

Abstract Abstract
Pulmonary hypertension is characterized by cellular and structural changes in the vascular wall of pulmonary arteries. We hypothesized that lysophosphatidic acid (LPA), a bioactive lipid, is implicated in this vascular remodeling in a rat model of hypoxic pulmonary hypertension. Exposure of Wistar rats to 10% O2 for 3 weeks induced an increase in the mean serum levels of LPA, to 40.9 (log-detransformed standard deviations: 23.4–71.7) μM versus 21.6 (11.0–42.3) μM in a matched control animal group (P = 0.037). We also observed perivascular LPA immunohistochemical staining in lungs of hypoxic rats colocalized with the secreted lysophospholipase D autotaxin (ATX). Moreover, ATX colocalized with mast cell tryptase, suggesting implication of these cells in perivascular LPA production. Hypoxic rat lungs expressed more ATX transcripts (2.4-fold) and more transcripts of proteins implicated in cell migration: β2 integrin (1.74-fold), intracellular adhesion molecule 1 (ICAM-1; 1.84-fold), and αM integrin (2.70-fold). Serum from the hypoxic group of animals had significantly higher chemoattractant properties toward rat primary lung fibroblasts, and this increase in cell migration could be prevented by the LPA receptor 1 and 3 antagonists. LPA also increased adhesive properties of human pulmonary artery endothelial cells as well as those of human peripheral blood mononuclear cells, via the activation of LPA receptor 1 or 3 followed by the stimulation of gene expression of ICAM-1, β-1, E-selectin, and vascular cell adhesion molecule integrins. In conclusion, chronic hypoxia increases circulating and tissue levels of LPA, which might induce fibroblast migration and recruitment of mononuclear cells in pulmonary vasculature, both of which contribute to pulmonary vascular remodeling.
Keywords: anti-lysophosphatidic acid antibody, fibroblast, circulating progenitor cells, cell migration and adhesion
Introduction
The chronic-hypoxia model of pulmonary hypertension (PH) is characterized by an important remodeling of the arteriolar wall. This PH model is very predictable and reproducible, and although implication of vasoconstriction and vasoproliferation of the different cell types constituting the vascular wall has been shown, all the factors involved in these mechanisms are not yet identified.1-4
Patients with PH can present with thrombotic pulmonary vascular lesions suggesting abnormalities of blood coagulation factors and platelet function. Hypoxia per se may accelerate blood coagulation5 and platelet adhesion and aggregation.6 The activation of platelets can result in the release of multiple and diverse soluble mediators such as interleukin-1, bioactive lipids, and chemokines with pleiotropic functions in inflammation.7,8 Platelets play a key role in thrombosis and inflammation, and several studies indicate a survival benefit in anticoagulated idiopathic PH patients.9 Activated platelets also generate lysophosphatidic acid (LPA), a bioactive lipid mediator. LPA binds to cell G-protein-coupled receptor to regulate cell growth, differentiation, and apoptosis and stimulates cell migration. Recently, it has been reported that a basal physiological level of LPA protects pulmonary vasculature in the mouse against remodeling induced by chronic hypoxia.10 These authors have shown that LPA deficiency alters endothelin signaling tremendously: it favors endothelin-A receptor pathways and downregulates endothelin-B receptor signaling.10 However, little is known about the effect of increased LPA concentration in the blood, although indirect data suggest that high LPA is deleterious.11,12 This lipid was also shown to enhance production and secretion of cytokines in lymphocytes.13 In the cardiovascular system, LPA alters endothelial barrier functions and initiates and perpetuates pathophysiological processes such as inflammation and atherogenesis.14 LPA also promotes fibroblast and monocyte differentiation into myofibroblast and fibrocyte, respectively,15,16 as well as dedifferentiation and migration of pulmonary smooth muscle cells.10
Several studies show the potential role of adventitial fibroblasts and of fibrocytes in PH.17 The classical “outside-in” hypothesis is that local interstitial fibroblasts migrate from adventitia into inflammatory areas and produce extracellular matrix proteins, leading to fibrosis.18 Adventitial fibroblasts respond to hormonal, inflammatory, and environmental stresses and present an activated phenotype. Such activation is characterized by increases in cellular proliferation and migration and by expression of α-smooth muscle actin, secretion of chemokines and cytokines, and growth and angiogenic factors contributing to the vascular inflammatory context, which is seen in PH.18 Chemokines and cytokines released by adventitial fibroblasts participate in the recruitment and activation of circulating leukocytes and progenitor cells, supporting the alternative “inside-out” hypothesis. This hypothesis suggests that circulating bone marrow–derived precursors are involved in vascular remodeling by infiltration and differentiation into fibroblast-like cells.17,18
Our study aimed at the investigation of the potential role of LPA in the pathogenesis of hypoxic PH by its action on adventitial fibroblasts and on the interaction of endothelial cells with circulating progenitor cells. We tested the hypothesis that exposure of rats to chronic hypoxia induces increased amounts of LPA (both circulating and tissular) and examined the effects of LPA in vitro on fibroblast migration and peripheral blood mononuclear cell (PBMC) adhesion to pulmonary endothelial cells.
Methods
Reagents
VPC-12249 was purchased from Avanti Polar Lipids (Alabaster, AL). Ki16425 was purchased from Selleck Chemicals (Houston, TX). All other reagents were purchased from Sigma-Aldrich (Haasrode, Belgium). LPA and VPC-12249 were dissolved in 0.1% bovine serum albumin in phosphate-buffered saline (PBS) at 1 mM, and Ki16425 was dissolved in dimethyl sulfoxide at 5 mM. All drugs were stored as frozen single-use aliquots.
Animal model
Wistar-Kyoto rats were purchased from Janvier Laboratories (Saint Berthevin, France). Rats were maintained under conditions of unlimited access to food and water and according to the guidelines issued by the local animal care ethics committee (Comité d’Ethique, reference 442N). Rats weighing 200 g were randomly assigned to the control or the hypoxic group. The hypoxic group (n = 10) was exposed to 10% O2 for 3 weeks. Exposure to hypoxia was achieved by using a large, sealed plastic cage (allowing ventilation) constantly flushed with nitrogen. The Fio2 was monitored constantly and adjusted with a PROOX-110 (Biospherix, Redfield, NY). Controls (n = 10) were kept in normoxic conditions. Animals were anesthetized by intraperitoneal Nembutal injection (sodium pentobarbital, 35 mg/kg body weight), serum samples were taken, and animals were killed by exsanguination. Tissue samples were collected for immunohistology, RNA isolation, and morphometry.
Tissue samples and pulmonary vascular remodeling analysis
After blood withdrawal, the heart and lungs were excised. The right lung was frozen in liquid nitrogen for molecular analyses. In half of the rats, the left lung was fixated by filling the airways with 4% formaldehyde for 24 hours and then embedded in paraffin. In the other half, lungs were inflated with 50% optimal cutting temperature (OCT-) reagent in PBS and frozen in isopentane for immunofluorescence studies. Transversally cut 5-μm-thick pulmonary sections were deparaffinized and then stained with hematoxylin-eosin or Miller’s elastic stain. Blood vessels with diameters greater than 60 μm and less than 200 μm were taken for vascular morphometric analysis. External and lumen perimeters were computed with the ImageJ program (ver. 1.46r). Wall thickness ratio was calculated by dividing the difference between the external and lumen perimeters by the external perimeter of the vessel.
Cell culture
Human primary cells and media were purchased from Lonza (Vervier, Belgium). Normal human lung fibroblasts were cultured in fibroblast growth medium-2 supplemented with 2% fetal bovine serum (FBS). Human pulmonary arterial endothelial cells (hPAECs) were cultured in endothelial cell growth medium (EGM-2) supplemented with 2% FBS. Rat lung fibroblasts were purchased from Cell Applications (via Tebu-bio, Belgium) and cultured in rat fibroblast growth medium. Cell culture flasks (70%–80% confluent) grown at 37ºC and 5% CO2 were passaged at a 1∶5 ratio and used for up to the eighth passage.
Cell migration
Human and rat lung fibroblast migration was assessed in the Boyden chamber system with BD Falcon HTS FluoroBlock 8-μm-pore 96-well filter plates (VWR, Leuven, Belgium). Cells in serum-free Dulbecco’s modified eagle medium (DMEM; 1 × 105 cells/cm2) were loaded into the upper compartments (0.08 cm2 effective growth area), together with the desired additives. The lower compartments were filled with DMEM containing chemoattractants (LPA or serum) and/or other additives. The chamber was incubated at 37ºC and 5% CO2 for 6 hours. After incubation, 2 μM of calcein-AM was added to the lower compartment. Fluorescently labeled cells, which migrated from the upper compartment, were viewed under an inverted microscope, whereas the filter itself blocked any light transmission from the upper compartment. A low-power field view of the filter accounting for 36% of its surface was taken with a charged-couple-device camera (Scion, Frederick, MD). Total cell numbers for each filter were then counted and calculated by 2 operators. Cell migration was expressed as the percent of seeded cells that migrated over the incubation time. Six inserts were prepared in each experiment for every condition.
PBMC isolation
Blood was collected from healthy adult volunteers in accordance with local ethical committee guidelines (agreement no. B2010/011); collection conformed to the Declaration of Helsinki on human experimentation. PBMCs were isolated from heparinized blood with Ficoll-Paque Plus (VWR) according to the manufacturer’s instructions.
PBMC adhesion
Ten thousand PAECs were grown in 96-well plates for 48–72 hours, until the endothelial cells formed a monolayer. PBMCs were harvested and prepared as a cell suspension at 0.5 × 106 cells/mL in serum-free EGM-2. In different experiments, PAECs or PBMCs were pretreated with LPA for 1 hour before the adhesion assay. PAECs were then washed with 250 μL serum-free medium, while pretreated PBMCs were spun down at 300 g for 5 minutes and resuspended in fresh serum-free EGM-2. Control cells were manipulated in the same manner in parallel. Culture medium was aspirated from endothelial cells, and 200 μL of the PBMC suspension was added to each well containing the endothelial monolayer. Cells were allowed to adhere for 30 minutes in a cell culture incubator. Medium from each well was discarded, and wells were washed 3 times with 250 μL of PBS. Cells were then fixed by methanol for 10 minutes at +4°C, stained with Giemsa stain for 40 minutes, washed several times with distilled water, and dried. Adherent PBMCs were counted under an inverted microscope. Counts from 4 separate fields per well accounting for 35% of the total surface were averaged. Experiments were performed on cells from at least 3 different passages, and 5–8 wells were used for every experimental condition.
Real-time RTq-PCR
Total RNA isolation from cultured cells or PBMCs was performed with the MagNA pure LC RNA isolation kit (Roche, Vilvoorde, Belgium). Lung tissue RNA isolation was performed with the RNeasy Mini Kit (Qiagen, Venlo, Netherlands). The complementary DNA (cDNA) synthesis was performed with transcriptor universal cDNA master (Roche). Real-time PCR was performed with a light-cycler detection system (Roche) using SYBR Green detection. The messenger RNA (mRNA) level was normalized to the geometric mean of several housekeeping genes (HPRT, UBC, GAPDH, B2M, and β-actin).
LPA dosage
Measurement of serum LPA concentration was performed with Lysophosphatidic Acid Assay Kit II (Echelon, Salt Lake City, UT).
Immunofluorescence
Frozen lung sections (6 μM) were rehydrated/permeabilized in 0.05% Tween/PBS for 20 minutes, blocked in 2% casein in PBS (pH = 7) for 20 minutes, and rinsed in PBS for 5 minutes. Slices were incubated overnight in a humid chamber at 4°C with rabbit anti-ATX (i.e., anti-autotaxin) antibody (Cayman Chemical) diluted at 1 μg/mL in 0.1% casein in PBS and mouse anti-LPA antibody, which recognizes the “head group” of LPA common to 14∶0, 16∶0, 18∶0, 18∶2, and 20∶4 species in particular, or with isotype control antibody (both kindly provided by L-Path, San Diego, CA), both diluted at 90 μg/mL, or with anti–mast cell tryptase antibody diluted at 4 μg/mL (Abcam, Cambridge, UK) and then washed 3 times for 5 minutes with PBS before incubation with anti-mouse-Alexa488 (1∶50; Invitrogen, Merelbeke, Belgium), anti-rabbit-Alexa594 (1∶100; Invitrogen) and 4′,6-diamidino-2-phenylindole (DAPI; 2 μg/mL) in PBS for 1 hour at room temperature. Slices were rinsed in PBS–0.05% Tween and mounted with Fluoromount liquid.
Statistics
Data on serum LPA concentration were tested for normality of the distribution with the Kolmogorov-Smirnov test. Since results obtained from the normoxic group of animals deviated significantly from the normal distribution, the values from both groups were log transformed before statistical analysis. Detransformation of lower and upper standard deviations was calculated as 10log(mean − SD) and 10log(mean + SD), respectively. The nonpaired t test or a one-way ANOVA, when appropriate, was performed to check for statistical significance between groups and treatments.
Results
We used a well-established rat model of chronic hypoxia to induce pulmonary vascular remodeling leading to PH. Figures 1A and 1B show the results of morphometric analysis of pulmonary blood vessels. Compared to control rats, vascular wall thickness increased by 27% in blood vessels with a diameter of 60–100 μm in rats subjected to chronic hypoxia for 3 weeks (open bars in Figure 1C; P = 0.002; ANOVA). The same analysis in blood vessels with a diameter of 100–200 μm demonstrated a 51% increase in vascular wall thickness in rats subjected to chronic hypoxia, compared to control rats (filled bars in Figure 1C; P = 0.0001; ANOVA). These results describe vascular remodeling in our chronic-hypoxia model of PH and thus support previously published observations.
Figure 1.
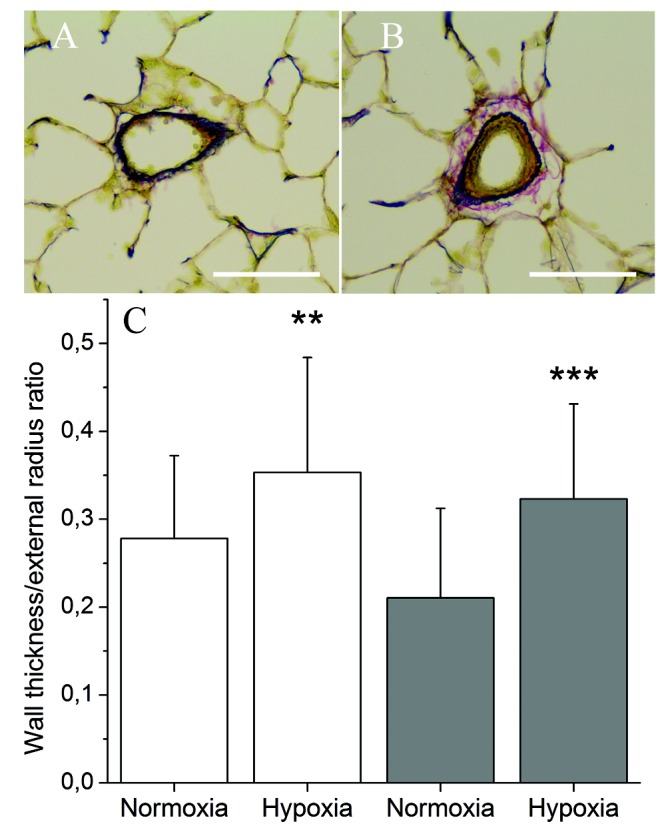
Morphometric analysis of pulmonary blood vessels. A, B, Representative paraffin-embedded lung sections stained using Miller’s technique from a normoxic rat (A) and a hypoxic rat (B). Elastic fibers are colored in dark blue, cytoplasm in yellow, and collagen in red. Scale bars: 50 μm. C, Analysis of the ratio between vessel lumen and external radii. Data represent mean + SD from blood vessels with a diameter of 60–100 μm (open bars) and vessels with a diameter of 100–200 μm (filled bars). Five normoxic and 5 hypoxic lungs were used, and 7–12 blood vessels were analyzed from each lung in each diameter category. Double asterisks indicate P = 0.002 and triple asterisks P = 0.0001 (one-way ANOVA).
We checked whether hypoxic rats presented higher LPA serum content, compared to normoxic animals. Results presented in Figure 2A show a statistically significant difference between hypoxic (40.9 [log-detransformed standard deviations: 23.4–71.7] μM, n = 10) and normoxic (21.6 [11.0–42.3] μM, n = 10) rats; P = 0.037. To check for tissue accumulation of LPA, we performed lung LPA immunofluorescent staining. Representative lung cryosections from normoxic and hypoxic rats are shown in Figure 2C–2F. They indicate increased perivascular LPA signal in hypoxic animals, which seems to be extracellular. Such extracellular LPA production might be largely determined by the secreted enzyme ATX. Therefore, we sought to determine its expression level. The results of RT-PCR show a 2.4-fold increase in expression of ATX mRNA in lung tissue from hypoxic rats (Figure 2B). We performed ATX-LPA co-immunostaining in lung cryosections. Figure 2C shows that ATX and LPA are colocalized in lungs. The segregation of ATX-LPA-positive cells in perivasculature in hypoxic lungs and the lack of such groups of cells in normoxia suggest that these cells might be of extrinsic origin. On the other hand, segregation of mast cells has been reported in the perivascular compartment in the lungs of hypoxic rats.19,20 We then performed ATX–mast cell tryptase co-immunostaining in lung cryosections. Figure 3 shows that ATX and mast cell tryptase are colocalized in lungs and suggests that lung tissue LPA accumulation in hypoxia is determined by recruitment of mast cells.
Figure 2.

A, Lysophosphatidic acid (LPA) content in serum from rats kept in ambient air and rats maintained in normobaric hypoxia (10% O2) for 3 weeks. Data obtained from normoxia group of animals deviated significantly from normal distribution (Kolmogorov-Smirnov test). Accordingly, the values from both groups were log transformed before statistical analysis. After detransformation, mean values were 21.6 (11.0–42.3) μM for the normoxia group and 40.9 (23.4–71.7) μM for the hypoxia group (P = 0.037 on lognormal data, nonpaired t test). B, Expression of lysophospholipase (autotaxin [ATX]) in lungs of normoxic and hypoxic rats. Data represent mean + SD of ATX messenger RNA (mRNA) expression normalized to geometric mean of mRNA expression of 2 housekeeping genes (GAPDH and HPRT). C–F, Immunohistochemical analysis of LPA and ATX expression in lungs of normoxic (C, D) and hypoxic (E, F) rats. Green, red, and blue channels are shown separately as thumbnails, and the large images show merged channels in the presence of immune antibodies (C, E) and isotype control antibody (D, F). DAPI: 4′,6-diamidino-2-phenylindole. Scale bars: 25 μm.
Figure 3.

Immunohystochemical analysis of mast cell tryptase (Tryp) and autotaxin (ATX) expression in lungs of normoxic (A, B) and hypoxic (C, D) rats. Green, red, and blue channels are shown separately as thumbnails, and the large images show merged channels in the presence of immune antibodies (A, C) and isotype control antibody (B, D). The arrow in A points to a single weakly ATX-positive cell in the lung of a normoxic rat. DAPI: 4′,6-diamidino-2-phenylindole. Scale bars: 25 μm.
In order to verify whether serum obtained from chronically hypoxic rats induces higher chemotaxis of rat primary pulmonary fibroblasts, we investigated cell migration by using the Boyden chamber assay. Figure 4A shows that hypoxic serum stimulated cell migration by 37%, as compared to serum from normoxic rats (P = 0.024; nonpaired t test). In addition, this figure demonstrates that a significant fraction of chemoattractant properties of the serum might be explained by the presence of LPA. The percentages of migrated cells were not significantly different when VPC-12249 (VPC), an antagonist of LPA receptors 1 and 3, was added to normoxic and hypoxic serum at a concentration of 10 μM (P = 0.059; nonpaired t test). Moreover, the difference between cell migration in the absence of VPC and that in the presence of VPC was significantly different between normoxia and hypoxia, 0.49% ± 0.21% and 0.71% ± 0.16%, respectively (mean ± SD, n = 10, P = 0.048). We concluded that the increase in cell migration observed for hypoxic serum could be attributed to the elevated serum LPA content.
Figure 4.

A, Chemotaxis of rat primary pulmonary fibroblasts toward serum from normoxic and hypoxic rats in the absence or presence of lysophosphatidic acid (LPA) receptor 1 and 3 antagonist VPC-12249 (VPC). Data represent mean + SE on 10 normoxic and 10 hypoxic rat serums. A single asterisk indicates P = 0.032 versus normoxia, double asterisks P = 0.024 versus normoxia, and triple asterisks P = 0.005 versus hypoxia and P = 0.059 versus normoxia + VPC; nonpaired t test. B, Chemotaxis of primary human pulmonary fibroblasts toward LPA. Data represent mean ± SE (n = 3). LPA increased cell migration in dose-dependent manner (P < 0.0001; one-way ANOVA), which was inhibited by VPC at all LPA concentrations; a section symbol (§) indicates P < 0.001 versus LPA alone (nonpaired t test).
We next sought to investigate the chemoattractant action of pure LPA in primary human pulmonary fibroblast cell migration assays. Figure 4B demonstrates that LPA increased cell migration in a dose-dependent manner (P < 0.0001; one-way ANOVA). Maximal effect was observed at a concentration of 1 μM. Moreover, this stimulation was significantly inhibited in the presence of the antagonist of LPA receptors 1 and 3, VPC-12249. These results demonstrate that LPA receptor 1 or 3 plays a role in the chemoattractant properties of LPA.
LPA has been shown to influence endothelial barrier function.9 We then looked at the interaction of PAECs with PBMCs. Figure 5A shows the effect of LPA on adhesive properties of PAECs and PBMCs. In these experiments, endothelial cells and PBMCs were pretreated separately. Our results demonstrate that LPA doubled the adhesion index of PAECs (P < 0.0001) and increased adhesion of PBMCs by approximately 50% (P < 0.001). The effect of LPA on the adhesive properties of PAECs was concentration dependent, which is demonstrated in Figure 5B. The threshold concentration is between 1 and 5 μM. Finally, the effect of LPA on PAEC and PBMC adhesion is significantly inhibited by the LPA receptor 1/3 antagonist VPC-12249 (P < 0.001), while VPC-12249 alone had a small agonist action on PAECs only (P = 0.008).
Figure 5.

Effect of lysophosphatidic acid (LPA) and LPA receptor 1 and 3 antagonist VPC-12249 (VPC) on adhesive properties of pulmonary arterial endothelial cells (PAECs; open bars) and peripheral blood mononuclear cells (filled bars). Data represent mean + SD of 3 independent experiments. Triple asterisks indicate P < 1 × 10−5 versus control, double asterisks P < 0.0004 versus control, a single asterisk P = 0.008 versus control, and a pound sign (#) P = 0.0005 versus LPA alone and P = 0.005 versus control, all by nonpaired t test. B, Concentration dependence of the effect of LPA on adhesive properties of PAECs. Data represent mean + SD of 3 independent experiments. Overall, P < 1 × 10−5 by one-way ANOVA. Triple asterisks indicate P < 1 × 10−5 versus control and double asterisks P < 0.0004 versus control.
Several proteins are implicated in the adhesion and migration processes. Changes in mRNA levels were analyzed by real-time PCR. We compared mRNA expression of β2 integrin, ICAM-1 (intracellular adhesion molecule 1), and αM integrin in normoxic and hypoxic rat lung tissue. Hypoxic rats presented an increased expression for the 3 genes: 1.74 fold (P < 0.001), 1.84 fold (P = 0.013), and 2.70 fold (P = 0.018) for genes coding for β2 integrin, ICAM-1, and αM integrin, respectively (Figure 6). In vitro, hPAECs treated with 10 μM of LPA for 2 hours showed increased gene expression of ICAM-1, VCAM-1 (vascular cell adhesion molecule 1), E-selectin, and β1 integrin (Figure 7). The effect of LPA was decreased or abolished by treatment of cells with Ki16425, a competitive and reversible antagonist of LPA receptors LPAR1, LPAR2, and LPAR3 (Figure 7).
Figure 6.
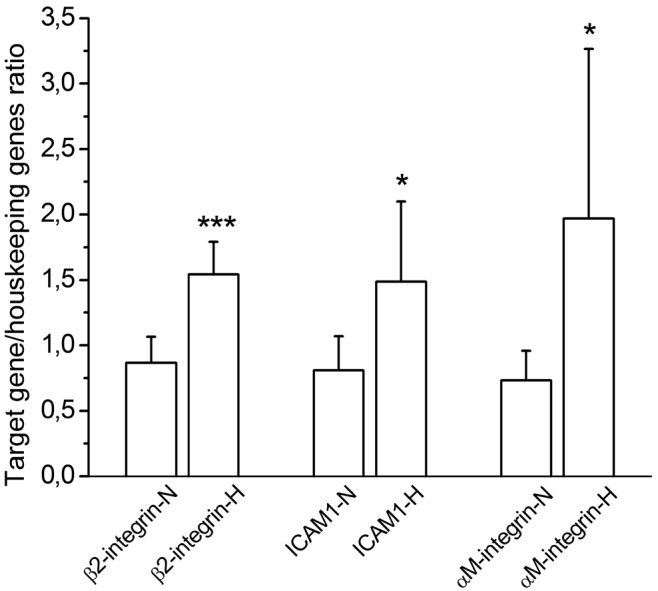
Messenger RNA (mRNA) expression of β2 integrin, intracellular adhesion molecule 1 (ICAM-1), and αM integrin in normoxic (N) and hypoxic (H) rat lung tissue (n = 10 in each group). Data represent mean + SD mRNA expression normalized to geometric mean of mRNA expression of 3 housekeeping genes (GAPDH, β-actin, and HPRT). Triple asterisks indicate P < 0.001 versus normoxic lung and a single asterisk P < 0.02 versus normoxic lung.
Figure 7.
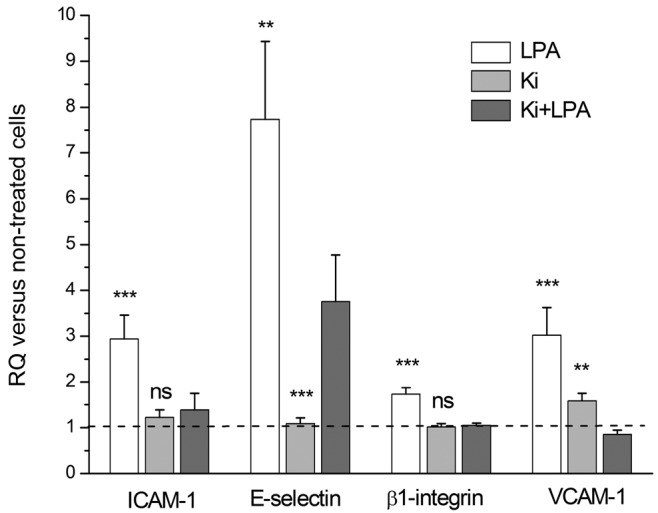
Messenger RNA (mRNA) expression of intracellular adhesion molecule 1 (ICAM-1), E-selectin, β1 integrin, and vascular cell adhesion molecule 1 (VCAM-1) in human pulmonary arterial endothelial cells treated in vitro with 10 μM of lysophosphatidic acid (LPA) and/or 10 μM of Ki16425 (Ki). Data represent mean + SD of mRNA expression normalized to geometric mean of mRNA expression of 3 housekeeping genes (HPRT, UBC, and B2M), in 3 independent experiments. Triple asterisks indicate P < 0.001 versus Ki+LPA, double asterisks P < 0.01 versus Ki+LPA, and “ns” P > 0.05 versus Ki+LPA. RQ: relative quantity.
Discussion
Our results confirm previous studies that showed pulmonary vascular remodeling in rats kept in hypoxia for 3 weeks. Compared with that from the normoxia group of rats, serum obtained from hypoxic rats contained a higher amount of LPA and pulmonary tissue expressed more transcripts and protein of ATX. We also show, in vitro, that serum from hypoxic rats enhanced pulmonary fibroblast migration and the interaction between PAECs and PBMCs. Our PCR analysis suggests that modification of adhesion and migration properties by LPA involves increased expression of genes coding for β2 integrin, ICAM-1, and αM integrin in hypoxic rat lung and genes coding for ICAM-1, E-selectin, β1 integrin, and VCAM-1 in PAECs. The in vitro effects of LPA could be prevented by use of an antagonist of LPA receptors.
Several studies have shown the role of bioactive lipids in mediating vascular physiology and pathophysiology.11,21-23 The major bioactive lipids that act on cell surface receptors are sphingosine-1-phosphate (S-1-P) and LPA, both of which circulate in the blood, but LPA seems to be more potent than S-1-P. LPA production in the circulation largely depends on platelet functioning.24 In this regard, it should be noted that pulmonary arterial hypertension (PAH) patients present endothelial dysfunction leading to the activation of platelets. Platelet activation is also involved in the systemic sclerosis (SSc) leading to vasculopathy, and serum levels of LPA are significantly increased in SSc patients.22 Interestingly, these patients often develop PH. Whether there is a link between establishment of PH and higher circulating LPA concentration remains to be determined. However, PAH patients are under multiple therapies, and unfortunately, measurement of their LPA serum concentration might not be accurate.
Our study was performed with ex vivo rat lung tissue and serum (hypoxic PAH model) and with in vitro normal human cells for specific effects of LPA. Our results with the rat hypoxic model of PAH demonstrate for the first time an increased LPA serum level, compared to that in a control normoxic group. LPA is continuously produced by ATX (lysophospholipase D) in the plasma25 and is directly consumed by cells or degraded by lipid phosphate phosphohydrolase.26 So plasma measurements would not always reflect increased levels of LPA. Serum collection, in our investigation, has been performed in the presence of exogenously added clotting factors to maximally stimulate platelets and coagulation, and the serum level gives the idea of the available total pool, releasable upon increased coagulation or thrombotic events. Accordingly, we suggest that the values reported in our study reflect the maximal possible LPA concentration in rat serum. Immunohistochemical analysis shows higher levels of LPA staining in hypoxic rat lung, which colocalizes with ATX, an enzyme involved in LPA production that promotes cell migration, metastasis, and angiogenesis. In different PH models, mast cells are found around the small pulmonary blood vessels.19,20 These cells have an important role in many inflammatory diseases and are involved in tissue remodeling. We assessed the cellular distribution of ATX in the lung, and our results show a colocalization of ATX and mast cells, similar to what has been observed in the gastrointestinal tract, where mast cells contribute to the production of LPA.27 These results suggest the possible involvement of LPA in hypoxic PAH pathogenesis. Indeed, LPA plays an important role in migration, adhesion, differentiation, and inflammation. These processes are all involved in the PAH pathobiology, and LPA could be implicated in the two current hypotheses of pulmonary remodeling: the “inside-out” hypothesis (interaction between monocytes and leukocytes) and the “outside-in” hypothesis (activation of adventitial fibroblasts) described by Stenmark.18
Differentiation of fibroblasts (activation of fibroblasts into myofibroblasts able to secrete collagen and express α-actin) is critical in the pathogenesis of PAH, but factors leading to these processes are not all identified. We and others have previously determined that LPA participates in the differentiation of lung fibroblasts into myofibroblasts.28-31 We have shown that treatment of fibroblasts with endothelin-1 induced their differentiation into myofibroblasts that express LPA-sensitive chloride channels, involved in cell migration.29 In the present study, we also show that higher numbers of rat pulmonary fibroblasts migrate toward serum obtained from chronically hypoxic rats, compared to serum from control animals. This observation could be explained in part by the increased LPA concentration, since there was no significant difference in cell migration when LPA receptors were blocked.
Several studies have shown, in rat or calf models of PH, an intense pulmonary adventitial remodeling due to recruitment of bone marrow–derived cells that can produce collagen and express smooth muscle cell α-actin (fibrocytes).17,18,32,33 Circulating monocytes are sensitive to LPA, which may shift them toward an activated state.32 Increased serum or local tissue level of LPA could induce the recruitment and adhesion of leukocytes to endothelial cells, allowing leukocyte migration to lead to fibrosis and vascular remodeling.33
Different proteins are involved in the adhesion and migration processes. Integrins play a key role in cell adhesion and cell motility. The ITGAM alpha subunit (CD11b) of integrin αMβ2 is directly involved in causing the adhesion and spreading of cells but cannot mediate cellular migration without the presence of the β2 (CD18) subunit. Integrin αMβ2 belongs to the leukocyte integrins. It is expressed on the surface of many leukocytes, including monocytes; it mediates inflammation by regulating leukocyte adhesion and migration. Perros et al34 showed an increased expression of CD11b in fibrocytes of PAH patients. CD11b, or αM integrin, is expressed on monocytes in an inactive conformation but can be rapidly activated by factors hitherto unknown. These authors hypothesized that the increased expression of CD11b in fibrocytes reflects their activation in the course of inflammatory response, as in patients with PAH.34 We report that hypoxic rat lung presents increased CD11b gene expression. Leukocyte recruitment occurs in several steps initiated by selectins. ICAM-1 and VCAM-1 are endothelial ligands for leukocytes’ β2 integrin, and leukocyte transmigration is dependent on expression of both adhesion receptors. Our results show that these molecules are upregulated by LPA in pulmonary endothelial cells.
In conclusion, in a chronic-hypoxia PH rat model, LPA is increased in both serum and pulmonary tissue. LPA in vitro is capable of inducing fibroblast migration as well as stimulating adhesion of circulating monocytes to endothelial cells. These observations suggest a causative role for LPA in the pathogenesis of hypoxic PAH. Further studies are required to explain how the LPA level changes during this disease.
Acknowledgments
We would like to thank Lpath for the kind gift of the anti-LPA antibody and Barbara Visentin personally for helpful advice on the use of this antibody. We would like also thank Arnaud Goolaerts for his help with animal surgical procedures. We gratefully acknowledge Roger Sabbadini and Sarah Sariban-Sohraby for their critical reading of the manuscript and invaluable comments.
Source of Support: This work was supported by research grant from Pfizer and FRSM (Fonds de la Recherche Scientifique Médicale) grant 3.4637.09.
Conflict of Interest: None declared.
References
- 1.Zhao L. Chronic hypoxia-induced pulmonary hypertension in rat: the best animal model for studying pulmonary vasoconstriction and vascular medial hypertrophy. Drug Disc Today Dis Models 2010;7(3–4):83–88.
- 2.Sylvester JT, Shimoda LA, Aaronson PI, Ward JPT. Hypoxic pulmonary vasoconstriction. Physiol Rev 2012;92(1):367–520. [DOI] [PMC free article] [PubMed]
- 3.Michelakis ED, McMurtry MS, Wu XC, Dyck JRB, Moudgil R, Hopkins TA, Lopaschuk GD, Puttagunta L, Waite R, Archer SL. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels. Circulation 2002;105(2):244–250. [DOI] [PubMed]
- 4.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 2006;99(7):675–691. [DOI] [PubMed]
- 5.O’Brodovich HM, Andrew M, Gray GW, Coates G. Hypoxia alters blood coagulation during acute decompression in humans. J Appl Physiol 1984;56(3):666–670. [DOI] [PubMed]
- 6.Dunleavy M, Dooley M, Cox D, Bradford A. Chronic intermittent asphyxia increases platelet reactivity in rats. Exp Physiol 2005;90(3):411–416. [DOI] [PubMed]
- 7.von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res 2007;100(1):27–40. [DOI] [PubMed]
- 8.Jurk K, Kehrel BE. Platelets: physiology and biochemistry. Semin Thromb Hemost 2005;31(4):381–392. [DOI] [PubMed]
- 9.Berger G, Azzam ZS, Hoffman R, Yigla M. Coagulation and anticoagulation in pulmonary arterial hypertension. Isr Med Assoc J 2009;11(6):376–379. [PubMed]
- 10.Cheng HY, Dong A, Panchatcharam M, Mueller P, Yang F, Li Z, Mills G, Chun J, Morris AJ, Smyth SS. Lysophosphatidic acid signaling protects pulmonary vasculature from hypoxia-induced remodeling. Arterioscler Thromb Vasc Biol 2012;32(1):24–32. [DOI] [PMC free article] [PubMed]
- 11.Tager AM, LaCamera P, Shea BS, Campanella GS, Selman M, Zhao Z, Polosukhin V, et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med 2008;14(1):45–54. [DOI] [PubMed]
- 12.Pradère JP, Gonzalez J, Klein J, Valet P, Grès S, Salant D, Bascands J-L, Saulnier-Blache J-S, Schanstra JP. Lysophosphatidic acid and renal fibrosis. Biochim Biophys Acta Mol Cell Biol Lipids 2008;1781(9):582–587. [DOI] [PMC free article] [PubMed]
- 13.Rubenfeld J, Guo J, Sookrung N, Chen R, Chaicumpa W, Casolaro V, Zhao Y, Natarajan V, Georas S. Lysophosphatidic acid enhances interleukin-13 gene expression and promoter activity in T cells. Am J Physiol Lung Cell Mol Physiol 2006;290(1):L66–L74. [DOI] [PubMed]
- 14.Lin CI, Chen CN, Lin PW, Chang KJ, Hsieh FJ, Lee H. Lysophosphatidic acid regulates inflammation-related genes in human endothelial cells through LPA1 and LPA3. Biochem Biophys Res Commun 2007;363(4):1001–1008. [DOI] [PubMed]
- 15.Tang N, Zhao Y, Feng R, Liu Y, Wang S, Wei W, Ding Q, An M, Wen J, Li L. Lysophosphatidic acid accelerates lung fibrosis by inducing differentiation of mesenchymal stem cells into myofibroblasts. J Cell Mol Med 2014;18(1):156–169. [DOI] [PMC free article] [PubMed]
- 16.Shao DD, Suresh R, Vakil V, Gomer RH, Pilling D. Pivotal advance: Th-1 cytokines inhibit, and Th-2 cytokines promote fibrocyte differentiation. J Leukocyte Biol 2008;83(6):1323–1333. [DOI] [PMC free article] [PubMed]
- 17.Stenmark KR, Frid MG, Yeager ME. Fibrocytes: potential new therapeutic targets for pulmonary hypertension? Eur Respir J 2010;36(6):1232–1235. [DOI] [PubMed]
- 18.Stenmark KR, Frid MG, Yeager M, Li M, Riddle S, McKinsey T, El Kasmi KC. Targeting the adventitial microenvironment in pulmonary hypertension: a potential approach to therapy that considers epigenetic change. Pulm Circ 2012;2(1):3–14. [DOI] [PMC free article] [PubMed]
- 19.Farhar S, Sharp J, Asosingh K, Park M, Comhair SAA, Tang WHW, Thomas J, et al. Mast cell number, phenotype, and function in human pulmonary arterial hypertension. Pulm Circ 2012;2(2):220–228. [DOI] [PMC free article] [PubMed]
- 20.Maxová H, Herget J, Vízek M. Lung mast cells and hypoxic pulmonary hypertension. Physiol Res 2012;61(1):1–11. [DOI] [PubMed]
- 21.van Nieuw Amerongen GP, Vermeer MA, van Hinsbergh VWM. Role of RhoA and Rho kinase in lysophosphatidic acid–induced endothelial barrier dysfunction. Arterioscler Thromb Vasc Biol 2000;20(12):e127–e133. [DOI] [PubMed]
- 22.Tokumura A, Carbone LD, Yoshioka Y, Morishige J, Kikuchi M, Postlethwaite A, Watsky MA. Elevated serum levels of arachidonoyl-lysophosphatidic acid and sphingosine 1-phosphate in systemic sclerosis. Int J Med Sci 2009;6(4):168–176. [DOI] [PMC free article] [PubMed]
- 23.Smyth SS, Cheng H-Y, Miriyala S, Panchatcharam M, Morris AJ. Roles of lysophosphatidic acid in cardiovascular physiology and disease. Biochim Biophys Acta Mol Cell Biol Lipids 2008;1781(9):563–570. [DOI] [PMC free article] [PubMed]
- 24.Aoki J, Taira A, Takanezawa Y, Kishi Y, Hama K, Kishimoto T, Mizuno K, Saku K, Taguchi R, Arai H. Serum lysophosphatidic acid is produced through diverse phospholipase pathways. J Biol Chem 2002;277(50):48737–48744. [DOI] [PubMed]
- 25.Albers HMHG, Dong A, van Meeteren LA, Egan DA, Sunkara M, van Tilburg EW, Schuurman K, et al. Boronic acid-based inhibitor of autotaxin reveals rapid turnover of LPA in the circulation. Proc Natl Acad Sci USA 2010;107(16):7257–7262. [DOI] [PMC free article] [PubMed]
- 26.Tomsig JL, Snyder AH, Berdyshev EV, Skobeleva A, Mataya C, Natarajan V, Brindley DN, Lynch KR. Lipid phosphate phosphohydrolase type 1 (LPP1) degrades extracellular lysophosphatidic acid in vivo. Biochem J 2009;419(3):611–618. [DOI] [PMC free article] [PubMed]
- 27.Mori K, Kitayama J, Aoki J, Kishi Y, Shida D, Yamashita H, Arai H, Nagawa H. Submucosal connective tissue-type mast cells contribute to the production of lysophosphatidic acid (LPA) in the gastrointestinal tract through the secretion of autotaxin (ATX)/lysophospholipase D (lysoPLD). Virchows Arch 2007;451(1):47–56. [DOI] [PubMed]
- 28.Watsky MA, Weber KT, Sun Y, Postlewaite A. New insights into the mechanism of fibroblast to myofibroblast transformation and associated pathologies. Int Rev Cell Mol Biol 2010;282:165–192. [DOI] [PubMed]
- 29.Shlyonsky V, Ben Soussia I, Naeije R, Mies F. Opposing effects of bone morphogenetic protein-2 and endothelin-1 on lung fibroblast chloride currents. Am J Respir Cell Mol Biol 2011;45(6):1154–1160. [DOI] [PubMed]
- 30.Yin Z, Watsky MA. Chloride channel activity in human lung fibroblasts and myofibroblasts Am J Physiol Lung Cell Mol Physiol 2005;288(6):L1110–L1116. [DOI] [PubMed]
- 31.Yin Z, Tong Y, Zhu H, Watsky MA. ClC-3 is required for LPA-activated Cl− current activity and fibroblast-to-myofibroblast differentiation. Am J Physiol Cell Physiol 2008;294(2):C535–C542. [DOI] [PubMed]
- 32.Fueller M, Wang DA, Tigyi G, Siess W. Activation of human monocytic cells by lysophosphatidic acid and sphingosine-1-phosphate. Cell Signal 2003;15(4):367–375. [DOI] [PubMed]
- 33.Vestweber D. Adhesion and signaling molecules controlling the transmigration of leukocytes through endothelium. Immunol Rev 2007;218:178–196. [DOI] [PubMed]
- 34.Gambaryan N, Cohen-Kaminsky S, Montani D, Girerd B, Huertas A, Seferian A, Humbert M, Perros F. Circulating fibrocytes and pulmonary arterial hypertension. Eur Respir J 2012;39(1):210–222. [DOI] [PubMed]