

Abstract Abstract
The extent to which pulmonary arterial hypertension (PAH) experts share common practice patterns that are in alignment with published expert consensus recommendations is unknown. Our objective was to characterize the clinical management strategies used by an international cohort of self-identified PAH experts. A 32-item questionnaire composed mainly of rank order or Likert scale questions was distributed via the Internet (August 5, 2013, through January 20, 2014) to four international pulmonary vascular disease organizations. The survey respondents (N = 105) were field experts reporting 11.6 ± 8.7 years of PAH experience. Likert scale responses (1 = disagree, 7 = agree) were 3.0–5.0, indicating a disparity in opinions, for 78% of questions. Respondent (dis)agreement scores were 4.4 ± 2.2 for use of expert recommendations to determine catheterization timing in PAH. For PAH patients without cardiogenic shock or known vasoreactivity status, the most and least preferred first-line therapies (1 = most preferred, 5 = least preferred) were phosphodiesterase type 5 inhibitors (PDE-Vi) and subcutaneous prostacyclin analogues, respectively (1.4 ± 0.8 vs. 4.0 ± 1.1; P < 0.05). Compared with US-practicing clinicians (N = 46), non-US-practicing clinicians (N = 57) favored collaboration between cardiology and pulmonary medicine for clinical decision making (1 = disagree, 7 = agree; 3.1 ± 2.2 vs. 4.8 ± 2.2; P < 0.0001) and PDE-Vi (6.5% vs. 22.4%) as first-line therapy for PAH patients with cardiogenic shock but were less likely to perform vasoreactivity testing in patients with lung disease–induced pulmonary hypertension (4.3 ± 2.1 vs. 2.2 ± 1.6; P < 0.0001). In conclusion, practice patterns among PAH experts diverge from consensus recommendations and differ by practice location, suggesting that opportunity may exist to improve care quality for this highly morbid cardiopulmonary disease.
Keywords: clinical care, survey, pulmonary arterial hypertension, right ventricle
Introduction
Adherence to standardized care is an established contributor to improved outcome in various cardiovascular and respiratory diseases, including coronary artery disease, genetic cardiomyopathies, and asthma, among others.1-3 The development of evidence-based expert consensus guidelines, which now exist across virtually all clinical scenarios in cardiopulmonary medicine,4 define disease-specific metrics for diagnosis, treatment, and long-term patient management. Consensus guideline recommendations, in turn, have evolved as a premium strategy by which to improve quality of care and clinical outcomes for patients with cardiopulmonary disease.5
Pulmonary arterial hypertension (PAH) is a severe cardiopulmonary disease associated with substantial morbidity and premature longevity.6,7 A range of pulmonary circulation–specific pharmacotherapeutic and surgical strategies has emerged recently to improve functional capacity, quality of life, and survival in PAH.8,9 Despite these advances, there is substantial underdiagnosis and inappropriate treatment of PAH in clinical practice.10,11 To address these issues, an international effort was undertaken in 2009 to establish the first expert consensus documents in the contemporary era for the diagnosis, treatment, and management of PAH patients.12-14 Since then, however, there have been numerous reports of ongoing heterogeneity in diagnostic and treatment strategies for patients with PAH.15-17 Practice patterns in PAH are reported to differ by institution,18 by era of practitioners,19 and even by geographic region.20,21
Taken together, these findings suggest that penetration of expert consensus recommendations into clinical practice in PAH may as yet be unrealized. However, there are few empirical data with which to address this concern. To fill this gap in our present understanding of treatment patterns in PAH and to identify areas in which a significant lack of consensus remains among experts regarding care, a survey containing questions relevant to assessment, diagnosis, and treatment of PAH was distributed to an international cohort of PAH and pulmonary vascular disease specialists.
Methods
This survey study was approved by the investigational review board for patient safety and privacy at Brigham and Women’s Hospital and complies with the Declaration of Helsinki. Written consent was implied by completion of the questionnaire.
Survey content
The complete survey is available at http://pvri.info/content/pulmonary-hypertension-clinician-survey. In general, surveys consisted of (1) a cover letter outlining the purpose of the survey and soliciting participation from self-identified PAH experts, (2) a general information respondent form, and (3) 28 multiple choice, rank order, or 7-point Likert scale questions (1 = disagree, 7 = agree; Table 1).22,23 The topics relevant to PAH clinical practice under investigation were practice patterns related to hemodynamic and pulmonary vasoreactivity testing, selection and implementation of PAH-specific drug therapies, and clinical management of PAH patients. In some cases, questions involving clinical practice preferences for pulmonary vascular diseases other than PAH were included, particularly with respect to patients with pulmonary hypertension due to lung disease (i.e., World Health Organization [WHO] group 3 pulmonary hypertension). The rationale for including questions addressing practice profile differences in these patients was based on overlap in pathophenotype10 and diagnostic approach12 reported by some for WHO group 3 and PAH patients and on ongoing debate within the larger pulmonary vascular disease community regarding optimal treatment of WHO group 3 pulmonary hypertension. An expert in the field of survey science performed survey validation, and a pilot trial of the survey was offered to selected members of the Pulmonary Vascular Research Institute (PVRI) prior to initiation of the study.
Table 1.
Survey outline
| Section focus | Items (N) | Question types | Topics addressed |
|---|---|---|---|
| PAH diagnosis | 10 | Likert: 5 Multiple choice: 5 |
Catheterization and vasoreactivity assessments for PAH diagnosis: • Timing • Indications • Methods |
| PAH drug therapy | 14 | Likert: 6 Rank order: 7 Multiple choice: 1 |
Drug therapy selection/preference: • By clinical scenario (PAH) • By clinical scenario (RV failure) • Barriers to drug selection Health insurance type in clinical practice: • National health plan vs. • Work insurance program vs. • Private health insurance vs. • Patient self-pay |
| PAH clinical assessment | 8 | Likert: 7 Rank order: 1 |
Clinical follow-up: • Ambulatory monitoring • Laboratory assessment • Imaging/catheterization Resources for clinical decision making: • Academic resources • Clinical collaborations with consultants |
PAH: pulmonary arterial hypertension; RV: right ventricle.
Survey distribution
Members of the PVRI, the European Respiratory Society, the Pulmonary Hypertension Association, and the American Thoracic Society received an e-mail announcing the survey. The message contained an electronic link to the survey on the PVRI website, which was available for completion between August 5, 2013, and January 20, 2014. These organizations were selected for participation in this study owing to a strong track record of PAH and pulmonary vascular disease publications authored by association members.12,24-26 Survey question responses were anonymously and automatically stored in the PVRI database, and data were transposed to Microsoft Excel prior to analysis.
Statistical analysis
All statistical analyses were performed using Origin (ver. 9.1; Northampton, MA). Categorical variables are reported as frequencies; unless otherwise indicated, continuous data are expressed as mean ± SD (range). The unpaired Student t test was used to compare two independent groups. Comparisons between multiple groups were made using one-way analysis of variance (ANOVA).
Results
Study population
A total of 105 anonymous individual surveys were completed and submitted during the study period (80 men; mean age, 47.3 years; age range, 29–68 years; Table 2). Survey respondents were from 25 countries and 6 continents (Fig. 1A) and were trained principally in pulmonary medicine (N = 63) and cardiology (N = 29), although a total of 6 clinical specialties and nursing were represented (Fig. 1B).
Table 2.
Survey cohort characteristics
| Profile of respondents (N = 105) | Result |
|---|---|
| Men, N | 80 |
| Age, mean (range), years | 47.3 (29–68) |
| PAH practice experience, years | 11.6 ± 8.7 |
| Total clinical hours/week | 36.7 ± 17.3 |
| Total PAH clinical hours/week | 15.8 ± 12.1 |
| Participants in PAH research, % | 100 |
| Research foci/focus, % | |
| Basic science | 48.5 |
| Preclinical studies | 30.4 |
| Clinical trials | 80.9 |
Data are mean ± SD, unless otherwise indicated. PAH: pulmonary arterial hypertension.
Figure 1.

Geographic distribution and clinical training expertise of survey respondents. Pulmonary vascular disease experts (N = 105) completed an Internet-based survey to characterize differences in pulmonary arterial hypertension clinical practice patterns. A, Respondents were from 25 countries on 6 continents. B, The survey cohort included healthcare professionals trained in 7 medical specialties and nursing.
Participants reported 11.6 ± 8.7 years of PAH clinical practice, with 43.1% of all clinical effort devoted to PAH patients. Clinicians’ PAH patient panels were generated by local and nonlocal referrals for 91.4% and 80.0% of respondents, respectively, and a majority (57.4%) reported employment at an institution with an active heart/lung transplant program. All survey respondents reported participation in PAH research, which involved basic science (48.5%), preclinical studies (30.4%), and/or clinical trials (80.9%). Involvement in ≥2 areas of research was reported by 38 (36.1%) survey participants, and 20 (19.0%) respondents reported participation in all research areas. Among 4 commonly available clinical practice resources, study participants rank ordered expert consensus recommendations as the most influential on clinical practice (1.6 ± 0.9 on a scale of 1 [most useful] to 5 [least useful]), followed by international scientific sessions (1.7 ± 0.9), personal clinical experience (3.5 ± 1.3), and information provided by pharmaceutical companies (4.5 ± 0.6; P < 0.05 for comparison across groups).
Invasive cardiopulmonary hemodynamic assessment and pulmonary vasoreactivity testing
Despite the importance of invasive cardiopulmonary hemodynamic assessment and vasoreactivity testing on diagnosis and prognosis in PAH,27 the application of right heart catheterization (RHC), left heart catheterization (LHC), and/or confrontational pulmonary vasodilator testing in clinical practice is controversial.28 Thus, the survey consisted of a series of questions relevant to RHC (Table 3) and LHC (Table 4) in PAH. Participants reported 4.4 ± 2.2 (1–7) on a 7-point Likert scale (1 = disagree, 7 = agree) in response to a statement assessing (dis)agreement with expert consensus recommendations as a key resource for determining the timing of RHC for diagnosis/prognosis of PAH. Similar trends were reported for the role of invasive cardiopulmonary hemodynamic assessment in PAH patient management beyond diagnosis: (dis)agreement scores for RHC as a routine method to assess treatment efficacy or to evaluate further clinical deterioration were 4.1 ± 2.0 (1–7) and 4.4 ± 2.1 (1–7), respectively. Increased parity was reported for the role of repeat vasoreactivity testing as a component of routine clinical care in PAH (2.9 ± 2.0 [1–7]), and agreement was reported in favor of echocardiography for the routine monitoring of right ventricular function in clinically stable patients (6.1 ± 1.4 [1–7]).
Table 3.
Responses to questions involving the relevance of cardiac catheterization and pulmonary vasoreactivity testing to the diagnosis/management of pulmonary arterial hypertension
| Survey question | Response (N = 105), mean ± SD (range) |
|---|---|
| A key guideline for determining the timing of invasive hemodynamic assessment for patients with pulmonary arterial hypertension (PAH) in your practice is the expert consensus guideline statement on the topic published in 200912 | 4.4 ± 2.2 (1–7) |
| It is reasonable to perform pulmonary vasoreactivity testing as part of the clinical evaluation for patients with suspected chronic lung disease–induced pulmonary hypertension (i.e., World Health Organization [WHO] group 3 pulmonary hypertension) | 3.1 ± 2.1 (1–7) |
| A substantial decrease in pulmonary hypertension severity on pulmonary vasoreactivity testing influences your choice of first-line pulmonary vasodilator therapy in patients requiring treatment of symptomatic PAH | 5.6 ± 1.7 (1–7) |
| Right heart catheterization is a routine strategy by which to monitor (i.e., follow-up) the treatment responsiveness of PAH patients to pulmonary vasodilator therapy | 4.1 ± 2.0 (1–7) |
| Among patients initiated on pulmonary circulation–specific therapy following invasive hemodynamic assessment, a repeat right heart catheterization is performed in your practice in patients with clinical deterioration only | 4.3 ± 2.1 (1–7) |
Answers are reported on the basis of a 7-point Likert scale (1 = disagree, 7 = agree).
Table 4.
Role of left heart catheterization (LHC) in the diagnosis of pulmonary arterial hypertension
| LHC survey question | Response, N (%) |
|---|---|
| LHC is not a part of the cardiopulmonary hemodynamic assessment in my practice | 58 (55.2) |
| LHC is always performed unless contraindicated | 30 (28.5) |
| LHC is performed only if the transpulmonary gradient is ≥10 mmHg | 6 (5.7) |
| LHC is performed only if the pulmonary capillary wedge pressure is <15 mmHg | 5 (4.7) |
Results are responses reported in answer to the following question: “Which (one) of the following best characterizes the circumstances under which LHC is performed at the time of right heart catheterization in your practice (choose 1)?”
(Dis)agreement scores for pulmonary vasoreactivity testing in patients with suspected WHO group 3 pulmonary hypertension (i.e., lung disease–induced pulmonary hypertension), for which evidence-based guidelines do not exist, were 3.1 ± 2.1 (1–7). However, it is worthwhile to note that differences with respect to this issue were observed according to the mode of healthcare reported for PAH patients by survey respondents. Compared with survey participants for whom health insurance of patients was reported to be mainly through a national health plan (N = 62), those survey participants for whom health insurance of patients was reported to be mainly through private health insurance (N = 17) tended to be more agreeable with vasoreactivity testing for WHO group 3 patients (2.8 ± 2.0 vs. 4.4 ± 2.1; P < 0.01; Fig. 2).
Figure 2.
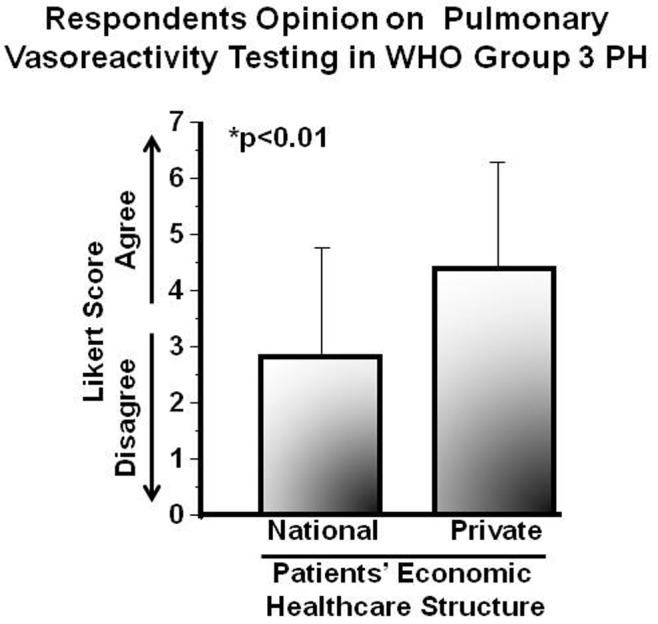
Patients’ economic healthcare status influences the opinion of pulmonary arterial hypertension (PAH) experts on pulmonary vasoreactivity testing in pulmonary hypertension due to lung disease. An international cohort of pulmonary vascular disease experts (N = 105) completed an Internet-based survey soliciting a response of (dis)agreement to the following statement: “It is reasonable to perform pulmonary vasoreactivity testing as part of the clinical evaluation for patients with suspected chronic lung disease–induced pulmonary hypertension (i.e., World Health Organization [WHO] group 3 pulmonary hypertension).” Results are presented by primary health insurance coverage (national healthcare system vs. private healthcare insurance) of PAH patients in the practice of each respondent, as reported in the survey by respondents. Data are expressed as mean ± SD.
PAH treatment
A key determination of the 2009 expert consensus guidelines in PAH was establishing the role of treatment with calcium channel antagonists as the primary pharmacotherapy for patients demonstrating pulmonary vasoreactivity in response to acute pulmonary vasodilator challenge in the absence of cardiogenic shock.29 In response to a survey question addressing (dis)agreement with this recommendation (1 = disagree, 7 = agree), results were 4.7 ± 2.3 (1–7). In turn, among PAH patients with unknown vasoreactivity status without cardiogenic shock, (dis)agreement with calcium channel antagonist therapy as first-line treatment was (1 = disagree, 7 = agree) 1.6 ± 1.5 (1–7). Furthermore, rank order analysis of first-line agents under these clinical conditions presented in order of most to least preferred was phosphodiesterase type 5 inhibitor (PDE-Vi), nonselective endothelin receptor antagonist (ERA), selective endothelin type A receptor antagonist (ETARA), inhaled prostacyclin analogue (PGI2), parenteral PGI2, and subcutaneous PGI2 (1.4 ± 0.8 vs. 2.3 ± 1.0 vs. 2.5 ± 1.1 vs. 3.4 ± 1.2 vs. 3.6 ± 1.3 vs. 4.0 ± 1.1; N = 105; P < 0.05 by ANOVA; Fig. 3A). Interestingly, preference for PDE-Vi as first-line therapy in WHO group 3 pulmonary hypertension was less well established, with respondents reporting a (dis)agreement score of 4.0 ± 2.1 (1–7). With respect to strategies involving initiation of dual pulmonary vasodilator therapy, a tendency toward agreement with maximizing the drug dose of a single pulmonary hypertension–specific therapy prior to initiation of an additional pulmonary hypertension-specific therapy was observed (4.9 ± 1.6 [1–7]).
Figure 3.

Pulmonary vasodilator therapy preference in pulmonary arterial hypertension (PAH). A, An international cohort of pulmonary vascular disease experts (N = 105) ranked first-line pulmonary vasodilator therapy by preference for the treatment of PAH patients without cardiogenic shock in whom the response to vasoreactivity testing was not established. 1 = most preferred, 5 = least preferred. B, Experts identified the most preferred drug therapy for the first-line treatment of PAH patients with cardiogenic shock. PDE-Vi: phosphodiesterase type 5 inhibitor; ERA: endothelin receptor antagonist; ETARA: selective endothelin type A receptor antagonist; PGI2: prostacyclin analogue.
In concert with expert consensus recommendations, parenteral PGI2 was identified by a majority (69.5%) of survey respondents as the preferred first-line treatment of PAH patients with cardiogenic shock, although PDE-Vi and ERA were selected by 16.5% and 10.5% of respondents, respectively (Fig. 3B). Greater disparity was observed for the preferred inotropic agent to support cardiac output in patients with right ventricular dysfunction due to PAH (Fig. 4).
Figure 4.
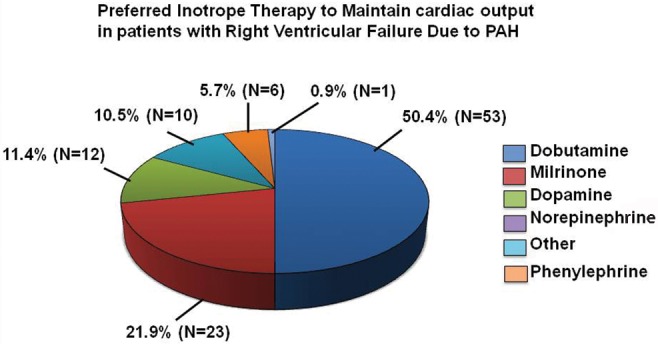
Inotropic therapy preference for patients with right ventricular failure due to pulmonary arterial hypertension (PAH). An international cohort of pulmonary vascular disease experts (N = 105) completed an Internet-based survey soliciting the selection of one of 6 provided answer options to the following question: “In patients with right ventricular failure secondary to PAH, which inotrope do you prefer to maintain sufficient cardiac output?” Data are expressed as percentage of responses.
Five common barriers to the implementation of preferred therapy in PAH across the study cohort were rank-ordered by survey participants (N = 105; 1 = most common, 5 = least common; Table 5). Overall, patients’ social circumstances preventing drug compliance was reported as the strongest factor to limit optimal treatment selection, while access to drug therapy was least likely to influence drug selection (2.3 ± 0.1 vs. 3.6 ± 0.1; P < 0.001).
Table 5.
Potential barriers to prescribing preferred therapy in pulmonary arterial hypertension (PAH)
| Potential treatment barrier | Rank order response (N = 105), mean ± SD |
|---|---|
| Social circumstances prevent drug therapy compliance | 2.3 ± 0.1 |
| Comorbid psychological/psychiatric disease prevents drug therapy compliance | 2.8 ± 0.1 |
| Patients’ financial circumstances prevent access to drug therapy | 3.2 ± 0.2 |
| Patient refuses drug therapy | 3.4 ± 0.1 |
| Drug therapy is not provided by the pharmacy accessed by the patient or the healthcare provider | 3.7 ± 0.1 |
Answers are reported on the basis of a rank order scale (1 = most common barrier to PAH treatment, 5 = least common barrier to PAH treatment).
Ambulatory care strategies
Periodically measuring biochemical evidence of heart failure and functional capacity is reported as an acceptable strategy by which to monitor the clinical trajectory of PAH in the ambulatory setting,30 although the extent to which this is applied in clinical practice is unknown. Responses indicating (dis)agreement with the routine measurement of 6-minute walk distance or plasma brain natriuretic peptide concentration were 5.0 ± 1.7 and 4.7 ± 1.6, respectively. Along these lines, an evolving trend in the management of patients with complex pulmonary vascular disease is through a multidisciplinary effort between pulmonary medicine and cardiology, among other disciplines.31 Whereas (dis)agreement scores were 4.2 ± 2.2 that key decisions for the management of PAH patients are achieved through a consensus decision between a pulmonary medicine and a cardiology expert, we observed a significant difference with respect to this issue by geographic location. Compared with US-practicing clinicians (N = 46), a greater tendency to report joint pulmonary-cardiology collaborations in PAH management was observed among non-US-practicing clinicians (N = 59; 3.1 ± 2.2 vs. 4.8 ± 2.2; P < 0.0001; Fig. 5).
Figure 5.
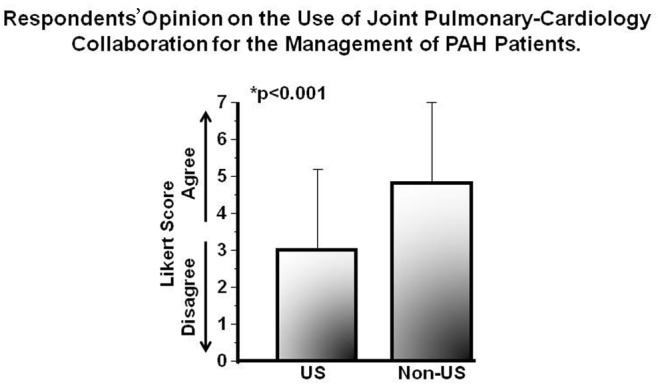
Regional differences and the use of a multidisciplinary approach to the management of pulmonary arterial hypertension (PAH) patients. An international cohort of pulmonary vascular disease experts (N = 105) completed an Internet-based survey soliciting a response of (dis)agreement to the following statement: “Key management decisions for PAH patients are achieved through a consensus decision between a pulmonary medicine and cardiology expert in pulmonary hypertension.” Results are presented according to geographic location of practice. Data are expressed as mean ± SD.
Regional differences in care and treatment patterns
Owing to our observation that use of joint pulmonary-cardiology collaborations in PAH differs between US and non-US experts, we next explored the effect of these regional differences on diagnostic and treatment strategies in PAH and other pulmonary vascular diseases. We observed no significant difference between US-practicing (N = 46) and non-US-practicing (N = 59) clinicians with respect to (dis)agreement scores (1 = disagree, 7 = agree) for use of 6-minute walk distance (4.7 ± 1.7 vs. 5.2 ± 1.8; P = 0.11), plasma B-type natriuretic peptide (4.8 ± 1.8 vs. 4.6 ± 1.3; P = 0.38), and echocardiography (6.0 ± 1.5 vs. 6.1 ± 1.2; P = 0.64) to monitor the clinical progress of PAH patients in the ambulatory setting. Compared with non-US-practicing clinicians, US-practicing clinicians reported a similar tendency to utilize invasive cardiopulmonary hemodynamic assessment as a routine test performed during follow-up of PAH patients prescribed vasodilator therapy (1 = disagree, 7 = agree; 4.1 ± 2.0 vs. 4.2 ± 2.0; P = 0.91); however, a significant difference was observed between groups with respect to performing vasoreactivity testing in the diagnostic evaluation of patients with WHO group 3 pulmonary hypertension (2.2 ± 1.6 vs. 4.3 ± 2.1; P < 0.0001; Fig. 6A).
Figure 6.

Differences in care patterns for patients with pulmonary vascular disease according to geographic location of clinicians. An international cohort of pulmonary vascular disease experts completed an Internet-based survey, and results were analyzed according to US-practice (N = 46) or non-US-practice (N = 57) location. A, Respondents’ (dis)agreement scores are reported in response to the following statements: “Right heart catheterization is a routine strategy by which to monitor (i.e., follow-up) the treatment responsiveness of PAH patients to pulmonary vasodilator therapy” (left bar graph) and “It is reasonable to perform pulmonary vasoreactivity testing as part of the clinical evaluation for patients with suspected chronic lung disease–induced pulmonary hypertension (i.e., World Health Organization [WHO] group 3 pulmonary hypertension)” (right bar graph). RHC: right heart catheterization; PAH: pulmonary arterial hypertension; VASO-R: vasoreactivity testing. Data are expressed as mean ± SD. B, Percentage of respondents identifying each of 6 potential treatments as the most preferred drug therapy for the first-line treatment of PAH patients with cardiogenic shock. PGI2: prostacyclin analogue; PDE-Vi: phosphodiesterase type 5 inhibitor; ERA: endothelin receptor antagonist; ETARA: selective endothelin type A receptor antagonist.
Rank order analysis of 6 potential first-line agents for the treatment of PAH patients without cardiogenic shock in the absence of known pulmonary vasoreactivity status revealed that PDE-Vi therapy is favored similarly by non-US-practicing and US-practicing clinicians (1 = most preferred, 5 = least preferred; 1.4 ± 0.9 vs. 1.5 ± 0.7; P = 0.71). However, whereas US-practicing physicians were more likely than non-US-practicing clinicians to favor first-line therapy in this clinical situation with subcutaneous PGI2 (3.7 ± 1.3 vs. 4.3 ± 0.9; P < 0.01) and parenteral PGI2 (3.2 ± 1.3 vs. 3.8 ± 1.2; P < 0.02), the opposite trend was observed for ERA use (2.7 ± 0.9 vs. 2.0 ± 1.0; P < 0.002). The percentage of respondents identifying each of 6 therapies as preferred for the first-line treatment of symptomatic PAH patients with cardiogenic shock according to practice location is provided in Figure 6B, which illustrates differences between non-US-practicing and US-practicing clinicians for the selection of PGI2 (56.9% vs. 86.9%), PDE-Vi (22.4% vs. 6.5%), and inhaled PGI2 (15.5% vs. 2.1%).
Discussion
This is the largest empirical report to date to characterize the opinions and clinical practice patterns of PAH experts, which was defined in this study as substantial PAH experience, academic focus, and international distribution of practice locations reported by the survey respondents. Our findings demonstrate important disparity among study participants in each of the measured facets of relevance to PAH clinically, including strategies for diagnosis, pharmacotherapy selection, and ambulatory patient management. Although participants identified expert consensus recommendations as the most valued resource for clinical decision making in PAH, results from this survey indicate that practice patterns diverge from these recommendations in several areas, particularly with respect to indications for RHC/pulmonary vasoreactivity testing as well as evidence-based algorithms for PAH therapy initiation.
Several recent publications have demonstrated that a mismatch between expert consensus recommendations and applied clinical care patterns exists for patients with pulmonary vascular disease and have shown that this trend is consistent across various patient populations, including community-based, referred, and military veteran cohorts.11,15,32 Failure to adhere to consensus recommendations in pulmonary vascular disease, in turn, is associated with suboptimal rates of diagnosis and treatment. In one study by Deaño and colleagues,11 pulmonary hypertension misdiagnosis rates were as high as 52%, which was associated with similar rates of inappropriate therapy. Similar findings are reported for evaluation completeness for PAH, in which appropriate screening for root cause of pulmonary vascular dysfunction, such as connective tissue disease or human immunodeficiency virus, may be as low as 29% in nonreferral populations. Our findings are in concert with these earlier observations by suggesting that variability in care patterns are observed in practice globally even among expert PAH clinicians.
These data identify differences in drug therapy preference for PAH patients irrespective of cardiogenic shock status and by practice location as well as for pharmacotherapeutic support in the setting of right ventricular failure. Although we did not investigate a rationale by which to account for survey answers, our findings suggest that patients’ social circumstances and mental health status may influence treatment selection. Our finding that health insurance status may affect diagnostic testing selection in WHO group 3 pulmonary hypertension is consistent with observations made in other diseases, indicating that nonclinical factors influence care patterns in the absence of standardized guidelines.33,34
Disparity was observed for drug class preference in PAH relative to consensus recommendations: whereas parenteral PGI2 therapy initiation is supported by a class 1 recommendation in current35 and recent12 guidelines for patients with PAH, cardiogenic shock, and unknown vasoreactivity status, several alternative therapies were identified in our survey as preferred first-line treatment under these clinical conditions. Disparity in opinion was also reported for the use of calcium channel antagonist therapy in PAH patients with preserved vasoreactivity despite reproducible clinical trial data29,36 and a class 1 recommendation35 in support of this drug class as an effective treatment for this PAH clinical profile. However, these findings may be accounted for by published expert recommendations indicating that PDE-Vi, in addition to calcium channel antagonist therapy, is an acceptable treatment option under these clinical conditions.37 No consensus was observed among experts for the use of PDE-Vi therapy in WHO group 3 pulmonary hypertension, which is in concert with varying opinions reported on this issue in the literature.32,38
There are several limitations to this study beyond factors intrinsic to survey research that merit discussion when interpreting our findings. First, our approach was to solicit survey responses from self-identified PAH experts belonging to medical organizations with an established commitment to pulmonary vascular disease. The precise survey response rate expressed as a fraction of participants relative to the overall universe of PAH experts, thus, cannot be calculated, nor were objective criteria for defining PAH expert used a priori to determine inclusion eligibility in this study. Taken together, the degree to which our study cohort was truly representative of the PAH expert community cannot be quantified beyond PAH-specific experience, PAH research involvement, and time commitment in clinical practice devoted to PAH patients (as provided in Table 2). Nevertheless, since survey completion alone may be indicative of participation bias among the study cohort, interpretation of our data also requires consideration of this potentially confounding effect.
Second, the survey was distributed internationally in English. Thus, it is not possible to characterize the potential confounding effect of language barriers to study participation and/or answer accuracy. Along these lines, the possibility remains that statement formatting resulted in ambiguous questions and/or answer selections, which would undoubtedly introduce a response bias to our results. Additionally, physicians practicing in the United States represented the largest geographical group of survey respondents, although the effect of this on the results of analyses involving the entire cohort was not addressed specifically.
Third, this survey was not intended to identify a rationale by which to account for respondents’ survey question answers. Therefore, it is conceivable that inaccessibility to resources (or clinical experiences) necessary to interpret question(s) accurately contributed to answer selection, rather than clinical acumen alone.
Conclusions
Despite the availability of a contemporary expert consensus statements, there is disparity in opinion regarding the clinical approach to PAH among experts in the field. Specifically, divergence from evidence-based recommendations was reported in this survey study for diagnostic strategies and application of drug therapy for patients, which was observed across different hypothetical clinical scenarios. These data illustrate the need for standardized and validated clinical guidelines in the field of pulmonary vascular disease, which is anticipated to improve outcome in patients afflicted with PAH and other diseases of similar pathophysiology.
Acknowledgments
We thank the pulmonary arterial hypertension healthcare providers who answered the survey and provided these data; Mark R. Zonfrillo, MD, MSCE, for his review of the survey; and Nikki Krol for her expert administrative assistance. We also acknowledge the expert advice and assistance in completing this project by participating medical societies, particularly Drs. Jason Yuan at the American Thoracic Society, Andrew Peacock at the European Respiratory Society, and Martin Wilkins at the Pulmonary Vascular Research Institute (PVRI) as well as Rino Aldrighetti at the Pulmonary Hypertension Association. This data set will be made available to members of the PVRI on request as part of an open access initiative.
Source of Support: This work was supported by a Young Investigator Grant from the Pulmonary Vascular Research Institute (to JRR and BAM), the National Institutes of Health (1K08HL111207-01A1), the Pulmonary Hypertension Association, and the Lerner and Klarman Foundations at Brigham and Women’s Hospital (to BAM).
Conflict of Interest: BAM receives funding from Gilead Sciences to research pulmonary arterial hypertension. GB receives consulting funding from Bayer, Novartis, Pfizer, and Ikaria.
References
- 1.Joynt KE, Blumenthal DM, Orav EJ, Resnic FS, Jha AK. Association of public reporting for percutaneous coronary intervention with utilization and outcomes among Medicare beneficiaries with acute myocardial infarction. JAMA 2012;308(14):1460–1468. [DOI] [PMC free article] [PubMed]
- 2.Maron BJ, Pfister GC, Puffer JC. Preparticipation cardiovascular screening for young athletes. JAMA 2000;284(8):957–958. [PubMed]
- 3.Braido F, Brusselle G, Ingrassia E, Nicolini G, Price D, Roche N, et al. InternationaL cross-sectIonAl and longItudinal assessment on aSthma cONtrol in European adult patients—the LIAISON study protocol. BMC Pulm Med 2013;13:18. [DOI] [PMC free article] [PubMed]
- 4.Parekh PJ, Buerlein RC, Shams R, Herre J, Johnson DA. An update on the management of implanted cardiac devices during electrosurgical procedures. Gastrointest Endosc 2013;78(6):836–841. [DOI] [PubMed]
- 5.Paterick TE, Paterick TJ, Fletcher GF, Maron BJ. Medical and legal issues in the cardiovascular evaluation of competitive athletes. JAMA 2005;294(23):3011–3018. [DOI] [PubMed]
- 6.Ling Y, Johnson MK, Kiely DG, Condliffe R, Elliot CA, Gibbs JS, et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med 2012;186(8):790–796. [DOI] [PubMed]
- 7.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med 2006;173(9):1023–1030. [DOI] [PubMed]
- 8.Pulido T, Adzerikho I, Channick RN, Delcroix M, Galiè N, Ghofrani HA, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013;369(9):809–818. [DOI] [PubMed]
- 9.Esch JJ, Shah PB, Cockrill BA, Farber HW, Landzberg MJ, Mehra MR, et al. Transcatheter Potts shunt creation in patients with severe pulmonary arterial hypertension: initial clinical experience. J Heart Lung Transplant 2013;32(4):381–387. [DOI] [PubMed]
- 10.Maron BA, Choudhary G, Khan UA, Jankowich MD, McChesney H, Ferrazzani SJ, et al. Clinical profile and underdiagnosis of pulmonary hypertension in US veteran patients. Circ Heart Fail 2013;6(5):906–912. [DOI] [PMC free article] [PubMed]
- 11.Deaño RC, Glassner-Kolmin C, Rubenfire M, Frost A, Visovatti S, McLaughlin VV, et al. Referral of patients with pulmonary hypertension diagnoses to tertiary pulmonary hypertension centers: the multicenter RePHerral study. JAMA Intern Med 2013;173(10):887–893. [DOI] [PubMed]
- 12.McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol 2009;53(17):1573–1619. [DOI] [PubMed]
- 13.Barbera JA, Escribano P, Morales P, Gomez MA, Oribe M, Martinez A, et al. Standards of care in pulmonary hypertension: consensus statement of the Spanish Society of Pulmonology and THoracic Surgery (SEPAR) and the Spanish Society of Cardiology (SEC). Rev Esp Cardiol 2008;61(2):170–184. [PubMed]
- 14.Montani D, O’Callaghan DS, Jais X, Savale L, Natali D, Redzepi A, et al. Implementing the ESC/ERS pulmonary hypertension guidelines: real-life cases from a national referral centre. Eur Respir Rev 2009;18(114):272–290. [DOI] [PubMed]
- 15.McLaughlin VV, Langer A, Tan M, Clements PJ, Oudiz RJ, Tapson VF, et al. Contemporary trends in the diagnosis and management of pulmonary arterial hypertension: an initiative to close the care gap. Chest 2013;143(2):324–332. [DOI] [PubMed]
- 16.Meyer S, McLaughlin VV, Seyfarth HJ, Bull TM, Vizza CD, Gomberg-Maitland M, et al. Outcomes of noncardiac, nonobstetric surgery in patients with PAH: an international prospective survey. Eur Respir J 2012;41(6):1302–1307. [DOI] [PubMed]
- 17.Waxman AB, Zamanian RT. Pulmonary arterial hypertension: new insights into the optimal role of current and emerging prostacyclin therapies. Am J Cardiol 2013;111(suppl. 5):1A–16A. [DOI] [PubMed]
- 18.Borrie AE, Ostrow DN, Levy RD, Swiston JR. Assessing response to therapy in idiopathic pulmonary arterial hypertension: a consensus survey of Canadian pulmonary hypertension physicians. Can Respir J 2011;18(4):230–234. [DOI] [PMC free article] [PubMed]
- 19.Taichman DB, McGoon MD, Harhay MO, Archer-Chicko C, Sager JS, Murugappan M, et al. Wide variation in clinicians’ assessment of New York Heart Association/World Health Organization functional class in patients with pulmonary arterial hypertension. Mayo Clin Proc 2009;84(7):586–592. [DOI] [PMC free article] [PubMed]
- 20.Harding S, Khimdas S, Bonner A, Baron M, Pope J. Best practices in scleroderma: an analysis of practice variability in SSc centers within the Canadian Scleroderma Research Group (CSRG). Clin Exp Rheumatol 2012;30(2 suppl. 71):S38–S42. [PubMed]
- 21.Avouac J, Huscher D, Furst DE, Opitz CF, Distler O, Allanore Y. Expert consensus for performing right heart catheterisation for suspected pulmonary arterial hypertension in systemic sclerosis: a Delphi consensus study with cluster analysis. Ann Rheum Dis 2013;73(1):191–197. [DOI] [PubMed]
- 22.Yeates P, O’Neill P, Mann K, Eva KW. Effect of exposure to good vs poor medical trainee performance on attending physician ratings of subsequent performances. JAMA 2012;308(21):2226–2232. [DOI] [PubMed]
- 23.Birkett NJ. Selecting the number of response categories for a Likert-type scale. Paper presented at: Proceedings of the American Statistical Association, 1986, pp. 488–492.
- 24.Yuan JX-Y, Morrell NW, Harikrishnan S, Butrous G. Pulmonary Circulation: a new venue for communicating your findings, ideas and perspectives. Pulm Circ 2011;1(1):1–2. [DOI] [PMC free article] [PubMed]
- 25.Peacock A, Naeije R, Galiè N, Reeves JT. End points in pulmonary arterial hypertension: the way forward. Eur Respir J 2004;23(6):947–953. [DOI] [PubMed]
- 26.Peacock AJ, Naeije R, Galiè N, Rubin L. End-points and clinical trial design in pulmonary arterial hypertension: have we made progress? Eur Respir J 2009;34(1):231–242. [DOI] [PubMed]
- 27.Malhotra R, Hess D, Lewis GD, Bloch KD, Waxman AB, Semigran MJ. Vasoreactivity to inhaled nitric oxide with oxygen predicts long-term survival in pulmonary arterial hypertension. Pulm Circ 2011;1(2):250–258. [DOI] [PMC free article] [PubMed]
- 28.Ryan JJ, Rich JD, Thiruvoipati T, Swamy R, Kim GH, Rich S. Current practice for determining pulmonary capillary wedge pressure predisposes to serious errors in the classification of patients with pulmonary hypertension. Am Heart J 2012;163(4):589–594. [DOI] [PubMed]
- 29.Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med 1992;327(2):76–81. [DOI] [PubMed]
- 30.Bernus A, Wagner BD, Accurso F, Doran A, Kaess H, Ivy DD. Brain natriuretic peptide levels in managing pediatric patients with pulmonary arterial hypertension. Chest 2009;135(3):745–751. [DOI] [PMC free article] [PubMed]
- 31.Vonk MC, van Dijk AP, Heijdra YF, van der Heijden HF, Bredie SJ, van den Hoogen FH. Pulmonary hypertension: its diagnosis and management, a multidisciplinary approach. Neth J Med 2005;63(6):193–198. [PubMed]
- 32.Maron BA, Goldstein RH, Rounds SI, Shapiro S, Jankowich M, Garshik E, et al. Study design and rationale for investigating phosphodiesterase type 5 inhibition for the treatment of pulmonary hypertension due to chronic obstructive pulmonary disease: the TADA-PHiLD (TADAlafil for Pulmonary Hypertension assocIated with chronic obstructive Lung Disease) trial. Pulm Circ 2013;3(4):889–897. [DOI] [PMC free article] [PubMed]
- 33.Carlisle DM, Leape LL, Bickel S, Bell R, Kamberg C, Genovese B, et al. Underuse and overuse of diagnostic testing for coronary artery disease in patients presenting with new-onset chest pain. Am J Med 1999;106(4):391–398. [DOI] [PubMed]
- 34.Woolf SH, Grol R, Hutchinson A, Eccles M, Grimshaw J. Clinical guidelines: potential benefits, limitations, and harms of clinical guidelines. BMJ 1999;318(7182):527–530. [DOI] [PMC free article] [PubMed]
- 35.Galiè N, Corris PA, Frost A, Girgis RE, Granton J, Jin Z-C, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol 2013;62(suppl. 25):D60–D72. [DOI] [PubMed]
- 36.Rich S, Brundage BH. High-dose calcium channel-blocking therapy for primary pulmonary hypertension: evidence from long-term reduction in pulmonary artery pressure and regression of right ventricular hypertrophy. Circulation 1987;76(1):135–141. [DOI] [PubMed]
- 37.Galiè N, Hoeper MM, Humbert M, Torbicki A, Vachiéry J-L, Barbera JA, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 2009;34(6):1219–1263. [DOI] [PubMed]
- 38.Blanco I, Santos S, Gea J, Guell R, Torres F, Gimeno-Santos E, et al. Sildenafil to improve respiratory rehabilitation outcomes in COPD: a controlled trial. Eur Respir J 2013;42(4):982–992. [DOI] [PubMed]