

Abstract Abstract
Pulmonary arterial hypertension (PAH) is a progressive disease characterized by severe remodeling of the pulmonary artery resulting in increased pulmonary artery pressure and right ventricular hypertrophy and, ultimately, failure. Bone marrow–derived progenitor cells play a critical role in vascular homeostasis and have been shown to be involved in the pathogenesis of PAH. A proliferation of c-Kit+ hematopoietic progenitors and mast cells has been noted in the remodeled vessels in PAH. Imatinib, a tyrosine kinase inhibitor that targets c-Kit, has been shown to be beneficial for patients with PAH. Here we hypothesize that the clinical benefit of imatinib in PAH could be related to c-Kit inhibition of progenitor cell mobilization and maturation into mast cells. As a corollary to the phase 3 study using imatinib in PAH, blood samples were collected from 12 patients prior to starting study drug (baseline) and while on treatment at weeks 4 and 24. Eight were randomized to imatinib and 4 to placebo. Circulating c-Kit+ and CD34+CD133+ hematopoietic progenitors as well as biomarkers of mast cell numbers and activation were measured. Circulating CD34+CD133+ and c-Kit+ progenitor cells as well as c-Kit+/CD34+CD133+ decreased with imatinib therapy (all P < 0.05). In addition, total tryptase, a marker of mast cell load, dropped with imatinib therapy (P = 0.02) and was related to pulmonary vascular resistance (R = 0.7, P = 0.02). The findings support c-Kit inhibition as a potential mechanism of action of imatinib in PAH and suggest that tryptase is a potential biomarker of response to therapy.
Keywords: pulmonary hypertension, imatinib, c-Kit, progenitor cells, mast cells
Introduction
Pulmonary arterial hypertension (PAH) is a progressive disease characterized by vasoconstriction, remodeling, and obliteration of the pulmonary vascular bed, leading to elevated pulmonary arterial pressure, right heart failure, and death. With available therapies, survival rates remain low, urging the need to develop more targeted therapies.
c-Kit is a transmembrane tyrosine kinase marker of bone marrow–derived hematopoietic stem cells and mast cells.1 Several studies have shown mobilization and accumulation of c-Kit+ cells in the pulmonary vasculature of patients with pulmonary hypertension as well as in murine models of pulmonary hypertension, suggesting that these cells contribute to the pathogenesis of pulmonary hypertension.2,3 Both myeloid hematopoietic progenitors and mast cells contribute to the c-Kit+ proangiogenic cells in the pulmonary vasculature in PAH.2 c-Kit-targeted therapy in murine models of chronic hypoxia has been shown to prevent pulmonary hypertension.3,4 In addition, use of the mast cell stabilizer cromolyn or antagonists of the chemokine receptors involved in recruitment of mast cell progenitors reduced the development of vascular remodeling and right ventricular hypertrophy in hypoxia-induced rodent pulmonary hypertension.5 In a small pilot study, tryptase, a marker of mast cell load, was found to be increased in PAH patients, and the levels dropped after treatment with mast cell blockade therapy.6 Mast cells are tissue resident cells and are not found circulating in peripheral blood. The increased number of mast cells is related to increased mobilization and recruitment of mast cell progenitors identified by c-Kit, CD133, and CD34 markers and their maturation into mast cells.7-9 As newer therapies targeting remodeling and proliferation are being investigated in PAH, inhibition of c-Kit+ progenitor and mast cells that may be contributing to the vasculogenesis in PAH seems promising. Imatinib is a broad-spectrum tyrosine kinase inhibitor that is used for the treatment of chronic myeloid leukemia and myeloproliferative/myelodysplastic disorders. It has inhibitory effects on both c-Kit and platelet-derived growth factor (PDGF) signaling, which play a role in the pathogenesis of PAH. Imatinib has been shown in several case reports to be beneficial for the treatment of PAH.10,11 In a phase 2 study, imatinib therapy was well tolerated by PAH patients and led to a significant reduction in pulmonary vascular resistance (PVR) in association with an increase in cardiac output (CO).12 A phase 3 study showed clinical benefits in patients treated with imatinib as add-on therapy for 24 weeks compared with placebo, mainly improved 6-minute walk distance, reduced PVR, and reduced pulmonary pressures with increased CO; however, serious adverse events were common.13 In parallel to the phase 3 trial, 7 centers in the United States participated in an ancillary study to assess whether imatinib provides clinical benefit to PAH patients through c-Kit+ blockade by inhibiting multipotent progenitor mobilization and differentiation, in particular into resident mast cells.
Methods
Samples were collected prior to starting the study drug (baseline) and during treatment at weeks 4 and 24. Blood samples were collected from 12 subjects. Circulating c-Kit+ and CD34+CD133+ hematopoietic progenitors were measured as previously described.14 Biomarkers of mast cell numbers and activation (total tryptase and both α- and β-tryptases as well as prostaglandins and leukotrienes [PGDM and LTE4]) were measured as previously described.6 In addition, since imatinib inhibits PDGF receptor (PDGFR) phosphorylation, the PDGF pathway was also investigated through measurement of PDGFR phosphorylation (Phospho-PDGFR Beta Cell-Based ELISA Kit; R&D Systems, Minneapolis, MN) as another beneficial mechanism of action for imatinib. Comparisons of patients receiving imatinib to those receiving placebo with respect to quantitative baseline characteristics were performed with two-sample t tests. Assessments of changes from baseline to either 4 or 24 weeks were performed with paired t tests within treatment groups. Pearson correlations were used to assess relationships between pairs of quantitative parameters. Analyses were performed using R (ver. 2.11.1; www.r-project.org).
Results and discussion
Of the 12 subjects enrolled in the ancillary study, 8 were randomized to imatinib and 4 to placebo. Three subjects in the imatinib group and 2 in the placebo group withdrew from the study before week 24. There were no significant differences in baseline characteristics ( Table S1). As expected with imatinib treatment, there were significant changes in the hematological parameters from baseline to week 24 (Table 1). There was a significant drop in red blood cell (RBC) count (P = 0.007), hematocrit (P = 0.02), and white blood cell count (P = 0.02), with an increase in mean corpuscular hemoglobin (P = 0.009). In parallel, circulating CD34+CD133+ and c-Kit+ progenitor cells as well as c-Kit+/CD34+CD133+ decreased with imatinib therapy (P < 0.05; Table 1). Total tryptase, an indicator of mast cell load, dropped significantly with imatinib therapy (P = 0.02; Fig. 1), whereas LTE4 and PGDM did not vary significantly. These changes were noted as early as at week 4 of treatment with imatinib ( Table S2). At baseline, RBC count, hematocrit, and hemoglobin were inversely related to CD34+CD133+ and c-Kit+ progenitor cells ( Table S3). There was no correlation between changes in RBC parameters and changes in progenitor cells with therapy.
Table 1.
Changes in variables from baseline to 24 weeks on treatment
| Imatinib (n = 8) | Placebo (n = 4) | |||
|---|---|---|---|---|
| Variable | Change | P | Change | P |
| RBC count, 106 cells/μL | −0.6 | 0.007 | −0.07 | 0.5 |
| Hemoglobin, g/dL | −0.8 | 0.07 | −0.1 | 0.7 |
| Hematocrit, % | −4.4 | 0.02 | −2.7 | 0.09 |
| MCH, pg | 1.8 | 0.009 | −0.7 | 0.5 |
| WBC count, 103 cells/μL | −0.9 | 0.02 | −0.7 | 0.5 |
| Total tryptase, ng/mL | −5.8 | 0.02 | 1.05 | 0.1 |
| %CD34+CD133+ | −0.02 | 0.04 | 0.01 | 0.7 |
| %c-Kit+ | −2.3 | 0.03 | 0.6 | 0.7 |
| %c-Kit+/CD34+CD133+ | −8 | 0.03 | −3.6 | 0.6 |
RBC: red blood cell; MCH: mean corpuscular hemoglobin; WBC: white blood cell.
Figure 1.
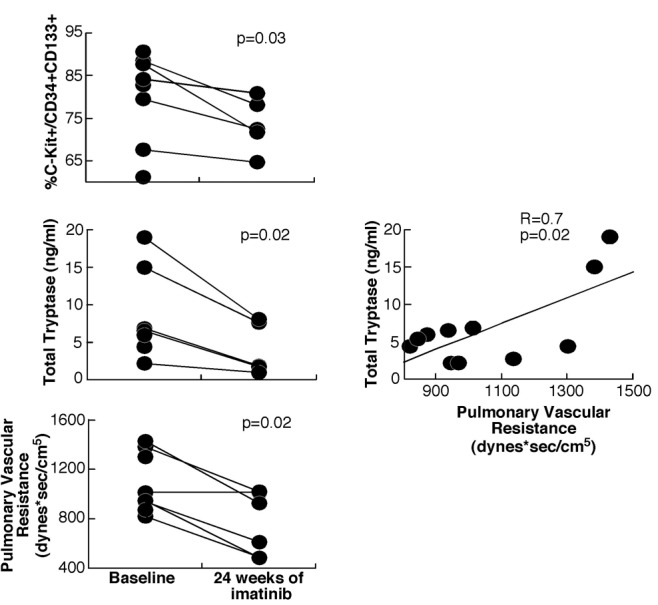
Mast cell markers and pulmonary vascular resistance decreased with imatinib therapy (3 subjects in the imatinib arm withdrew from the study by week 24). Tryptase levels correlated with disease severity in all subjects.
To assess whether imatinib acts through its inhibition of PDGF signaling, PDGFR phosphorylation of mononuclear cells was measured using the Phospho-PDGFR Beta Cell-Based ELISA Kit (R&D Systems). There was a trend toward an increase in PDGFR-α in the imatinib group (P = 0.06) but no change in PDGFR-β and phosphorylated PDGFR (P = 0.7).
The main clinical parameter that changed significantly in our small subgroup was PVR, with a significant drop noted in the imatinib group (change in PVR: −329 dyn s/cm5; P = 0.02; Fig. 1). At baseline, PVR was inversely related to hematocrit (R = −0.7, P = 0.03) and hemoglobin (R = −0.8, P = 0.005) and positively related to total tryptase (R = 0.7, P = 0.02; Fig. 1). However, changes in PVR with imatinib treatment did not correlate with RBC markers or any of the measured biomarkers.
In this small ancillary study, we have shown that treatment with imatinib in PAH led to a reduction in circulating proangiogenic and mast cell progenitors in association with clinical improvement. Total tryptase, a marker of mast cell load, decreased with imatinib treatment and correlated with disease severity. Our findings support c-Kit inhibition as a potential mechanism of action of imatinib in PAH and suggest that tryptase is a potential biomarker of response to therapy. The small sample size is a major limitation of the study and limits data interpretation. Nevertheless, the findings provide rationale that therapies targeting the c-Kit pathway should be further investigated in PAH.
Appendix. Supplemental material
Table S1.
Characteristics of subjects
| Age | Sex, F∶M | Race, C∶B | mPAP, mmHg | Cardiac output, L/min | PVR, dyn s/cm5 | RVSP, mmHg | NT-proBNP, pg/mL | 6MWD, m | |
|---|---|---|---|---|---|---|---|---|---|
| Imatinib group | 55 ± 6 | 6∶2 | 7∶1 | 57 ± 4 | 3.7 ± 0.4 | 1,088 ± 86 | 93 ± 10 | 1,296 ± 466 | 303 ± 25 |
| Placebo group | 50 ± 6 | 4∶0 | 4∶0 | 56 ± 11 | 3.9 ± 0.5 | 981 ± 87 | 102 ± 6 | 6,445 ± 3,454 | 349 ± 5 |
| P | 0.5 | 0.2 | 0.4 | 0.9 | 0.8 | 0.5 | 0.6 | 0.02 | 0.3 |
Data are mean ± SE, unless otherwise indicated. F: female; M: male; C: Caucasian; B: black; mPAP: mean pulmonary arterial pressure; PVR: pulmonary vascular resistance; RVSP: right ventricular systolic pressure; NT-proBNP: N-terminal prohormone of brain natriuretic peptide; 6MWD: 6-minute walk distance.
Table S2.
Changes in variables from baseline to 4 weeks on treatment
| Imatinib (n = 8) | Placebo (n = 4) | |||
| Variable | Change | P | Change | P |
| RBC count, 106 cells/μL | −0.3 | 0.03 | −0.15 | 0.7 |
| Hemoglobin, g/dL | −0.4 | 0.08 | −0.4 | 0.8 |
| Hematocrit, % | −2.5 | 0.009 | −2.5 | 0.5 |
| MCH, pg | 0.6 | 0.1 | 0.5 | 0.5 |
| WBC count, 103 cells/μL | 1 | 0.3 | −0.3 | 0.4 |
| Total tryptase, ng/mL | −4.2 | 0.002 | 0.9 | 0.4 |
| %CD34+CD133+ | −0.02 | 0.05 | 0.01 | 0.8 |
| %c-Kit+ | −1.6 | 0.05 | 0.3 | 0.9 |
| %c-Kit+/CD34+CD133+ | −1.5 | 0.7 | −8.5 | 0.02 |
| PDGFR-α, ng/mL | 26 | 0.04 | 6 | 0.4 |
RBC: red blood cell; MCH: mean corpuscular hemoglobin; WBC: white blood cell; PDGFR: platelet-derived growth factor receptor.
Table S3.
Significant correlations
| Variable | Variable | Pearson R | P |
|---|---|---|---|
| RBC count | %CD34+CD133+ | −0.8 | 0.003 |
| Hematocrit | %CD34+CD133+ | −0.85 | 0.001 |
| Hemoglobin | %CD34+CD133+ | −0.77 | 0.006 |
| RBC count | %c-Kit+ | −0.81 | 0.003 |
| Hematocrit | %c-Kit+ | −0.75 | 0.008 |
| Hemoglobin | %c-Kit+ | −0.65 | 0.03 |
| PVR | Hematocrit | −0.65 | 0.03 |
| PVR | Hemoglobin | −0.78 | 0.005 |
| PVR | Tryptase | 0.7 | 0.02 |
| LTE4 | PDGFR-α | 0.71 | 0.015 |
RBC: red blood cell; PVR: pulmonary vascular resistance; LTE4: leukotriene E4; PDGFR: platelet-derived growth factor receptor.
Source of Support: This study was funded by a Novartis Pharmaceuticals Corporation investigator-initiated study. KA is a scholar of the International Society for Advancement of Cytometry (ISAC).
Conflict of Interest: SF, RD, RF, SC, and SE have no conflicts of interest pertinent to this study. RB received grant support from United Therapeutics, Bayer, Gilead, Ikaria, and GeNO and speaking honoraria from United Therapeutics, Bayer, and Gilead. PH is supported by National Heart, Lung, and Blood Institute, National Institutes of Health, grants P01HL84946-01 and R01HL114910; he has also served on advisory boards for Novartis, Merck, Pfizer, and Gilead. FR received grant support form Actelion, Bayer, Novartis, Gilead, United Therapeutics, and Lung LLC; received speaking honoraria from Actelion, Gilead, United Therapeutics, Lung LLC, and Bayer; and served as a consultant for Actelion, Bayer, Novartis, Gilead, United Therapeutics, and Lung LLC. FT participated as speaker/consultant for or received research funding from Actelion, United Therapeutics, Lung LLC, Bayer, Novartis, Gilead, Medtronic, Akaria, and GeNO. DAQ is employed by Novartis and owns Novartis stock worth more than $50,000. KA received grant support from Novartis and GlaxoSmithKline.
References
- 1.Ogawa M, Matsuzaki Y, Nishikawa S, et al. Expression and function of c-kit in hemopoietic progenitor cells. J Exp Med 1991;174:63–71. [DOI] [PMC free article] [PubMed]
- 2.Montani D, Perros F, Gambaryan N, et al. C-kit-positive cells accumulate in remodeled vessels of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2011;184:116–123. [DOI] [PubMed]
- 3.Gambaryan N, Perros F, Montani D, et al. Targeting of c-kit+ haematopoietic progenitor cells prevents hypoxic pulmonary hypertension. Eur Respir J 2011;37:1392–1399. [DOI] [PubMed]
- 4.Gambaryan N, Perros F, Montani D, Cohen-Kaminsky S, Mazmanian GM, Humbert M. Imatinib inhibits bone marrow–derived c-kit+ cell mobilisation in hypoxic pulmonary hypertension. Eur Respir J 2010;36:1209–1211. [DOI] [PubMed]
- 5.Banasova A, Maxova H, Hampl V, et al. Prevention of mast cell degranulation by disodium cromoglycate attenuates the development of hypoxic pulmonary hypertension in rats exposed to chronic hypoxia. Respiration 2008;76:102–107. [DOI] [PubMed]
- 6.Farha S, Sharp J, Asosingh K, et al. Mast cell number, phenotype, and function in human pulmonary arterial hypertension. Pulm Circ 2012;2:220–228. [DOI] [PMC free article] [PubMed]
- 7.Rastogi P, White MC, Rickard A, McHowat J. Potential mechanism for recruitment and migration of CD133 positive cells to areas of vascular inflammation. Thromb Res 2008;123:258–266. [DOI] [PMC free article] [PubMed]
- 8.Radinger M, Jensen BM, Kuehn HS, Kirshenbaum A, Gilfillan AM. Generation, isolation, and maintenance of human mast cells and mast cell lines derived from peripheral blood or cord blood. Curr Protoc Immunol 2010:7.37.1–7.37.12. [DOI] [PMC free article] [PubMed]
- 9.Kirshenbaum AS, Goff JP, Semere T, Foster B, Scott LM, Metcalfe DD. Demonstration that human mast cells arise from a progenitor cell population that is CD34+, c-kit+, and expresses aminopeptidase N (CD13). Blood 1999;94:2333–2342. [PubMed]
- 10.Ghofrani HA, Seeger W, Grimminger F. Imatinib for the treatment of pulmonary arterial hypertension. N Engl J Med 2005;353:1412–1413. [DOI] [PubMed]
- 11.Patterson KC, Weissmann A, Ahmadi T, Farber HW. Imatinib mesylate in the treatment of refractory idiopathic pulmonary arterial hypertension. Ann Intern Med 2006;145:152–153. [DOI] [PubMed]
- 12.Ghofrani HA, Morrell NW, Hoeper MM, et al. Imatinib in pulmonary arterial hypertension patients with inadequate response to established therapy. Am J Respir Crit Care Med 2010;182:1171–1177. [DOI] [PMC free article] [PubMed]
- 13.Hoeper MM, Barst RJ, Bourge RC, et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation 2013;127:1128–1138. [DOI] [PubMed]
- 14.Farha S, Asosingh K, Xu W, et al. Hypoxia-inducible factors in human pulmonary arterial hypertension: a link to the intrinsic myeloid abnormalities. Blood 2011;117:3485–3493. [DOI] [PMC free article] [PubMed]