathogenesis of PPHN and further link impaired angiogenesis with abnormal distal lung structure in severe PPHN.
Figure 2.

Hemodynamic stress inhibits angiogenesis and alveolar growth in fetal sheep. Intrauterine pulmonary hypertension due to closure of the ductus arteriosus in utero causes decreased lung size by appearance (top left) and lung weight (top right; *P < 0.05 vs. control) and reduces vessel density (bottom left) and alveolarization (bottom right). PPHN: persistent pulmonary hypertension of the newborn; hpf: high-powered field. Reproduced with permission from Grover et al.48
Additional studies of isolated endothelial cells from control and PPHN sheep lungs further demonstrate that phenotypic abnormalities persist in vitro. Impaired growth and angiogenesis have been demonstrated from studies of endothelial cell isolates from PPHN sheep.73 In these studies, pulmonary artery studies of endothelial cells (PAECs) proliferate at slower rates and form fewer tubular networks in vitro when compared with age-matched controls, partly as a result of impaired VEGF-NO signaling and increased rho kinase (ROCK) activity.73,74 More recent studies have shown marked upregulation of endothelin-1 (ET-1) expression in isolated PPHN PAECs.75 In PAECs from PPHN sheep, ET-1 inhibition restores PAEC growth and tube formation, partly as a result of reduction of ET-1-induced activation of ROCK signaling.75
These findings suggest that ET-ROCK interactions play a key role in impaired endothelial function and angiogenesis in PPHN. Interestingly, findings in this perinatal model of PAH are strikingly different from the results of studies in adult rodent models of PAH, in which increased ROCK activity apparently enhanced vascular growth.76 We speculate that, in addition to beneficial effects on smooth muscle cell tone and proliferation, ET-1 receptor blockade may also enhance lung vascular growth in severe PPHN.
Decreased lung vascular growth, severe PAH, and marked lung hypoplasia are especially striking and increase mortality in infants with CDH (Figure 3). In an experimental model of CDH induced by surgical disruption of the diaphragm in fetal sheep, isolated PAECs had persistent abnormalities of growth and tube formation in vitro, as observed in PAECs from experimental PPHN.77 In addition to endothelial dysfunction, pulmonary arteries from CDH sheep were further characterized by the loss of the highly proliferative population of PAECs, as assessed by single-cell assay (Figure 4).77 Overall, these findings suggest that abnormal lung vascular growth in experimental CDH is associated with altered PAEC phenotype in vitro, with a striking loss of resident progenitor-like endothelial cells from conduit artery vessel walls.
Figure 3.
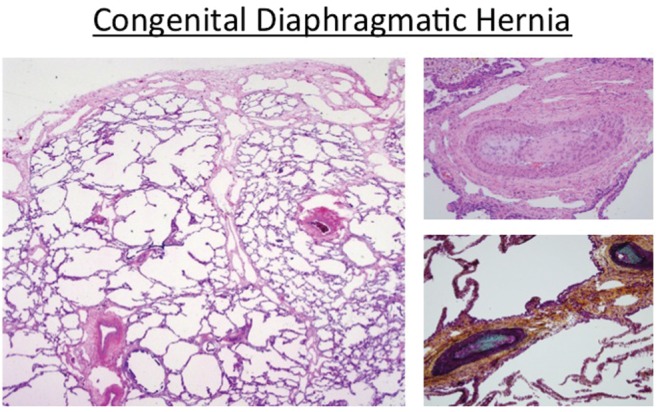
Histologic features of congenital diaphragmatic hernia, including decreased alveolarization and marked hypertensive arterial remodeling.
Figure 4.
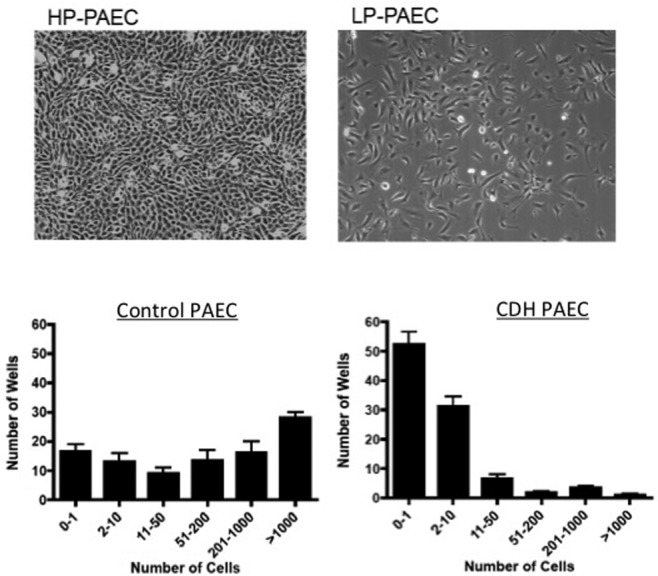
Loss of highly proliferative pulmonary artery endothelial cells (PAECs) from fetal pulmonary arteries in experimental congenital diaphragmatic hernia (CDH). Top, micrographs illustrating the markedly increased cell proliferation by highly proliferative (HP) PAECs in comparison with more typical growth of lowly proliferative (LP) PAECs. Bottom, findings from single-cell assay show the marked decrease in HP-PAEC populations from CDH isolates in comparison with controls. Reproduced with permission from Figures 8 and 9 of Acker et al.77
PVD in BPD
BPD is the chronic lung disease of infancy that follows mechanical ventilation and oxygen therapy of premature newborns for acute respiratory insufficiency at birth.78 BPD is often complicated by sustained oxygen dependency, prolonged need for ventilator support, recurrent respiratory exacerbations with frequent rehospitalizations, exercise intolerance, and high risk for PAH. PAH is common in infants with BPD, with an estimated incidence of 18%–25% of preterm infants,79-81 and is associated with high mortality (30%–40%), especially if sustained beyond the first months of life.82-85
Although long-term studies of BPD have primarily focused on late abnormalities of airway function, PVD can also persist into childhood and adult life.79,86-89 Tepper and colleagues88 have reported that infants with even mild BPD have decreased diffusion capacity when corrected for lung volume, in comparison with age-matched term controls, suggesting that BPD infants may have decreased alveolar surface area available for gas exchange. These differences in diffusion capacity for carbon monoxide were also found to persist in children at 11 years of age who had been born extremely preterm, and striking decreases in diffusion capacity can persist in adult survivors of BPD.87,89
Disruption of angiogenesis after endothelial injury and dysfunction contributes to the pathophysiology of BPD but also contributes to its pathogenesis.6-8 Endothelial cells are particularly susceptible to oxidant injury from intrauterine stress, postnatal hyperoxia, inflammation, and mechanical ventilation. Decreased angiogenesis reduces lung vascular surface area, causing further elevations of PVR, especially in response to high cardiac output with exercise, infection, or stress or with increased flow due to cardiac shunt lesions in infants with BPD (Figure 5).
Figure 5.

Pulmonary vascular disease in bronchopulmonary dysplasia (BPD). SMC: smooth muscle cell.
Preclinical data strongly support the hypothesis that impaired angiogenesis decreases alveolarization and that strategies that preserve endothelial cell survival, growth, and function may provide novel strategies for the prevention of BPD.6 Antiangiogenesis agents, such as thalidomide and fumagillin, reduced lung vascular density, alveolar number, and lung weight in infant rats.11 Similar effects were found after treatment of newborn rats with SU5416, a VEGF receptor inhibitor.12,40 In these studies, a single injection of SU5416 immediately after birth caused progressive PAH and reduced lung vascular density and alveolar growth in infant rats. Early endothelial cell apoptosis without inflammation preceded changes in vascular and alveolar structure.49 These findings suggest that angiogenesis is necessary for alveolarization during lung development and that disruption of lung vascular growth impedes alveolar growth after premature birth.
Premature birth with exposure to increased oxygen tension, inflammatory cytokines, and other adverse stimuli decrease VEGF expression and signaling, thereby altering lung vascular and alveolar structure.90 Lung VEGF mRNA and protein expression are decreased in diverse animal models of BPD, including large-animal models in primates and sheep as well as rodents (as reviewed by Abman90). In human studies, Bhatt and coworkers91 first demonstrated decreased VEGF and VEGFR-1 mRNA and protein in the lungs of premature infants who died with BPD. That VEGF levels are decreased in tracheal fluid samples from premature neonates who subsequently develop BPD provides further evidence that early downregulation of VEGF contributes to the pathogenesis of clinical BPD.92
Mechanisms through which impaired VEGF signaling inhibits vascular growth and alveolarization are uncertain, but they include decreased NO production. Past in vitro and in vivo studies have shown that VEGF stimulates endothelial eNOS expression in endothelial cells from the systemic circulation. NO mediates the angiogenic effects of VEGF on fetal PAECs,73 likely through activation of VEGFR-2 and stimulation of the Akt-PI3K pathway.93 Lung eNOS expression is decreased in rodent, primate, and ovine models of BPD. However, rhVEGF therapy can restore lung function after hyperoxia in eNOS−/− mice, suggesting that VEGF acts through non-eNOS-related pathways as well, such as HGF.43
The therapeutic potential for angiogenic growth factor therapy in experimental lung disease was illustrated by studies on the effects of systemic recombinant human VEGF165 (rhVEGF) treatment of newborn rats during or after exposure to hyperoxia, which showed enhanced vessel and alveolar growth.94,95 Intratracheal adenovirus-mediated VEGF gene therapy improves survival, promotes lung capillary formation, preserves alveolar development, and regenerates new alveoli in this same model of lung injury.40 In these studies, VEGF stimulated sprouting of immature and leaky capillaries, leading to lung edema. Combined lung VEGF and Ang-1 gene transfer preserves alveolarization and enhances angiogenesis with more mature capillaries that are less permeable, reducing the vascular leakage seen in VEGF-induced capillaries.40
The pathogenesis of PVD in BPD is complex and multifactorial, often resulting from interactions of genetic and epigenetic susceptibility factors with environmental exposures, including hyperoxia, hypoxia, hemodynamic stress, infection, and inflammation, among others.96,97 Several recent studies have shown that preeclampsia is a risk factor for BPD and PVD98 and that disruption of the VEGF and other vascular signaling pathways, including soluble endoglin associated with preeclampsia, may contribute to impaired angiogenesis in preterm infants born to these mothers.98-101 Placental overproduction of soluble VEGF receptor-1 (soluble fms-like tyrosine kinase-1 [sFlt-1]), which inhibits VEGF signaling through trapping free VEGF,102 causes maternal endothelial dysfunction and plays a central role in the pathogenesis of preeclampsia.99,103,104 Intra-amniotic sFlt-1 administration during late gestation impairs lung VEGF signaling and increases apoptosis of endothelial cells in newborn rats, which is followed by reductions in alveolarization and vascular growth, along with biventricular hypertrophy during infancy.98 Similarly, intrauterine growth restriction has been associated with an increased risk for PH in BPD infants105 and has also been associated with disruption of VEGF signaling and pulmonary vascular growth in experimental intrauterine growth restriction.106 Thus, disruption of vascular growth due to interactive prenatal and postnatal mechanisms impairs growth factor signaling and causes sustained reductions of alveolocapillary surface area, leading to abnormal gas exchange and long-lived respiratory impairment.107-111
Endogenous, umbilical cord–derived, and bone marrow–derived progenitor cells may play a role both in normal pulmonary angiogenesis and vasculogenesis and in the response to lung injury after birth.112,113 Several studies have revealed alterations in EPCs and mesenchymal stromal cells in experimental models of BPD.112,114,115 Late-outgrowth endothelial colony-forming cells (ECFCs), a specific subset of EPCs, are increased in the cord blood of preterm infants.116 However, among preterm infants, those who subsequently developed moderate or severe BPD at 36 weeks (corrected age) had markedly decreased cord blood ECFCs.117,118 Conflicting reports also exist about mesenchymal stromal cells, depending on the source.119-121 These findings highlight the importance of appropriately defining these progenitor cells and whether their function is altered because of their origin (lung, umbilical cord, bone marrow). Yet therapeutic administration of progenitor cells for the prevention or treatment of BPD holds significant promise, as several studies have shown benefit in experimental models.121-125
The existence of intrapulmonary arteriovenous connections (IAVCs) has been established by past studies utilizing differential injection methods, serial histologic sectioning, and fluorescent bead injection in human subjects and animal models.126-128 Physiologic demonstration of intrapulmonary shunt in fetal sheep has been shown by echocardiography, but shunting is less prominent in newborns.129 Late persistence or enhancement of IAVCs has been previously reported in pathologic settings as neonatal heart disease requiring cavopulmonary shunt operations. More recently, the presence of striking IAVCs, or “shunt” vessels, has been confirmed by histology and 3-dimensional reconstruction methods in autopsy lung tissue from preterm infants who died with severe BPD and alveolar capillary dysplasia with misalignment of pulmonary veins (Figure 6).130,131 Prominent, thin-walled, and centrally located abnormal intrapulmonary vessels were shown to connect small pulmonary arteries and veins with microvascular plexuses surrounding pulmonary arteries and airways. Vessels with such distinct appearance and course were not present in lungs of age-matched controls. We speculate that persistence of these “fetal shunt vessels” may contribute to the pathophysiology of neonatal lung diseases and perhaps intrapulmonary shunt throughout adulthood, as previously noted.132 Mechanisms that regulate the growth and development of IAVCs during development and with disease remain unknown.
Figure 6.
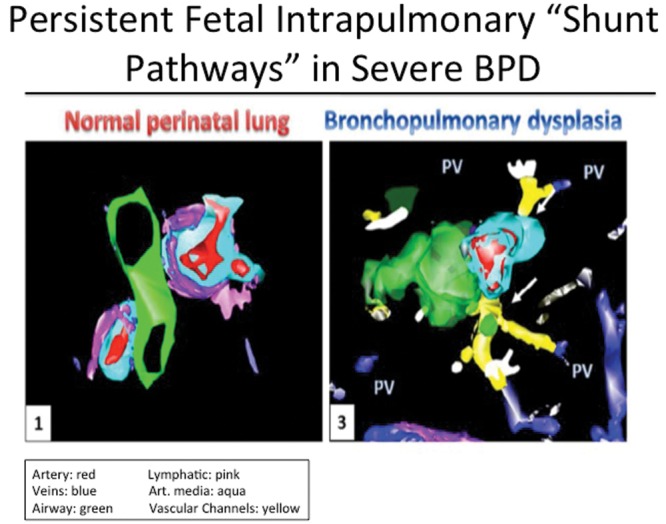
Three-dimensional reconstruction of distal lung tissue, illustrating intrapulmonary arteriovenous communications (“shunt vessels”) in infant with severe bronchopulmonary dysplasia (BPD). PV: pulmonary vein. Reproduced with permission from Galambos et al.130 Reprinted with permission of the American Thoracic Society. Copyright © 2014 American Thoracic Society. Galambos C Sims Lucas, Abman SH. (2013) Histologic evidence of intrapulmonary anastomoses by three-dimensional reconstruction in severe BPD. Ann Am Thorac Soc. 10:474–81. Official Journal of the American Thoracic Society.
In summary, reduced arterial number, structural abnormalities of the vessel wall, and abnormal vascular function, together with impairments in respiratory mechanics and cardiac function, contribute to increased PVR and PVD in BPD.8 Future work is needed to better define basic mechanisms of lung vascular growth and development, which will likely lead to novel therapeutic approaches to diseases associated with impaired vascular growth and PVD.
Abnormal lung vascular and alveolar development in Down syndrome
Down syndrome (DS) is associated with significant cardiovascular and pulmonary mortality and morbidity of neonates, infants, and children.133-136 Although it is well recognized that infants with DS are at marked risk for developing respiratory disease and severe PAH, the underlying mechanisms that contribute to cardiopulmonary diseases remain poorly understood. Newborns with DS are at higher risk for PPHN after birth, often requiring prolonged therapy. In addition, DS infants have worse PAH in conjunction with anatomic cardiac disease than non-DS infants with similar lesions. Finally, late-onset PAH in DS is often associated with such common problems as intermittent hypoxia due to obstructive sleep apnea, poor upper airway tone, tracheomalacia, and aspiration.136 The basis for this increased risk of PAH in DS has not been well understood; however, past pathologic findings have shown that infants who die with DS often have an abnormal lung structure that is characterized by decreased alveolarization and an abnormal lung vasculature, including persistence of the double capillary network (Figure 7). These features suggest that lung function of infants with DS is limited by decreased surface area, limiting gas exchange and increasing the risk of developing severe PAH at birth or during early childhood, especially in response to hemodynamic stress or chronic hypoxia. This suggests that even relatively small left-to-right shunts in congenital heart disease may represent a significantly higher level of hemodynamic stress in the setting of the underdeveloped lung vasculature in DS than is expected in normal infants.
Figure 7.

Lung histology from an infant with Down syndrome who died at 4 months, demonstrating enlarged and simplified alveolar spaces (left), a persistent double capillary layer (arrows) lining the alveolar spaces (middle), and hypertensive arterial remodeling (right).
Since lung histology of infants with DS often shares features of decreased lung vascular and alveolar growth as observed in infants with BPD, CDH, and PPHN, we suspect that abnormal lung angiogenesis causes decreased alveolarization during fetal and early postnatal life in infants with DS and that this alteration of lung structure contributes to the severity of early cardiovascular and pulmonary disease in children with DS. Subjects with DS have a decreased incidence of angiogenesis-related diseases, including solid tumors and vascular anomalies, as further reflected in a mouse model of DS.
DS patients have 3 copies of chromosome 21 that include at least 3 genes that have been shown to have potent antiangiogenic properties, including endostatin. Recently, endostatin and related DS genes have been shown to downregulate VEGF signaling through inhibition of calcium-calcineurin NF-AT (nuclear factor of activated T cells) activities, which attenuate endothelial proliferation and angiogenesis.137-143 Whereas these factors may be beneficial for protecting against tumor angiogenesis, we hypothesized that upregulation of such factors as endostatin may adversely affect lung structure through impaired angiogenesis, which further decreases alveolar growth and increases the risk for PAH. Using banked tissues of prenatal human DS and age-matched control lungs, we found a 3-fold increase of endostatin mRNA expression in prenatal DS lungs.144 Furthermore, these lung sections showed reduced microvascular density and thickened large and small pulmonary artery walls. These results strongly suggest a key role for genetically driven antiangiogenic signals in the pathogenesis of DS-related impaired lung vascular development and PAH.
Implications of fetal programming
Ongoing vascular growth and remodeling during infancy and childhood remain critical for the development and maintenance of normal lung architecture later in life. In addition, VEGF continues to function throughout postnatal life as an endothelial “maintenance” or “survival” factor, thereby sustaining normal vascular function through the expression of such key enzymes as eNOS and prostacyclin synthase. For example, chronic treatment of adult rats with the VEGF receptor inhibitor SU5416 causes enlargement of distal airspaces, indicative of emphysema, suggesting that VEGF is required for maintenance of distal lung structure throughout adulthood.12 This concept is further supported by the observation that angiocrine signals derived from distal lung endothelial cells induce and sustain regenerative lung alveolarization after pneumonectomy.13
Strong preclinical data suggests that early fetal events that disrupt angiogenesis may lead to sustained structural abnormalities of vascular and airspace growth, increasing susceptibility to late presentations of PAH and emphysema. For example, the effects of VEGF receptor inhibition in neonatal rats are sustained throughout infancy and persist into adulthood.12 Similarly, perinatal hypoxia increases susceptibility to late PAH after brief reexposure to hypoxia during adulthood (Figure 8, left).145 Clinical studies further support these laboratory findings. Young adult survivors of respiratory distress and PPHN have enhanced pulmonary vasoconstriction during acute exposure to hypoxia at altitude, despite the apparent absence of clinical signs of cardiopulmonary disease (Figure 8, right).146 Older children who were born at near-term gestation from pregnancies complicated by preeclampsia have increased basal PVR, by echocardiogram, and impaired flow-mediated dilation in the systemic circulation.147 These reports support overall concepts regarding the potential roles of fetal programming in determining late pulmonary vascular health.148 A better understanding of mechanisms that link perinatal events with adult disease may provide new insights into novel mechanisms underlying adult-onset PAH (Figure 9).
Figure 8.

Perinatal hypoxia increases risk of late pulmonary hypertension. Left, brief perinatal exposure to hypoxia increases susceptibility to PAH after late exposure to hypoxia in rats (RVSP: right ventricular systolic pressure; NN: normoxic control; NH: perinatal normoxia, postnatal hypoxia; HH: perinatal and postnatal hypoxia; HN: perinatal hypoxia, postnatal normoxia; NS: not significant; *P < 0.05 vs. NH; #P < 0.05 vs. NN); reproduced with permission from Figures 3 and 7 of Tang et al.145 Right, young adults with a history of neonatal distress and persistent pulmonary hypertension of the newborn have marked acute hypoxic pulmonary vasoreactivity response at altitude in humans; reproduced with permission from Sartori et al.146
Figure 9.

Schematic illustrating perinatal mechanisms contributing to sustained pulmonary vascular disease (PVD). IUGR: intrauterine growth restriction. Based on Figure 2 of Robbins et al.4
Conclusions
Despite sharing features of adult PAH, many disorders in children with PAH have unique characteristics that are distinctly different. Clearly, the pathobiology of pediatric PVD often includes impaired or dysregulated growth of the developing lung circulation, which plays an especially critical role in such pediatric disorders as PPHN, CDH, BPD, DS, and other diseases. Disruption of normal angiocrine signaling between endothelial and epithelial cells decreases lung airspace growth and surface area for gas exchange and increases the risk of PAH. Finally, these changes may further play a role in determining risk of or susceptibility to PVD and PAH throughout adulthood. Further studies are needed to better explore the impact of fetal programming and perinatal events on PVD and PAH during the “life course.”
Dedication
This article resulted from a presentation at the 2013 Grover Conference and is dedicated to the memory of Dr. Robyn Barst. There has never been a stronger advocate for improving outcomes of children with PAH than Dr. Barst. Her contributions to our understanding of mechanisms underlying PAH and its treatment were extraordinary and have had a long-lasting impact on our care of both children and adults with PAH. Throughout her career, from her early work establishing the key role for prostacyclin therapy in idiopathic PAH to her recent leadership that led to development of a Pediatric Task Force as part of the World Symposium on Pulmonary Hypertension, Dr. Barst was deeply involved in most of the major therapeutic advances in this field. Her passion, devotion, and tireless work ethic were extraordinary. Those who knew Robyn best speak of her outstanding mentorship and commitment to her family. Her leadership and drive to improve the outcomes of children with PAH will be missed, but her career serves as an exemplary model for current and future clinician-scientists caring for children with PAH. This article reflects on unique features of pediatric PAH and serves as our tribute to Dr. Barst’s career and many accomplishments in the field.
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Archer SL, Weir EK, Wilkins MR. Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation 2010;121(18):2045–2066. [DOI] [PMC free article] [PubMed]
- 2.del Cerro MJ, Abman S, Diaz G, Freudenthal AH, Freudenthal F, Harikrishnan S, Haworth SG, et al. A consensus approach to the classification of pediatric pulmonary hypertensive vascular disease: report from the PVRI Pediatric Taskforce, Panama 2011. Pulm Circ 2011;1(2):286–298. [DOI] [PMC free article] [PubMed]
- 3.Abman SH, Raj U. Towards improving the care of children with pulmonary hypertension: the rationale for developing a Pediatric Pulmonary Hypertension Network. Prog Pediatr Cardiol 2009;27(1–2):3–6. [DOI] [PMC free article] [PubMed]
- 4.Robbins IM, Moore TM, Blaisdell CJ, Abman SH. Improving outcomes for pulmonary vascular disease. Am J Respir Crit Care Med 2012;185(9):1015–1020. [DOI] [PMC free article] [PubMed]
- 5.Barst RJ, Ertel SI, Beghetti M, Ivy DD. Pulmonary arterial hypertension: a comparison between children and adults. Eur Respir J 2011;37(3):665–677. [DOI] [PMC free article] [PubMed]
- 6.Abman SH. Bronchopulmonary dysplasia: a vascular hypothesis. Am J Respir Crit Care Med 2001;164(10):1755–1756. [DOI] [PubMed]
- 7.Thébaud B, Abman SH. Bronchopulmonary dysplasia: where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am J Respir Crit Care Med 2007;175(10):978–985. [DOI] [PMC free article] [PubMed]
- 8.Stenmark KR, Abman SH. Lung vascular development: implications for the pathogenesis of bronchopulmonary dysplasia. Annu Rev Physiol 2005;67:623–661. [DOI] [PubMed]
- 9.Morrisey EE, Hogan BLM. Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev Cell 2010;18(1):8–23. [DOI] [PMC free article] [PubMed]
- 10.Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman S, Hirth PK, Waltenberger J, Voelkel NF. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest 2000;106(11):1311–1319. [DOI] [PMC free article] [PubMed]
- 11.Jakkula M, Le Cras TD, Gebb S, Hirth KP, Tuder RM, Voelkel NF, Abman SH. Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am J Physiol Lung Cell Mol Physiol 2000;279(3):L600–L607. [DOI] [PubMed]
- 12.Le Cras TD, Markham NE, Tuder RM, Voelkel NF, Abman SH. Treatment of newborn rats with a VEGF receptor inhibitor causes pulmonary hypertension and abnormal lung structure. Am J Physiol Lung Cell Mol Biol 2002;283(3):L555–L562. [DOI] [PubMed]
- 13.Ding BS, Nolan DJ, Guo P, Babazadeh AO, Cao Z, Rosenwaks Z, Crystal RG, et al. Endothelial-derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell 2011;147(3):539–553. [DOI] [PMC free article] [PubMed]
- 14.Voelkel NF, Gomez-Arroyo J, Mizuno S. COPD/emphysema: the vascular story. Pulm Circ 2011;1(3):320–326. [DOI] [PMC free article] [PubMed]
- 15.Farkas L, Gauldie J, Voelkel NF, Kolb M. Pulmonary hypertension and idiopathic pulmonary fibrosis: a tale of angiogenesis, apoptosis, and growth factors. Am J Respir Cell Mol Biol 2011;45(1):1–15. [DOI] [PubMed]
- 16.deMello DE, Sawyer D, Galvin N, Reid LM. Early fetal development of lung vasculature. Am J Respir Cell Mol Biol 1997;16(5):568–581. [DOI] [PubMed]
- 17.Hall SM, Hislop AA, Pierce CM, Haworth SG. Prenatal origins of human intrapulmonary arteries: formation and smooth muscle maturation. Am J Respir Cell Mol Biol 2000;23(2):194–203. [DOI] [PubMed]
- 18.Parera MC, van Dooren M, van Kempen M, de Krijger R, Grosveld F, Tibboel D, Rottier R. Distal angiogenesis: a new concept for lung vascular morphogenesis. Am J Physiol Lung Cell Mol Physiol 2005;288(1):L141–L149. [DOI] [PubMed]
- 19.Ingram DA, Mead LE, Moore DB, Woodard W, Fenoglio A, Yoder MC. Vessel wall-derived endothelial cells rapidly proliferate because they contain a complete hierarchy of endothelial progenitor cells. Blood 2005;105(7):2783–2786. [DOI] [PubMed]
- 20.Yoder MC, Ingram DA. Endothelial progenitor cell: ongoing controversy for defining these cells and their role in neoangiogenesis in the murine system. Curr Opin Hematol 2009;16(4):269–273. [DOI] [PubMed]
- 21.Zengin E, Chalajour F, Gehling UM, Ito WD, Treede H, Lauke H, Weil J, Reichenspurner H, Kilic N, Ergün S. Vascular wall resident progenitor cells: a source for postnatal vasculogenesis. Development 2006;133(8):1543–1551. [DOI] [PubMed]
- 22.Baker CD, Seedorf GJ, Wisnewski BL, Black CP, Ryan SL, Balasubramaniam V, Abman SH. Endothelial colony-forming cell conditioned media promotes angiogenesis in vitro and prevents pulmonary hypertension in experimental BPD. Am J Physiol Lung Cell Mol Physiol 2013;305(1):L73–L81. [DOI] [PMC free article] [PubMed]
- 23.Matthay MA, Anversa P, Bhattacharya J, Burnett BK, Chapman HA, Hare JM, Hei DJ, et al. Cell therapy for lung diseases: report from an NIH-NHLBI workshop, November 13–14, 2012. Am J Respir Crit Care Med 2013;188(3):370–375. [DOI] [PMC free article] [PubMed]
- 24.Lee C, Mitsialis SA, Aslam M, Vitali SH, Vergadi E, Konstantinou G, Sdrimas K, Fernandez-Gonzalez A, Kourembanas S. Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension. Circulation 2012;126(22):2601–2611. [DOI] [PMC free article] [PubMed]
- 25.Burri PH, Moschopulos M. Structural analysis of fetal rat lung development. Anat Rec 1992;234(3):399–418. [DOI] [PubMed]
- 26.Burri P. Lung development and pulmonary angiogenesis. In: Gaultier C, Bourbon JR, Post M, eds. Lung development. New York: Oxford University Press, 1999:122–151.
- 27.Djonov V, Schmid M, Tschanz SA, Burri PH. Intussusceptive angiogenesis: its role in embryonic vascular network formation. Circ Res 2000;86(3):286–292. [DOI] [PubMed]
- 28.Ben-Shlomo Y, Kuh D. A life course approach to chronic disease epidemiology: conceptual models, empiric challenges and interdisciplinary perspectives. Int J Epidemiol 2002;31(2):285–293. [PubMed]
- 29.Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J 1999;13(1):9–22. [PubMed]
- 30.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 1996;380(6573):439–442. [DOI] [PubMed]
- 31.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996;380(6573):435–439. [DOI] [PubMed]
- 32.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995;376(6535):66–70. [DOI] [PubMed]
- 33.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 1995;376(6535):62–66. [DOI] [PubMed]
- 34.Gerber HP, Hillan KJ, Ryan AM, Kowalski J, Keller GA, Rangell L, Wright BD, Radtke F, Aguet M, Ferrara N. VEGF is required for growth and survival in neonatal mice. Development 1999;126(6):1149–1159. [DOI] [PubMed]
- 35.Healy AM, Morgenthau L, Zhu X, Farber HW, Cardoso WV. VEGF is deposited in the subepithelial matrix at the leading edge of branching airways and stimulates neovascularization in the murine embryonic lung. Dev Dyn 2000;219(3):341–352. [DOI] [PubMed]
- 36.Gebb SA, Shannon JM. Tissue interactions mediate early events in pulmonary vasculogenesis. Dev Dyn 2000;217(2):159–169. [DOI] [PubMed]
- 37.Zhao L, Wang K, Ferrara N, Vu TH. Vascular endothelial growth factor co-ordinates proper development of lung epithelium and vasculature. Mech Dev 2005;122(7–8):877–886. [DOI] [PubMed]
- 38.Ng YS, Rohan R, Sunday ME, deMello DE, D’Amore PA. Differential expression of VEGF isoforms in mouse during development and in the adult. Dev Dyn 2001;220(2):112–121. [DOI] [PubMed]
- 39.Galambos C, Ng YS, Ali A, Noguchi A, Lovejoy S, D’Amore PA, deMello DE. Defective pulmonary development in the absence of heparin-binding vascular endothelial growth factor isoforms. Am J Respir Cell Mol Biol 2002;27(2):194–203. [DOI] [PubMed]
- 40.Thébaud B, Ladha F, Michelakis ED, Sawicka M, Thurston G, Eaton F, Hashimoto K, et al. Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: evidence that angiogenesis participates in alveolarization. Circulation 2005;112(16):2477–2486. [DOI] [PubMed]
- 41.Akeson AL, Cameron JE, Le Cras TD, Whitsett JA, Greenberg JM. Vascular endothelial growth factor-A induces prenatal neovascularization and alters bronchial development in mice. Pediatr Res 2005;57(1):82–88. [DOI] [PubMed]
- 42.Le Cras TD, Spitzmiller RE, Albertine KH, Greenberg JM, Whitsett JA, Akeson AL. VEGF causes pulmonary hemorrhage, hemosiderosis, and air space enlargement in neonatal mice. Am J Physiol Lung Cell Mol Physiol 2004;287(1):L134–L142. [DOI] [PubMed]
- 43.Yamamoto H, Yun EJ, Gerber HP, Ferrara N, Whitsett JA, Vu TH. Epithelial-vascular cross talk mediated by VEGF-A and HGF signaling directs primary septae formation during distal lung morphogenesis. Dev Biol 2007;308(1):44–53. [DOI] [PubMed]
- 44.Abman S, Kinsella J, Mercier J. Nitric oxide and endothelin in the developing pulmonary circulation: physiologic and clinical implications. In: Gaultier C, Bourbon JR, Post M, eds. Lung development. New York: Oxford University Press, 1999:196–220.
- 45.Han RNN, Babaei S, Robb M, Lee T, Ridsdale R, Ackerley C, Post M, Stewart DJ. Defective lung vascular development and fatal respiratory distress in endothelial NO synthase-deficient mice: a model of alveolar capillary dysplasia? Circ Res 2004;94(8):1115–1123. [DOI] [PubMed]
- 46.Balasubramaniam V, Maxey AM, Morgan DB, Markham NE, Abman SH. Inhaled NO restores lung structure in eNOS-deficient mice recovering from neonatal hypoxia. Am J Physiol Lung Cell Mol Physiol 2006;291(1):L119–L127. [DOI] [PubMed]
- 47.Balasubramaniam V, Tang JR, Maxey A, Plopper CG, Abman SH. Mild hypoxia impairs alveolarization in the endothelial nitric oxide synthase (eNOS) deficient mouse. Am J Physiol Lung Cell Mol Physiol 2003. doi:10.1152/ajplung.00421.2002. [DOI] [PubMed]
- 48.Grover TR, Parker TA, Balasubramaniam V, Markham NE, Abman SH. Pulmonary hypertension impairs alveolarization and reduces lung growth in the ovine fetus. Am J Physiol Lung Cell Mol Physiol 2005;288(4):L648–L654. [DOI] [PubMed]
- 49.Tang JR, Balasubramaniam V, Maxey A, Markham N, Abman SH. Early inhaled nitric oxide treatment decreases apoptosis of endothelial cells in neonatal rat lungs after vascular endothelial growth factor receptor inhibition. Am J Physiol Lung Cell Mol Physiol 2007;293(5):L1271–L1280. [DOI] [PubMed]
- 50.Schwartz SM, Benditt EP. Clustering of replicating cells in aortic endothelium. Proc Natl Acad Sci USA 1976;73(2):651–653. [DOI] [PMC free article] [PubMed]
- 51.Bondjers G, Glukhova M, Hansson GK, Postnov YV, Reidy MA, Schwartz SM. Hypertension and atherosclerosis: cause and effect, or two effects with one unknown cause? Circulation 1991;84(6 suppl.):VI2–V16. [PubMed]
- 52.Schwartz SM, Heimark RL, Majesky MW. Developmental mechanisms underlying pathology of arteries. Physiol Rev 1990;70:1177–1209. [DOI] [PubMed]
- 53.Solodushko V, Fouty B. Proproliferative phenotype of pulmonary microvascular endothelial cells. Am J Physiol Lung Cell Mol Physiol 2007;292(3):L671-L677. [DOI] [PubMed]
- 54.Alvarez DF, Huang L, King JA, ElZarrad MK, Yoder MC, Stevens T. Lung microvascular endothelium is enriched with progenitor cells that exhibit vasculogenic capacity. Am J Physiol Lung Cell Mol Physiol 2008;294(3):L419–L430. [DOI] [PubMed]
- 55.Clark J, Alvarez DF, Alexeyev MF, King JA, Huang L, Yoder MC, Stevens T. Regulatory role for nucleosome assembly protein-1 in the proliferative and vasculogenic phenotype of pulmonary endothelium. Am J Physiol Lung Cell Mol Physiol 2008;294(3):L431–L439. [DOI] [PubMed]
- 56.Levin DL, Heymann MA, Kitterman JA, Gregory GA, Phibbs RH, Rudolph AM. Persistent pulmonary hypertension of the newborn infant. J Pediatr 1976;89(4):626–633. [DOI] [PubMed]
- 57.Kinsella JP, Abman SH. Recent developments in the pathophysiology and treatment of persistent pulmonary hypertension of the newborn. J Pediatr 1995;126(6):853–864. [DOI] [PubMed]
- 58.Geggel RL, Reid LM. The structural basis of PPHN. Clin Perinatol 1984;11(3):525–549. [PubMed]
- 59.Murphy JD, Rabinovitch M, Goldstein JD, Reid LM. The structural basis of persistent pulmonary hypertension of the newborn infant. J Pediatr 1981;98(6):962–967. [DOI] [PubMed]
- 60.Murphy JD, Vawter GF, Reid LM. Pulmonary vascular disease in fatal meconium aspiration. J Pediatr 1984;104(5):758–762. [DOI] [PubMed]
- 61.Van Marter LJ, Leviton A, Allred EN, Pagano M, Sullivan KF, Cohen A, Epstein MF. Persistent pulmonary hypertension of the newborn and smoking and aspirin and nonsteroidal antiinflammatory drug consumption during pregnancy. Pediatrics 1996;97(5):658–663. [PubMed]
- 62.Levin DL, Hyman AI, Heymann MA, Rudolph AM. Fetal hypertension and the development of increased pulmonary vascular smooth muscle: a possible mechanism for persistent pulmonary hypertension of the newborn infant. J Pediatr 1978;92(2):265–269. [DOI] [PubMed]
- 63.Abman SH, Shanley PF, Accurso FJ. Failure of postnatal adaptation of the pulmonary circulation after chronic intrauterine pulmonary hypertension in fetal lambs. J Clin Invest 1989;83(6):1849–1858. [DOI] [PMC free article] [PubMed]
- 64.Morin FC III. Ligating the ductus arteriosus before birth causes persistent pulmonary hypertension in the newborn lamb. Pediatr Res 1989;25(3):245–250. [DOI] [PubMed]
- 65.Williams MC, Wyble LE, O’Brien WF, Nelson RM, Schwenke JR, Casanova C. Persistent pulmonary hypertension of the neonate and asymmetric growth restriction. Obstet Gynecol 1998;91(3):336–341. [DOI] [PubMed]
- 66.Delaney C, Gien J, Roe G, Isenberg N, Kailey J, Abman SH. Serotonin contributes to high pulmonary vascular tone in a sheep model of persistent pulmonary hypertension of the newborn. Am J Physiol Lung Cell Mol Physiol 2013;304(12):L894–L901. [DOI] [PMC free article] [PubMed]
- 67.Haworth SG, Reid LM. Persistent fetal circulation: newly recognized structural features. J Pediatr 1976;88(4):614–620. [DOI] [PubMed]
- 68.Stenmark KR, Abman SH, Accurso FJ. Etiologic mechanisms of persistent pulmonary hypertension of the newborn. In: Weir EK, Reeves JT, eds. Pulmonary vascular physiology and pathophysiology. New York: Dekker, 1989:355–402.
- 69.Goldberg SJ, Levy RA, Siassi B, Betten J. The effects of maternal hypoxia and hyperoxia upon the neonatal pulmonary vasculature. Pediatrics 1971;48(4):528–533. [PubMed]
- 70.Murphy JD, Aronovitz MJ, Reid LM. Effects of chronic in utero hypoxia on the pulmonary vasculature of the newborn guinea pig. Pediatr Res 1986;20(4):292–295. [DOI] [PubMed]
- 71.Villamor E, Le Cras TD, Horan MP, Halbower AC, Tuder RM, Abman SH. Chronic intrauterine pulmonary hypertension impairs endothelial nitric oxide synthase in the ovine fetus. Am J Physiol Lung Cell Mol Physiol 1997;272(5):L1013–L1020. [DOI] [PubMed]
- 72.Shaul PW, Yuhanna IS, German Z, Chen Z, Steinhorn RH, Morin FC 3rd. Pulmonary endothelial NO synthase gene expression is decreased in fetal lambs with pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 1997;272(5):L1005–L1012. [DOI] [PubMed]
- 73.Gien J, Seedorf GJ, Balasubramaniam V, Markham N, Abman SH. Chronic intrauterine pulmonary hypertension impairs endothelial cell growth and angiogenesis in vitro. Am J Respir Crit Care Med 2007;176(11):1146–1153. [DOI] [PMC free article] [PubMed]
- 74.Gien J, Seedorf GJ, Balasubramaniam V, Markham N, Abman SH. Chronic intrauterine pulmonary hypertension increases endothelial rho-kinase activity and impairs angiogenesis in vitro. Am J Physiol Lung Cell Mol Physiol 2008;295(4):L680–L687. [DOI] [PMC free article] [PubMed]
- 75.Gien J, Tseng N, Seedorf G, Roe G, Abman SH. Endothelin-1 impairs angiogenesis in vitro through Rho-kinase activation after chronic intrauterine pulmonary hypertension in fetal sheep. Pediatr Res 2013;73(3):252–262. [DOI] [PMC free article] [PubMed]
- 76.Hyvelin JM, Howell K, Nichol A, Costello CM, Preston RJ, McLoughlin P. Inhibition of rho-kinase attenuates hypoxia-induced angiogenesis in the pulmonary circulation. Circ Res 2005;97:185–191. [DOI] [PubMed]
- 77.Acker SN, Seedorf GJ, Abman SH, Nozik-Grayck E, Partrick DA, Gien J. Pulmonary artery endothelial cell dysfunction and decreased populations of highly proliferative endothelial cells in experimental congenital diaphragmatic hernia. Am J Physiol Lung Cell Mol Physiol 2013;305(12):L943–L952. [DOI] [PMC free article] [PubMed]
- 78.Kinsella JP, Greenough A, Abman SH. Bronchopulmonary dysplasia. Lancet 2006;367(9520):1421–1431. [DOI] [PubMed]
- 79.Mourani PM, Abman SH. Pulmonary vascular disease in bronchopulmonary dysplasia: physiology, diagnosis, and treatment. In: Abman SH, ed. Bronchopulmonary dysplasia. New York: Informa, 2010:347–363.
- 80.An HS, Bae EJ, Kim GB, Kwon BS, Beak JS, Kim EK, Kim HS, Choi JH, Noh CI, Yun YS. Pulmonary hypertension in preterm infants with bronchopulmonary dysplasia. Korean Circ J 2010;40(3):131–136. [DOI] [PMC free article] [PubMed]
- 81.Bhat R, Salas AA, Foster C, Carlo WA, Ambalavanan N. Prospective analysis of pulmonary hypertension in extremely low birth weight infants. Pediatrics 2012;129(3):e682–e689. [DOI] [PMC free article] [PubMed]
- 82.Khemani E, McElhinney DB, Rhein L, Andrade O, Lacro RV, Thomas KC, Mullen MP. Pulmonary artery hypertension in formerly premature infants with bronchopulmonary dysplasia: clinical features and outcomes in the surfactant era. Pediatrics 2007;120(6):1260–1269. [DOI] [PubMed]
- 83.Slaughter JL, Pakrashi T, Jones DE, South AP, Shah TA. Echocardiographic detection of pulmonary hypertension in extremely low birth weight infants with bronchopulmonary dysplasia requiring prolonged positive pressure ventilation. J Perinatol 2011;31(10):635–640. [DOI] [PubMed]
- 84.Kim DH, Kim HS, Choi CW, Kim EK, Kim BI, Choi JH. Risk factors for pulmonary artery hypertension in preterm infants with moderate or severe bronchopulmonary dysplasia. Neonatology 2012;101(1):40–46. [DOI] [PubMed]
- 85.Gorenflo M, Vogel M, Obladen M. Pulmonary vascular changes in bronchopulmonary dysplasia: a clinicopathologic correlation in short- and long-term survivors. Pediatr Pathol 1991;11(6):851–866. [DOI] [PubMed]
- 86.Yee M, White RJ, Awad HA, Bates WA, McGrath-Morrow SA, O’Reilly MA. Neonatal hyperoxia causes pulmonary vascular disease and shortens life span in aging mice. Am J Pathol 2011;178(6):2601–2610. [DOI] [PMC free article] [PubMed]
- 87.Wong PM, Lees AN, Louw J, Lee FY, French N, Gain K, Murray CP, Wilson A, Chambers DC. Emphysema in young adult survivors of moderate-to-severe bronchopulmonary dysplasia. Eur Respir J 2008;32(2):321–328. [DOI] [PubMed]
- 88.Balinotti JE, Chakr VC, Tiller C, Kimmel R, Coates C, Kisling J, Yu Z, Nguyen J, Tepper RS. Growth of lung parenchyma in infants and toddlers with chronic lung disease of infancy. Am J Respir Crit Care Med 2010;181(10):1093–1097. [DOI] [PMC free article] [PubMed]
- 89.Lum S, Kirkby J, Welsh L, Marlow N, Hennessy E, Stocks J. Nature and severity of lung function abnormalities in extremely pre-term children at 11 years of age. Eur Respir J 2011;37(5):1199–1207. [DOI] [PubMed]
- 90.Abman SH. Impaired VEGF signaling in the pathogenesis of neonatal pulmonary vascular disease. In: Yuan JXJ, Ward JPT, eds. Membrane receptors, channels and transporters in the pulmonary circulation. Advances in Experimental Medicine and Biology 661. New York: Springer, 2010:323–335. [DOI] [PubMed]
- 91.Bhatt AJ, Pryhuber GS, Huyck H, Watkins RH, Metlay LA, Maniscalco WM. Disrupted pulmonary vasculature and decreased vascular endothelial growth factor, Flt-1, and TIE-2 in human infants dying with bronchopulmonary dysplasia. Am J Respir Crit Care Med 2001;164(10):1971–1980. [DOI] [PubMed]
- 92.Lassus P, Turanlahti M, Heikkilä P, Andersson LC, Nupponen I, Sarnesto A, Andersson S. Pulmonary vascular endothelial growth factor and Flt-1 in fetuses, in acute and chronic lung disease, and in persistent pulmonary hypertension of the newborn. Am J Respir Crit Care Med 2001;164(10):1981–1987. [DOI] [PubMed]
- 93.Grover TR, Zenge JP, Parker TA, Abman SH. Vascular endothelial growth factor causes pulmonary vasodilation through activation of the phosphatidylinositol-3-kinase–nitric oxide pathway in the late-gestation ovine fetus. Pediatr Res 2002;52(6):907–912. [DOI] [PubMed]
- 94.Kunig A, Balasubramaniam V, Markham N, Morgan D, Montgomery G, Grover TR, Abman SH. Recombinant human VEGF treatment enhances alveolarization during recovery after hyperoxic lung injury in neonatal rats. Am J Physiol Lung Cell Mol Physiol 2005;289(4):L529–L535. [DOI] [PubMed]
- 95.Kunig AM, Balasubramaniam V, Markham NE, Seedorf G, Gien J, Abman SH. Recombinant human VEGF treatment transiently increases lung edema but enhances lung structure after neonatal hyperoxia. Am J Physiol Lung Cell Mol Physiol 2006;291(5):L1068–L1078. [DOI] [PubMed]
- 96.Hilgendorff A, Reiss I, Ehrhradt H, Eickelberg O, Alvira CM. Chronic lung disease in the preterm infant: lessons learned from animal models. Am J Respir Cell Mol Biol 2014;50(2):233–245. [DOI] [PMC free article] [PubMed]
- 97.Madurga A, Mižiková I, Ruiz-Camp J, Morty RE. Recent advances in late lung development and the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 2013;305(12):L893–L905. [DOI] [PubMed]
- 98.Tang JR, Karumanchi SA, Seedorf G, Markham N, Abman SH. Excess soluble vascular endothelial growth factor receptor-1 in amniotic fluid impairs lung growth in rats: linking preeclampsia with bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 2012;302(1):L36–L46. [DOI] [PMC free article] [PubMed]
- 99.Foidart JM, Schaaps JP, Chantraine F, Munaut C, Lorquet S. Dysregulation of anti-angiogenic agents (sFlt-1, PLGF, and sEndoglin) in preeclampsia—a step forward but not the definitive answer. J Reprod Immunol 2009;82(2):106–111. [DOI] [PubMed]
- 100.Lapaire O, Shennan A, Stepan H. The preeclampsia biomarkers soluble fms-like tyrosine kinase-1 and placental growth factor: current knowledge, clinical implications and future application. Eur J Obstet Gynecol Reprod Biol 2010;151(2):122–129. [DOI] [PubMed]
- 101.Li F, Hagaman JR, Kim HS, Maeda N, Jennette JC, Faber JE, Karumanchi SA, Smithies O, Takahashi N. eNOS deficiency acts through endothelin to aggravate sFlt-1–induced pre-eclampsia–like phenotype. J Am Soc Nephrol 2012;23(4):652–660. [DOI] [PMC free article] [PubMed]
- 102.Shibuya M. Structure and function of VEGF/VEGF-receptor system involved in angiogenesis. Cell Struct Funct 2001;26(1):25–35. [DOI] [PubMed]
- 103.Levine RJ, Lam C, Qian C, Yu KF, Maynard SE, Sachs BP, Sibai BM, et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med 2006;355(10):992–1005. [DOI] [PubMed]
- 104.Wang CN, Chang SD, Peng HH, Lee YS, Chang YL, Cheng PJ, Chao AS, Wang TH, Wang HS. Change in amniotic fluid levels of multiple anti-angiogenic proteins before development of preeclampsia and intrauterine growth restriction. J Clin Endocrinol Metab 2010;95(3):1431–1441. [DOI] [PubMed]
- 105.Check J, Gotteiner N, Liu X, Su E, Porta N, Steinhorn R, Mestan KK. Fetal growth restriction and pulmonary hypertension in premature infants with bronchopulmonary dysplasia. J Perinatol 2013;33(7):553–557. [DOI] [PMC free article] [PubMed]
- 106.Rozance PJ, Seedorf GJ, Brown A, Roe G, O’Meara MC, Gien J, Tang JR, Abman SH. Intrauterine growth restriction decreases pulmonary alveolar and vessel growth and causes pulmonary artery endothelial cell dysfunction in vitro in fetal sheep. Am J Physiol Lung Cell Mol Physiol 2011;301(6):L860–L871. [DOI] [PMC free article] [PubMed]
- 107.De Paepe ME, Mao Q, Powell J, Rubin SE, DeKoninck P, Appel N, Dixon M, Gundogan F. Growth of pulmonary microvasculature in ventilated preterm infants. Am J Respir Crit Care Med 2006;173(2):204–211. [DOI] [PMC free article] [PubMed]
- 108.Janer J, Andersson S, Kajantie E, Lassus P. Endostatin concentration in cord plasma predicts the development of bronchopulmonary dysplasia in very low birth weight infants. Pediatrics 2009;123(4):1142–1146. [DOI] [PubMed]
- 109.Mitchell SH, Teague WG. Reduced gas transfer at rest and during exercise in school-age survivors of bronchopulmonary dysplasia. Am J Respir Crit Care Med 1998;157(5):1406–1412. [DOI] [PubMed]
- 110.Hakulinen AL, Järvenpää AL, Turpeinen M, Sovijärvi A. Diffusing capacity of the lung in school-aged children born very preterm, with and without bronchopulmonary dysplasia. Pediatr Pulmonol 1996;21(6):353–360. [DOI] [PubMed]
- 111.Allen J, Zwerdling R, Ehrenkranz R, Gaultier C, Geggel R, Greenough A, Kleinman R, et al. Statement on the care of the child with chronic lung disease of infancy and childhood. Am J Respir Crit Care Med 2003;168(3):356–396. [DOI] [PubMed]
- 112.van Haaften T, Byrne R, Bonnet S, Rochefort GY, Akabutu J, Bouchentouf M, Rey-Parra GJ, et al. Airway delivery of mesenchymal stem cells prevents arrested alveolar growth in neonatal lung injury in rats. Am J Respir Crit Care Med 2009;180(11):1131–1142. [DOI] [PMC free article] [PubMed]
- 113.O’Reilly M, Thébaud B. Cell-based strategies to reconstitute lung function in infants with severe bronchopulmonary dysplasia. Clin Perinatol 2012;39(3):703–725. [DOI] [PMC free article] [PubMed]
- 114.Irwin D, Helm K, Campbell N, Imamura M, Fagan K, Harral J, Carr M, et al. Neonatal lung side population cells demonstrate endothelial potential and are altered in response to hyperoxia-induced lung simplification. Am J Physiol Lung Cell Mol Physiol 2007;293(4):L941–L951. [DOI] [PubMed]
- 115.Balasubramaniam V, Mervis CF, Maxey AM, Markham NE, Abman SH. Hyperoxia reduces bone marrow, circulating, and lung endothelial progenitor cells in the developing lung: implications for the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 2007;292(5):L1073–L1084. [DOI] [PubMed]
- 116.Baker CD, Ryan SL, Ingram DA, Seedorf GJ, Abman SH, Balasubramaniam V. Endothelial colony-forming cells from preterm infants are increased and more susceptible to hyperoxia. Am J Respir Crit Care Med 2009;180(5):454–461. [DOI] [PMC free article] [PubMed]
- 117.Baker CD, Balasubramaniam V, Mourani PM, Sontag MK, Black CP, Ryan SL, Abman SH. Cord blood angiogenic progenitor cells are decreased in bronchopulmonary dysplasia. Eur Respir J 2012;40(6):1516–1522. [DOI] [PMC free article] [PubMed]
- 118.Borghesi A, Massa M, Campanelli R, Bollani L, Tzialla C, Figar TA, Ferrari G, et al. Circulating endothelial progenitor cells in preterm infants with bronchopulmonary dysplasia. Am J Respir Crit Care Med 2009;180(6):540–546. [DOI] [PubMed]
- 119.Paviotti G, Fadini GP, Boscaro E, Agostini C, Avogaro A, Chiandetti L, Baraldi E, Filippone M. Endothelial progenitor cells, bronchopulmonary dysplasia and other short-term outcomes of extremely preterm birth. Early Hum Dev 2011;87(7):461–465. [DOI] [PubMed]
- 120.Popova AP, Bozyk PD, Bentley JK, Linn MJ, Goldsmith AM, Schumacher RE, Weiner GM, Filbrun AG, Hershenson MB. Isolation of tracheal aspirate mesenchymal stromal cells predicts bronchopulmonary dysplasia. Pediatrics 2010;126(5):e1127–e1133. [DOI] [PMC free article] [PubMed]
-
121.Popova AP, Bozyk PD, Goldsmith AM, Linn MJ, Lei J, Bentley JK, Hershenson MB. Autocrine production of TGF-/article/back/ref-list/ref/mixed-citation/inline-formula
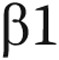 promotes myofibroblastic differentiation of neonatal lung mesenchymal stem cells. Am J Physiol Lung Cell Mol Physiol 2010;298(6):L735–L743. [DOI] [PMC free article] [PubMed]
promotes myofibroblastic differentiation of neonatal lung mesenchymal stem cells. Am J Physiol Lung Cell Mol Physiol 2010;298(6):L735–L743. [DOI] [PMC free article] [PubMed] - 122.Pierro M, Thébaud B. Mesenchymal stem cells in chronic lung disease: culprit or savior? Am J Physiol Lung Cell Mol Physiol 2010;298(6):L732–L734. [DOI] [PubMed]
- 123.Tropea KA, Leder E, Aslam M, Lau AN, Raiser DM, Lee JH, Balasubramaniam V, et al. Bronchioalveolar stem cells increase after mesenchymal stromal cell treatment in a mouse model of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 2012;302(9):L829–L837. [DOI] [PMC free article] [PubMed]
- 124.Zhang X, Wang H, Shi Y, Peng W, Zhang S, Zhang W, Xu J, Mei Y, Feng Z. Role of bone marrow-derived mesenchymal stem cells in the prevention of hyperoxia-induced lung injury in newborn mice. Cell Biol Int 2012;36(6):589–594. [DOI] [PubMed]
- 125.Chang YS, Choi SJ, Sung DK, Kim SY, Oh W, Yang YS, Park WS. Intratracheal transplantation of human umbilical cord blood-derived mesenchymal stem cells dose-dependently attenuates hyperoxia-induced lung injury in neonatal rats. Cell Transplant 2011;20(11–12):1843–1854. [DOI] [PubMed]
- 126.Robertson B. Postnatal formation and obliteration of arterial anastomoses in the human lung: a microangiographic and histologic study. Pediatrics 1969;43(6):971–979. [PubMed]
- 127.Wilkinson MJ, Fagan DG. Postmortem demonstration of intrapulmonary arteriovenous shunting. Arch Dis Child 1990;65(4):435–437. [DOI] [PMC free article] [PubMed]
- 128.McMullan DM, Hanley FL, Cohen GA, Portman MA, Riemer RK. Pulmonary arteriovenous shunting in the normal fetal lung. J Am Coll Cardiol 2004;44(7):1497–1500. [DOI] [PubMed]
- 129.Tobin CE. Arteriovenous shunts in the peripheral pulmonary circulation in the human lung. Thorax 1966;21(3):197–204. [DOI] [PMC free article] [PubMed]
- 130.Galambos C, Sims-Lucas S, Abman SH. Histologic evidence of intrapulmonary anastomoses by three-dimensional reconstruction in severe bronchoplumonary dysplasia. Ann Am Thorac Soc 2013;10(5):474–481. [DOI] [PMC free article] [PubMed]
- 131.Galambos C, Sims-Lucas S, Abman SH. Three-dimensional reconstruction identifies misaligned pulmonary veins as intrapulmonary shunt vessels in alveolar capillary dysplasia. J Pediatr 2014;164(1):192–195 [DOI] [PMC free article] [PubMed]
- 132.Lovering AT, Romer LM, Haverkamp HC, Pegelow DF, Hokanson JS, Eldridge MW. Intrapulmonary shunting and pulmonary gas exchange during normoxic and hypoxic exercise in healthy humans. J Appl Physiol 2008;104(5):1418–1425. [DOI] [PubMed]
- 133.Cua CL, Blankenship A, North AL, Hayes J, Nelin LD. Increased incidence of idiopathic pulmonary hypertension in Down syndrome neonates. Pediatr Cardiol 2007;28(4):250–254. [DOI] [PubMed]
- 134.Hawkins A, Langton-Hewer S, Henderson J, Tulloh RM. Management of pulmonary hypertension in Down syndrome. Eur J Pediatr 2011;170(7):915–921. [DOI] [PubMed]
- 135.Weijerman ME, van Furth AM, van der Mooren MD, van Weissenbruch MM, Rammeloo L, Broers CJM, Gemke RJBJ. Prevalence of congenital heart defects and persistent pulmonary hypertension of the neonate with Down syndrome. Eur J Pediatr 2010;169(10):1195–1199. [DOI] [PMC free article] [PubMed]
- 136.McDowell KM, Craven DI. Pulmonary complications of Down syndrome during childhood. J Pediatr 2011;158(2):319–325. [DOI] [PubMed]
- 137.Zorick TS, Mustacchi Z, Bando SY, Zatz M, Moreira-Filho CA, Olsen B, Passos-Bueno MR. High serum endostatin levels in Down syndrome: implications for improved treatment and prevention of solid tumours. Eur J Hum Gen 2001;9(11):811–814. [DOI] [PubMed]
- 138.Kim YM, Hwang S, Kim YM, Pyun BJ, Kim TY, Lee ST, Gho YS, Kwon YG. Endostatin blocks vascular endothelial growth factor–mediated signaling via direct interaction with KDR/Flk-1. J Biol Chem 2002;277(31):27872–27879. [DOI] [PubMed]
- 139.Yang A, Reeves RH. Increased survival following tumorigenesis in Ts65Dn mice that model Down syndrome. Cancer Res 2011;71(10):3573–3581. [DOI] [PMC free article] [PubMed]
- 140.Ryeom S, Folkman J. Role of endogenous angiogenesis inhibitors in Down syndrome. J Craniofac Surg 2009;20(suppl. 1):595–596. [DOI] [PubMed]
- 141.Baek K-H, Zaslavsky A, Lynch RC, Britt C, Okada Y, Siarey RJ, Lensch MW, et al. Down’s syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature 2009;459(7250):1126–1130. [DOI] [PMC free article] [PubMed]
- 142.Minami T, Horiuchi K, Miura M, Abid MR, Takabe W, Noguchi N, Kohro T, et al. VEGF- and thrombin-induced termination factor, DSCR-1, attenuates endothelial cell proliferation and angiogenesis. J Biol Chem 2004:279(48):50537–50554. [DOI] [PubMed]
-
143.Patel NS, Mathura VS, Bachmeier C, Beaulieu-Abdelahad D, Laporte V, Weeks O, Mullan M, Paris D. Alzheimer’s /article/back/ref-list/ref/mixed-citation/inline-formula
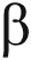 -amyloid peptide blocks VEGF mediated signaling via direct interaction with VEGFR2. J Neurochem 2010;112(1):66–76. [DOI] [PubMed]
-amyloid peptide blocks VEGF mediated signaling via direct interaction with VEGFR2. J Neurochem 2010;112(1):66–76. [DOI] [PubMed] - 144.Galambos C, Minic A, Nguyen D, Seedorf G, Abman SH. Increased lung endostatin expression impairs perinatal lung development in Down Syndrome. Pediatr Dev Pathol 2013;16(6):464–476.
- 145.Tang JR, Le Cras TD, Morris KG, Abman SH. Brief perinatal hypoxia increases severity of pulmonary hypertension after reexposure to hypoxia in infant rats. Am J Physiol Lung Cell Mol Physiol 2000;278(2):L356–L364. [DOI] [PubMed]
- 146.Sartori C, Alleman Y, Trueb L, Delabays A, Nicod P, Scherrer U. Augmented vasoreactivity in adult life associated with perinatal vascular insult. Lancet 1999;353(9171):2205–2207. [DOI] [PubMed]
- 147.Jayet PY, Rimoldi SF, Stuber T, Salmòn CS, Hutter D, Rexhaj E, Thalmann S, et al. Pulmonary and systemic vascular dysfunction in young offspring of mothers with preeclampsia. Circulation 2010;122(5):488–494. [DOI] [PubMed]
- 148.Rexhaj E, Bloch J, Jayet PY, Rimoldi SF, Dessen P, Mathieu C, Tolsa JF, Nicod P, Scherrer U, Sartori C. Fetal programming of pulmonary vascular dysfunction in mice: role of epigenetic mechanisms. Am J Physiol Heart Circ Physiol 2011;301(1):H247–H252. [DOI] [PubMed]