

Abstract Abstract
Initiation, progression, and resolution of vaso-occlusive pain episodes in sickle cell disease (SCD) have been recognized as reperfusion injury, which provokes an inflammatory response in the pulmonary circulation. Some 5-lipoxygenase (5-lox) metabolites are potent vasoconstrictors in the pulmonary circulation. We studied stimulation of production of the inflammatory eicosanoids leukotrienes (LTs) and prostaglandin E2 (PGE2) by isolated rat lungs perfused with sickle (HbSS) erythrocytes. Our hypothesis is that HbSS erythrocytes produce more LTs than normal (HbAA) erythrocytes, which can induce vaso-occlusive episodes in SCD patients. Lung perfusates were collected at specific time points and purified by high-pressure liquid chromatography, and LTC4 and PGE2 contents were measured by enzyme-linked immunosorbent assay (ELISA). Rat lung explants were also cultured with purified HbAA and HbSS peptides, and 5-lox, cyclooxygenase 1/2, and platelet-activating factor receptor (PAFR) proteins were measured by Western blotting, while prostacyclin and LTs produced by cultured lung explants were measured by ELISA. Lung weight gain and blood gas data were not different among the groups. HbSS-perfused lungs produced more LTC4 and PGE2 than HbAA-perfused lungs: 10.40 ± 0.62 versus 0.92 ± 0.2 ng/g dry lung weight (mean ± SEM; P = 0.0001) for LTC4. Inclusion of autologous platelets (platelet-rich plasma) elevated LTC4 production to 12.6 ± 0.96 and 7 ± 0.60 ng/g dry lung weight in HbSS and HbAA perfusates, respectively. HbSS lungs also expressed more 5-lox and PAFR. The data suggest that HbSS erythrocytes and activated platelets in patient’s pulmonary microcirculation will enhance the synthesis and release of the proinflammatory mediators LTC4 and PGE2, both of which may contribute to onset of the acute chest syndrome in SCD.
Keywords: pulmonary circulation, 5-lipoxygenase, leukotriene receptors, PAF receptor, cyclooxygenase, prostaglandins
Introduction
Eicosanoids, products of arachidonic acid if synthesized and released in abnormal amounts, increase pulmonary vascular pressure and promote pulmonary edema.1,2 Leukotriene (LT)B4 and the cysteinyl leukotrienes (CystLTs), such as LTC4, are produced in rat lung during hypoxia and can induce pulmonary hypertension.3 CystLTs such as LTC4, LTD4, and LTE4 are produced by alveolar macrophages, basophils, eosinophils, and mast cells as well as by lung vascular endothelium and lung parenchyma.4-7 Stimulation of inflammatory cells results in the conversion of arachidonic acid to 5-hydroperoxyeicosatetraenoic acid(5-HPETE) and then to the epoxide LTA4 through the joint activities of the non-heme iron dioxygenase 5-lipoxygenase and 5-lipoxygenase-activating protein. LTA4 is further transformed to LTB4 by LTA4 hydrolase. LTC4 synthase further transforms LTA4 to LTC4, which can be metabolized to LTD4 by γ-glutamyl-transpeptidase and to LTE4 by cysteinyl-glycine dipeptidase. LTC4 and other CystLTs function through their respective receptors, CystLT1 and CystLT2, to induce their physiologic responses (Figure 1). CystLT receptors mediate a range of proinflammatory effects, such as increased endothelial membrane permeability leading to plasma exudation and pulmonary edema; bronchoconstriction; and vascular smooth muscle contraction of the pulmonary microvasculature.4-7 LTC4 synthase has been measured in human lung membranes and human platelet homogenates.8,9
Figure 1.

Scheme of leukotriene biosynthesis and physiological effects in the lung. Arachidonic acid released by action of phospholipase A2 (PLA2) is acted upon by 5-lipoxygenase (5-Lox) producing 5-HPETE (5-hydroperoxyeicosatetraenoic acid), which is metabolized to various leukotrienes (LT), including LTC4, which binds to its CystLT1 receptors to evoke some physiological responses, including inflammation and vasoconstriction. FLAP: 5-Lox activating protein; 5-HETE: 5-hydroxyeicosatetraenoic acid; CystLT: cysteinyl leukotriene.
Sickle cell disease (SCD) chronic lung disease and the acute chest syndrome are two of the notable causes of death in adult patients with sickle cell disease.10-12 Some lung complications in SCD result from direct or indirect effects of pulmonary microvascular occlusion by sickled erythrocytes and their adhesion to the vascular endothelium,13,14 leading to release of potent vasoactive molecules, including prostacyclin, thromboxane A2, and LTC4.15,16 Indeed, the initiation, progression, and resolution of vaso-occlusive pain episodes due to red blood cell sickling and adhesion to endothelium in the affected tissues have been characterized as “reperfusion injury.”17 Homozygous hemoglobin SS (HbSS) disease patients in steady state have higher circulating thromboxane B2 and LTC4 than do control (HbAA) individuals, suggesting a greater expression and activity of LTC4 synthase.18 However, there is no experimental evidence indicating a direct effect of sickle erythrocyte production of 5-lipoxygenase (5-lox) metabolites from the lung vasculature. Furthermore, the role and significance of the leukotrienes as inflammatory mediators in the pathogenesis of acute or chronic lung disease in SCD patients are not well understood. We employed an in vitro model of isolated, perfused rat lung to investigate the effect of the interaction of sickle erythrocytes with the pulmonary microvasculature on the production of LTC4. Also, using purified HbSS and HbAA peptides, we studied expression of some inflammatory proteins by adult rat lungs stimulated with these peptides. We hypothesized that HbSS erythrocytes, compared to control HbAA erythrocytes, will augment synthesis and release of LTC4 and that HbSS peptide will stimulate expression of inflammatory proteins while downregulating expression of anti-inflammatory proteins by the rat lungs.
Methods
Blood samples from patients and volunteers
The study was approved by the Institutional Review Board of the Committee on Human Research of the Los Angeles Biomedical Research Institute at Harbor-UCLA Medical Center, Torrance, California, and informed consent was obtained from all participants. Venous blood samples were obtained from 6 healthy control subjects (students and laboratory staff) with HbAA, aged 18–39 years, and 12 adult SCD (HbSS disease) patients, aged 18–39 years, who were in steady state. The patients had no manifestation of painful episodes and had not been hospitalized in the 6 months before the study. Also, the patients had no documented infection at the time of study, had not received a blood transfusion within the preceding 2 months, and were not receiving hydroxyurea therapy. The study subjects were not on any nonsteroidal anti-inflammatory medications. Free-flowing venous blood was withdrawn with minimum trauma into 3.8% sodium citrate solution.
Preparation of erythrocytes and platelet-rich plasma
Platelet-rich plasma (PRP) was prepared from the blood of every participant by centrifugation at 150 g for 10 minutes at 37°C. The PRP was pipetted into a polypropylene plastic tube prerinsed with 13% acid-citrate dextrose. The buffy coat of white blood cells was removed carefully and discarded.
The red blood cells of control HbAA or SCD HbSS participants were resuspended in Krebs bicarbonate buffer, pH 7.4, containing 119 mM NaCl, 4.7 mM KCl, 22.6 mM NaHCO3, 1.17 mM MgSO4, 1.18 mM KH2PO4, 1.6 mM CaCl2, 5.5 mM glucose, and 1% bovine serum albumin (BSA); layered onto 3 mL of Ficoll-Paque (LKB-Pharmacia, San Francisco); and centrifuged at 360 g for 15 minutes to remove extra platelets, mononuclear cells, and granulocytes. The erythrocytes were then washed by resuspension in 5 volumes of phosphate-buffered saline (PBS) containing 131 mM NaCl, 5.1 mM Na2HPO4, 1.5 mM KH2PO4, 1% BSA, and 8.3 mM glucose (GPBS); centrifuged at 200 g for 10 minutes to remove the Ficoll-Hypaque; and then resuspended to a hematocrit of 7% in 40–50 mL of GPBS. The erythrocyte suspension was then transferred to a recirculating perfusion circuit maintained at 37°C, as reported previously.19
Isolation of rat lungs
Lungs of adult rats, body weight 350–450 g, were isolated, cannulated, and perfused as previously described.19 Briefly, the rats were sedated with intramuscular injection of ketamine, 25 mg/kg, after which anesthesia was supplemented with pentobarbital sodium (25 mg/kg) via the femoral vein. An endotracheal tube was inserted into the trachea and tied, and the rat was allowed to breathe 100% oxygen spontaneously. After infusion with heparin sodium (1,000 IU/kg), an overdose of pentobarbital sodium (100 mg/kg) was given intravenously. The superior and inferior venae cavae were ligated, and the heart and lung were removed en bloc via a midline sternotomy. Cannulas filled with saline were placed into the main pulmonary artery and left atrium via the right and left ventricles, respectively, being careful that no air bubbles entered the pulmonary artery. Cannulated lungs were placed on their dorsal surfaces on a moistened heating pad and covered with a humidified, nonevacuated Plexiglas chamber. The lungs were then ventilated via the tracheal cannula with a gas mixture containing 30% O2, 8% CO2, and the balance N2, using an anesthesia bag (INTEC, Englewood, CO), and washed with a Masterflex roller pump at 50 mL/kg/min in a non-recirculating system with PBS, pH 7.4, via the pulmonary artery cannula until the effluent was clear of the rats’ red blood cells and the lung appeared bleached.
The washed lungs were then connected to the perfusion circuit, which included a Harvard rat ventilator, and ventilated at 25 breaths/min, with a tidal volume of 2.5 mL and an end-expiratory pressure of 5 cmH2O, with a gas mixture of 30% O2, 8% CO2, and the balance N2. All lungs were ventilated with peak airway pressure between 15 and 25 cmH2O and kept distended at a constant airway pressure of 7 cmH2O. Other components of the perfusion circuit were the Masterflex roller pump that pumped the perfusate, a physiograph to determine pressure, and the calibrated perfusion reservoir. The perfusate comprised the GPBS and a suspension of HbAA erythrocytes or a suspension of HbSS erythrocytes, perfused into the pulmonary artery at 40 mL/kg/min through a bubble trap placed in a heat exchanger. To abolish prostanoid production by active vasomotion of the rat lung during perfusion, papaverine (70 mg/mL) was added to the perfusion reservoir to paralyze the lungs.19 The perfusate drained from the left atrium into a calibrated venous reservoir that contained the perfusate. The pulmonary arterial pressure was controlled by adjusting the flow rate and left atrial pressure by the height of the venous reservoir. By adjustment of the inflow, outflow, and airway pressures, the lungs were maintained and perfused in zone 3 conditions.
Determination of lung pressure
All lungs were perfused at a constant pressure. However, pulmonary arterial and left atrial pressures were monitored continuously via small polyethylene tubing (PE60) placed in the main pulmonary artery and the left atrium and connected to pressure transducers (Gould Statham P23). A data acquisition board (DT2821; Data Translation, Marlboro, MA) was used for analog input and digital output. In the course of the experiment, pulmonary arterial and left atrial pressure signals were simultaneously digitized with a data acquisition software package, Global Lab (Data Translation), at 500 samples per second and displayed on a monitor. Once the data were collected and stored on the computer’s hard drive, they were retrieved and analyzed with another signal-processing software package, DADiSP (DSP Development, Cambridge, MA). In all instances, the pulmonary arterial and left atrial pressures did not change during the duration of the perfusion study. Airway pressure was also measured continuously with a water manometer. The zero reference level for vascular pressures was the base of the lung.
Experimental design
The experimental animals were divided into 3 groups. The lungs of each group were perfused as described below.
Experiment 1 (group 1; n = 6). Lungs were perfused at a flow rate of 40 mL/kg/min with the GPBS buffer for 15 minutes, followed by addition of 15 mL of GPBS, equivalent to the volume of PRP used in groups 2 and 3, and then perfusion continued for 15 more minutes. The flow rate was then increased to 80 mL/kg/min, and the perfusion continued for another 15 minutes.
Experiment 2 (group 2; n = 6). Lungs were perfused at a flow rate of 40 mL/kg/min with control HbAA erythrocytes washed and resuspended in GPBS at a hematocrit (Hct) of 7% for 15 minutes, followed by addition of 15 mL of PRP, and then perfusion continued for 15 more minutes. The flow rate was then increased to 80 mL/kg/min, and the perfusion continued for another 15 minutes.
Experiment 3 (group 3; n = 12). Lungs were perfused at a flow rate of 40 mL/kg/min with HbSS erythrocytes washed and resuspended in GPBS at a Hct of 7% for 15 minutes, followed by addition of 15 mL of PRP, and then perfusion continued for 15 more minutes. The flow rate was then increased to 80 mL/kg/min, and the perfusion continued for another 15 minutes to assess the effect of increased rheology on erythrocytes in the microcirculation.
The pH, carbon dioxide and oxygen partial pressures (pco2 and po2, respectively), and dextrose concentration of the perfusates were monitored at regular intervals and adjusted to physiological values with NaHCO3, by bubbling O2 and/or adding dextrose if necessary. At the end of each perfusion, the volume of perfusate was determined, the exact flow rate was determined, and the change in lung weight was calculated.
Sample collection
One-milliliter samples of the perfusing fluids were withdrawn from the left atrial cannula into aspirin-treated tubes at the following time points: 0 minutes (baseline) and 15 minutes later; 0 (baseline) and 15 minutes after addition of PRP; and finally 15 minutes after the 2-fold increase of the perfusion flow rate. The baseline sample was collected immediately after connecting the lung to the perfusion circuit but before the flow rate was set at the desired value of 40 mL/kg/min. In order to establish consistency for the sampling, the 0-minute time points were taken to be 60 seconds after the initiation of flow and 60 seconds after the addition of PRP. Each sample was spun at 1,500 g at 4°C for 10 minutes. The supernatants were transferred into fresh tubes, frozen in liquid nitrogen, and stored at −80°C. Samples were usually analyzed within 4 weeks. Storage at −80°C for 4 weeks did not alter the amount of arachidonic acid metabolites measured from each sample.
Metabolite extraction, high-pressure liquid chromatography (HPLC), and enzyme-linked immunosorbent assay (ELISA) for LTC4 and prostaglandin E2 (PGE2)
Samples were extracted for 5-lox products before purification by HPLC and quantification by ELISA, as previously reported.15,19 The ELISA kits were purchased from Neogen (Louisville, KY). Curve fitting of the standard and calculation of the amount of metabolite were performed with a weighted nonlinear ELISA program. Final results of metabolite concentration were corrected for recovery, normalized to the volume of perfusate at the time of sample collection, with the baseline value subtracted from each time point, and the concentration was expressed as nanograms of metabolite per gram of lung dry weight.
In vitro studies with HbAA and HbSS peptides on lung explants
Stimulation of protein expression by HbAA and HbSS peptides
Lung explants of adult rats (350 ± 25 g) were isolated, and the vasculature was washed to remove rat blood components by perfusion with PBS. Washed lungs were cut into 0.3–0.5-mg wet weight sections; treated separately with 1 μg/mL normal hemoglobin A2 (HbAA peptide; Sigma-Aldrich, catalog no. H0267) and sickle cell hemoglobin S (HbSS peptide; Sigma-Aldrich catalog no. H0392), each dissolved in Krebs bicarbonate buffer, pH 7.4, supplemented with 5% serum; and incubated for 24 hours in an incubator aerated with 5% CO2 in air. Controls were lung explants cultured with the Krebs buffer alone. Lung tissues were homogenized with 40 mM HEPES buffer, pH 7.4, containing protease inhibitors.20,12 Proteins were subjected to Coomassie blue analysis before being used for sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), as we have reported.21 Protein expression was quantified against β-actin expression.
Stimulation of eicosanoid production by stimulated lungs
Lung explants were incubated for 4 hours in a 5% CO2 incubator. Control lung explants were incubated in culture buffer alone or with 1.0 μM of calcium ionophore A23187, a phospholipase A2 activator, so as to enable comparison of the effects of HbAA and HbSS peptides on arachidonic acid release from membrane phospholipids. Metabolites were purified by HPLC, measured by ELISA, and normalized to protein concentration.
Western blotting/SDS-PAGE
SDS-PAGE was performed on 4%–12% Tris-glycine gradient gels (Lonza, Rockland, ME), as previously reported.21 Membranes were probed for 5-lox, platelet-activating factor receptor (PAFR), cyclooxygenase (COX)1, and COX2 with anti-5-lox antibody (a generous donation by Dr. Peters-Golden, University of Michigan). Antibodies to PAFR (polyclonal), COX1, and COX2 were purchased from Cayman Chemical (Ann Arbor, MI), with an anti-rabbit Ig (immunoglobulin) HRP (horseradish peroxidase)-linked secondary antibody. Signals were captured with Amersham ECL Western blot detection kit on X-ray film. Each protein was quantified against expression of β-actin standard.
Data analysis
The numerical data are reported as means ± SD for blood gas indexes, but amounts of metabolites measured are reported as means ± SEM of rat dry lung weight. To compare data within groups—for example, 0 minute, 15 minute, etc.—a paired repeated-measures statistic was used, applying Dunn’s post hoc test. Data from HbSS erythrocytes or peptides were compared with data from HbAA erythrocytes or peptides and data from GPBS perfusions, using Student’s t test and ANOVA with the Tukey post hoc test. Differences were considered significant at P < 0.05.
Results
Characteristics of patients with SCD and normal control volunteers
Mean age, weight, hemoglobin density, platelet volume, platelet and white blood cell counts, and Hct of the SCD patients and normal controls are shown in Table 1. The ages of the SCD patients and normal controls and the platelet counts were not different; however, the weight of SCD patients and their hemoglobin density, platelet volume, and Hct were less than those of the normal controls, while the white blood cell count of the SCD patients was higher than that of the normal controls.
Table 1.
Characteristics of patients with sickle cell disease and normal control volunteers
| Subjects | Age, years | Weight, kg | Hb density, g/dL | MPV | Plt count, ×109/L | Hct, % | WBC, ×106/L |
|---|---|---|---|---|---|---|---|
| Controls (HbAA) | 33.5 ± 6.8 | 79.7 ± 3.1 | 13.1 ± 0.9 | 11.3 ± 1.8 | 199.3 ± 29.5 | 40.3 ± 2.8 | 5.7 ± 0.68 |
| Sickle cell patients (HbSS) | 32.8 ± 3.4 | 50.1 ± 3.4* | 7.2 ± 0.6* | 8.7 ± 0.1 | 206.5 ± 2.6 | 20.2 ± 2.0* | 8.1 ± 1.5* |
All data are expressed as means ± SEM. There was no difference in age and platelet (Plt) count between the groups. However, the weight, hemoglobin (Hb) density, mean platelet volume (MPV), hematocrit (Hct), and white blood cell (WBC) count were different.
P < 0.05.
Acid-base status and lung weight during the perfusion
The blood-gas indexes (mean ± SD) of perfusates at time of sample withdrawal were pH of 7.38 ± 0.09, pco2 of 37 ± 10 Torr, and po2 of 105 ± 16 Torr for the 24 lungs studied (Table 2). All lungs had gained weight by the end of the perfusion. However, lungs that developed airway edema during the study were not included in the data analyses presented here. Weight gain (mean ± SEM) was as follows: GPBS lungs: 176% ± 44%, n = 6 (range: 100%–400%); HbAA erythrocyte lungs: 156% ± 47%, n = 6 (range: 30%–300%); and HbSS erythrocytes lungs: 160% ± 32%, n = 12 (range: 20%–300%). There was no difference in percent weight gain between the 3 groups of lungs (GPBS vs. HbAA, P = 0.77; GPBS vs. HbSS, P = 0.81; HbAA vs. HbSS, P = 0.95), but GPBS lungs appeared to gain more weight than either HbAA- or HbSS-erythrocyte lungs (Table 2).
Table 2.
Weight gain in isolated perfused rat lungs, lung pressure, and averaged acid-base indexes of the effluents at the beginning and end of perfusion
| Perfusate | N | Lung weight, % change | pH | pco2, mmHg | po2, mmHg |
|---|---|---|---|---|---|
| GPBS | 6 | 176 ± 36 | 7.38 ± 0.09 | 37 ± 10 | 105 ± 16 |
| HbAA RBCs | 6 | 156 ± 38 | 7.33 ± 0.06 | 39 ± 8 | 110 ± 12 |
| HbSS RBCs | 12 | 160 ± 26 | 7.41 ± 0.05 | 38 ± 7 | 114 ± 12 |
Data are means ± SD. All the lungs gained weight at the end of the perfusion. Lungs that developed airway edema during the study were discarded. There was no significant difference in the percent gain in weight among the 3 groups of lungs. GPBS-perfused lungs tended to gain a little more weight than lungs perfused with hemoglobin SS (HbbSS) or control hemoglobin AA (HbAA) erythrocytes; GPBS versus HbAA: P = 0.77; GPBS versus HbSS: P = 0.81; HbAA versus HbSS: P = 0.95. pco2: partial pressure of CO2; po2: partial pressure of O2; GBPS: phosphate-buffered saline containing 131 mM NaCl, 5.1 mM Na2HPO4, 1.5 mM KH2PO4, 1% bovine serum albumin, and 8.3 mM glucose; RBCs: red blood cells.
HbSS erythrocytes stimulate greater LTC4 production by perfused lungs
Amounts of all metabolites (LTC4 and PGE2) measured from the perfusates are shown as means ± SEM in nanograms per gram lung dry weight. Changes in LTC4 levels produced with GPBS buffer alone and with buffer plus HbAA or HbSS erythrocytes are shown in Figure 2 and Table 3. At the baseline (0 minutes), production of LTC4 by lungs perfused with GPBS was 2.9 ± 0.3. During perfusion with GPBS, the amount of LTC4 measured was not different from that from the initial 15 minutes of perfusion. At 15 minutes, the amount of LTC4 in GPBS-perfused lungs was 3.15 ± 0.40, which is not different from the value at the baseline (P = 0.85).
Figure 2.
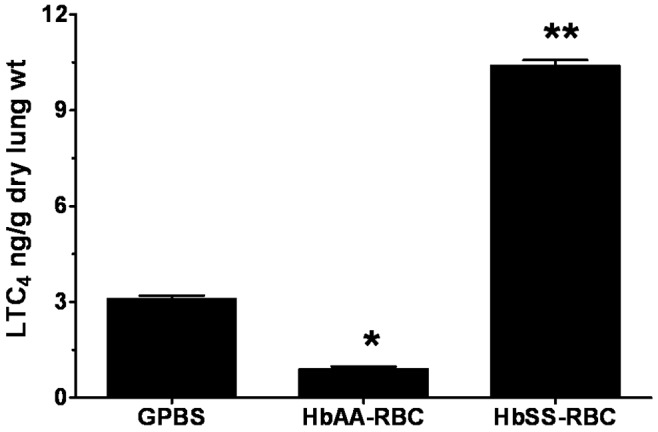
Profile of leukotriene C4 (LTC4) produced by isolated rat lungs perfused with buffer alone or with erythrocytes, with metabolites measured at 15 minutes of perfusion. Three separate groups of lungs were perfused with buffer (GPBS; n = 6), normal erythrocytes (HbAA-RBC; n = 6), or sickle cell erythrocytes (HbSS-RBC; n = 12). Perfusates were collected as described in “Methods,” and the LTC4 was extracted, purified by high-pressure liquid chromatography, and measured by enzyme-linked immunosorbent assay. Data are means + SEM. A single asterisk indicates significant (P < 0.05) difference from LTC4 production in GPBS control; double asterisks indicate significant (P < 0.05) difference from both GPBS controls and the HbAA-RBC group. GPBS: phosphate-buffered saline containing 131 mM NaCl, 5.1 mM Na2HPO4, 1.5 mM KH2PO4, 1% bovine serum albumin, and 8.3 mM glucose.
Table 3.
Percent change in leukotriene LTC4 levels, relative to baseline levels, measured in effluents of isolated rat lung perfused with GPBS, HbAA-RBCs, and HbSS-RBCs and the perfusates combined with autologous platelet-rich plasma (PRP)
| Perfusate | 15 min | +PRP, 0 min | +PRP, 15 min | +PRP, 30 min |
|---|---|---|---|---|
| GPBS | 120 | 41 | 12 | 24 |
| HbAA RBCs | 29.5 | 27.9 | −63.9 | −68.9 |
| HbSS RBCs | 63.8 | 67.5 | 77.4 | 103.1 |
Measurements were taken after 15 minutes of perfusion with perfusate alone and 3 times after addition of PRP: immediately (0 min), 15 minutes later (15 min), and after 15 more minutes, during which the perfusion rate was twice the initial rate (30 min); see “Experimental design.” GBPS: phosphate-buffered saline containing 131 mM NaCl, 5.1 mM Na2HPO4, 1.5 mM KH2PO4, 1% bovine serum albumin, and 8.3 mM glucose; HbAA: hemoglobin AA; HbSS: hemoglobin SS; RBCs: red blood cells.
Perfusion with HbAA erythrocytes produced relatively very low and consistent amounts of LTC4 during the initial 15 minutes, 0.92 ± 0.22. In contrast, 15 minutes after the perfusion was initiated, the lungs perfused with HbSS erythrocytes produced 10.40 ± 0.62 of LTC4, more than 10 times the amount produced by HbAA erythrocytes and about 3 times the amount produced by the GPBS buffer alone. LTC4 production by isolated lungs perfused with HbSS erythrocytes during the initial 15 minutes was different from that of either GPBS- or HbAA-perfused lungs (P < 0.0001; Figure 2).
Combined perfusion of erythrocytes and autologous PRP increased LTC4 production
LTC4 production by lungs perfused with GPBS buffer alone, a mixture of GPBS buffer plus platelets, or a mixture of HbSS or HbAA erythrocytes plus autologous PRP is shown in Figure 3. All data are shown as means ± SEM in nanograms per gram lung dry weight. Perfusion of lungs with GPBS alone increased LTC4 production from the initial 3.1 ± 0.40 to 4.4 ± 0.72. However, after 15 minutes of continuous perfusion with this mixture, LTC4 production decreased to 1.5 ± 0.32 (Figure 3). Perfusion of lungs with a mixture of GPBS plus platelets (GPBS+Plts) produced a nonsignificant increase in LTC4 production both at 0 minutes (5.3 ± 1.3) and at 15 minutes (2.0 ± 0.5) after perfusion, compared to effect of GPBS alone. Likewise, coperfusion with the mixture of HbAA erythrocytes and autologous PRP increased LTC4 production from the initial 0.92 ± 0.22 to 1.8 ± 0.61. After 15 minutes of continuous perfusion with the mixture of HbAA and autologous PRP—LTC4 produced 15 minutes after addition of autologous PRP, i.e., HbAA + PRP (P = 0.12; AA-RBC-Plts in Figure 3)—the amount of LTC4 produced did not change (1.7 ± 0.53). In contrast, perfusion of lungs for 15 minutes with the mixture of HbSS erythrocytes and autologous PRP increased LTC4 production, compared to HbAA lungs with same treatment (12.58 ± 0.96 vs. 1.7 ± 0.53; P = 0.0001). LTC4 production by the mixture of HbSS erythrocytes and autologous PRP did not change during the 15-minute perfusion, compared to amount produced at the beginning of the perfusion (12.58 ± 0.96 vs. 11.58 ± 0.60; P = 0.309; Figure 3). The lack of significant increase in LTC4 production after perfusion with GPBS+Plts suggests that the interaction of HbSS erythrocytes and autologous PRP is necessary to activate the platelets and thence stimulate more LTC4 production.
Figure 3.
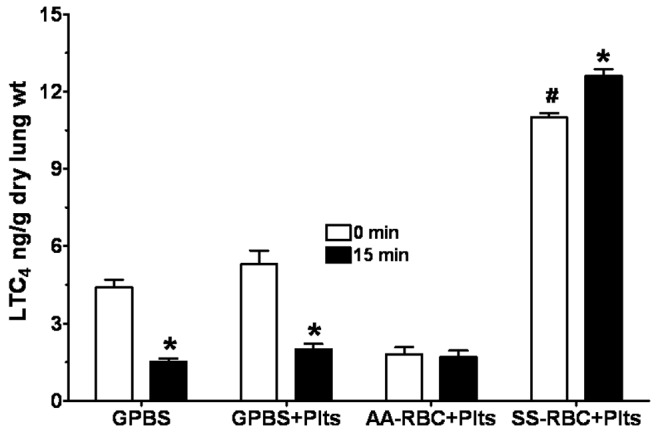
Profile of leukotriene C4 (LTC4) produced by isolated rat lungs perfused with GPBS or with HbAA (AA) or HbSS (SS) erythrocytes plus autologous platelets (Plts), measured at 0 and 15 minutes of perfusion. See Figure 2 for experimental details and other abbreviations. Data are means + SEM. An asterisk indicates significant (P < 0.05) difference from LTC4 production in the same group at 0 minutes. A pound sign (#) indicates significant (P < 0.05) difference from LTC4 production in all other groups.
Increasing the perfusion rate with mixtures of erythrocytes and autologous PRP to 80 mL/kg/min augments LTC4 production by perfused lungs
The effect on LTC4 production of increasing perfusion rate to 80 mL/kg/min is shown in Figure 4. Perfusion of GPBS at 80 mL/kg/min increased the production of LTC4 by 80% over production at 40 mL/kg/min. As was observed with the 15-minute perfusion with GPBS+Plts at 40 mL/kg/min, perfusion with GPBS+Plts produced a nonsignificant increase in LTC4 production at 80 mL/kg/min. Perfusion with the mixture of HbAA erythrocytes and autologous PRP decreased LTC4 measured in effluents at 80 mL/kg/min. In contrast, perfusion with the mixture of HbSS erythrocytes and autologous PRP increased LTC4 measured at 80 mL/kg/min compared to that at 40 mL/kg/min, 16.8 ± 3.1 versus 12.6 ± 0.96 (Figure 4).
Figure 4.
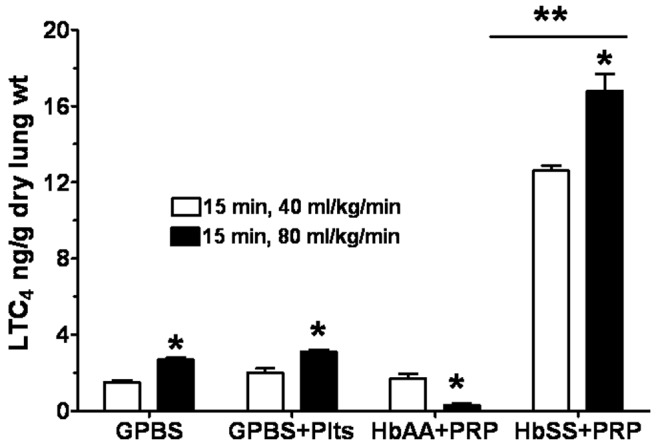
Profile of leukotriene C4 (LTC4) produced by isolated rat lungs perfused with GPBS or with HbAA or HbSS erythrocytes plus autologous platelets (Plts or PRP [platelet-rich plasma]) for 15 minutes at the initial perfusion rate (open bars) and for 15 more minutes at twice that rate (filled bars). See Figure 2 for experimental details and other abbreviations. Data are means + SEM. A single asterisk indicates significant (P < 0.05) difference from LTC4 production at the initial perfusion rate; double asterisks indicate significant (P < 0.05) difference from both GPBS controls and the HbAA group.
Production of PGE2 by lungs during the perfusion
Production of PGE2 measured from the perfusates, presented as means ± SEM in nanograms per gram lung dry weight, is shown in Figure 5 and Table 4. In general, perfusion of lungs with GPBS+Plts produced a release of PGE2 that was, like LTC4 release, not different from the effect of GPBS alone. At the beginning of the perfusion (time 0 minutes), PGE2 production by GPBS-perfused lungs was 0.014 ± 0.02. PGE2 production by lungs perfused with GPBS alone or with GPBS+Plts did not change during the study. For instance, 15 minutes after the initial flow rate was doubled, the PGE2 produced by GPBS lungs was 0.011 ± 0.02 (Figure 5), which is not different from the value at 0 minutes (P = 0.9). For lungs perfused with HbAA erythrocytes, PGE2 production was not detected at any of the time points or under any of the experimental conditions. On the other hand, HbSS erythrocyte–perfused lungs produced 0.37 ± 0.08 of PGE2 at 0 minutes, which increased to 0.96 ± 0.09 after 15 minutes of perfusion (Figure 5). Perfusion with the mixture of HbSS erythrocytes and autologous PRP increased PGE2 production to 1.52 ± 0.24, but production decreased by 18% 15 minutes after the addition of PRP to the HbSS erythrocytes (Figure 6). When the flow rate was doubled, PGE2 production after 15 minutes of perfusion increased to 1.66 ± 0.19 (Figure 7), which was 350% higher than the PGE2 produced at 0 minutes (0.37 ± 0.08).
Figure 5.
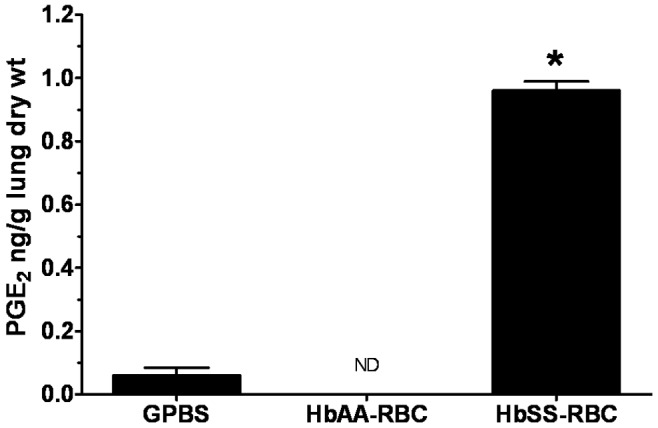
Profile of prostaglandin E2 (PGE2) produced by isolated rat lungs perfused with buffer alone or with erythrocytes, with metabolites measured at 15 minutes of perfusion. See Figure 2 for experimental details and abbreviations. Data are means + SEM. ND: none detected. An asterisk indicates significant (P < 0.05) difference from PGE2 production in GPBS control.
Table 4.
Percent change in prostaglandin E2 (PGE2) levels, relative to baseline levels, measured in effluents of isolated rat lung perfused with GPBS, HbAA-RBCs, and HbSS-RBCs and the perfusates combined with autologous platelet-rich plasma (PRP)
| Perfusate | 15 min | +PRP, 0 min | +PRP, 15 min | +PRP, 30 min |
|---|---|---|---|---|
| GPBS | NC | NC | NC | NC |
| HbAA RBCs | ND | ND | ND | ND |
| HbSS RBCs | 160 | 314 | 240 | 349 |
Measurements were taken after 15 minutes of perfusion with perfusate alone and 3 times after addition of PRP: immediately (0 min), 15 minutes later (15 min), and after 15 more minutes, during which the perfusion rate was twice the initial rate (30 min); see “Experimental design.” GBPS: phosphate-buffered saline containing 131 mM NaCl, 5.1 mM Na2HPO4, 1.5 mM KH2PO4, 1% bovine serum albumin, and 8.3 mM glucose; HbAA: hemoglobin AA; HbSS: hemoglobin SS; RBCs: red blood cells; NC: no change; ND: none detected in the assay.
Figure 6.
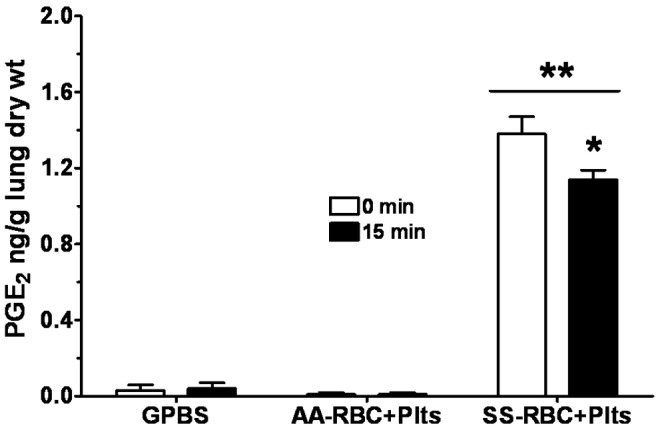
Profile of prostaglandin E2 (PGE2) produced by isolated rat lungs perfused with GPBS or with HbAA (AA) or HbSS (SS) erythrocytes plus autologous platelets (Plts), measured at 0 and 15 minutes of perfusion. See Figure 2 for experimental details and other abbreviations. Data are means + SEM. A single asterisk indicates significant (P < 0.05) difference from PGE2 production in the same group at 0 minutes; double asterisks indicate significant (P < 0.05) difference from PGE2 production in all other groups.
Figure 7.
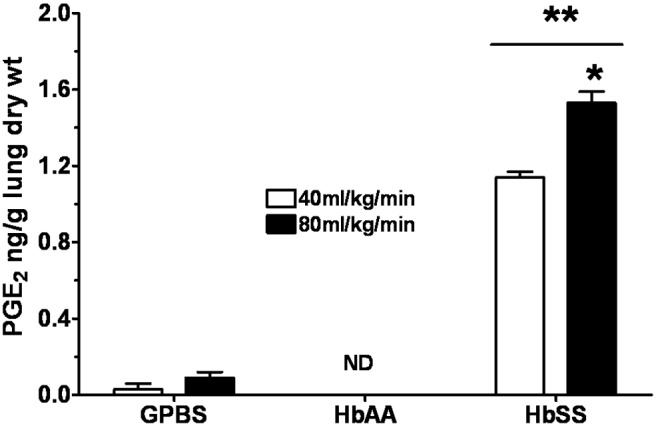
Profile of prostaglandin E2 (PGE2) produced by the isolated rat lungs perfused with GPBS or with HbAA or HbSS erythrocytes for 15 minutes at the initial perfusion rate (open bars) and at twice that rate (filled bars). See Figure 2 for experimental details and other abbreviations. Data are means + SEM. A single asterisk indicates significant (P < 0.05) difference from PGE2 production at the initial perfusion rate; double asterisks indicate significant (P < 0.05) difference from PGE2 production in both GPBS controls and the HbAA group.
Production of PGE2 increased during the initial 15 minutes of perfusion. Lungs perfused with HbSS erythrocytes produced significantly more PGE2 than HbAA- or GPBS-perfused lungs (P < 0.0003). Lungs perfused with HbAA erythrocytes did not produce detectable levels of PGE2. Coperfusion of the lungs with the GPBS or HbAA erythrocytes plus PRP did not affect lung production of PGE2.
Expression of the inflammatory proteins 5-lox and PAFR and COX proteins
Figures 8 and 9 show expression of the inflammatory proteins 5-lox and PAFR, respectively, by lung explants stimulated with HbSS and HbAA peptides and Krebs buffer control. The 5-lox protein expression by HbAA lungs was not different from that by Krebs control lungs. Lungs incubated with HbSS peptides expressed significantly more (20% more) 5-lox protein than either Krebs control or HbAA-peptide lungs (Figure 8). With respect to PAFR protein expression (Figure 9), lungs stimulated with HbAA peptide expressed the lowest amount of PAFR, compared to expression by Krebs buffer control and HbSS-peptide lungs. Expression by HbAA peptide–treated lungs was 40% less than that by Krebs control lungs and 80% less than that by HbSS peptide–treated lungs. Therefore, lungs exposed to HbSS peptide expressed significantly more PAFR protein than either Krebs control or HbAA-peptide lungs.
Figure 8.
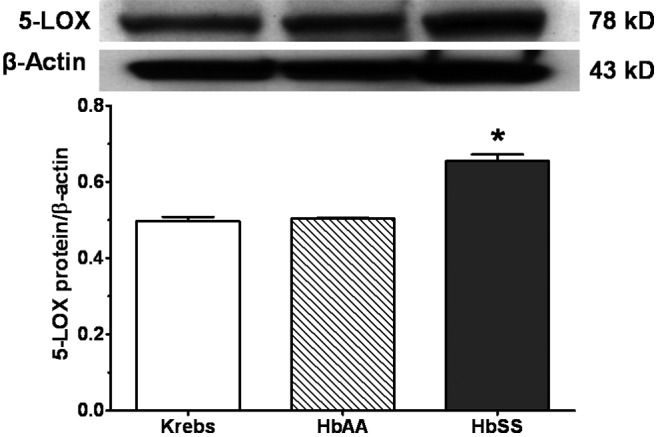
Expression of inflammatory protein 5-lipoxygenase (5-LOX) by rat lung explants stimulated in vitro with normal (HbAA) or sickle cell (HbSS) peptides. Adult rat lungs were prepared for studies as described in “Methods.” Proteins were prepared, and 5-LOX was measured by Western blotting. HbSS peptide increased expression of 5-LOX. Data are means + SEM. An asterisk indicates significant (P < 0.05) difference from Krebs buffer control.
Figure 9.
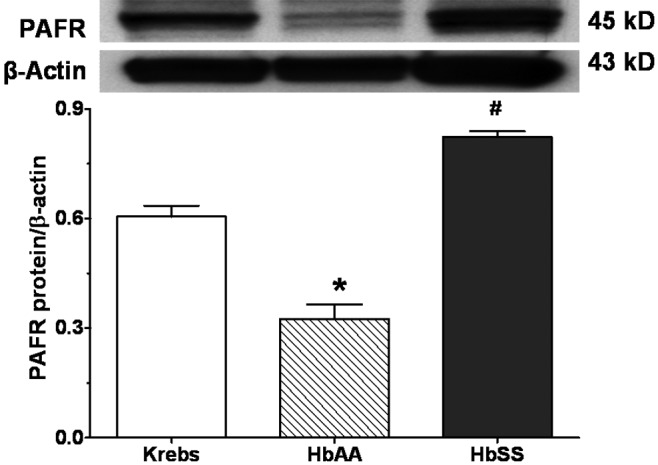
Expression of inflammatory protein platelet-activating factor receptor (PAFR) by rat lung explants stimulated in vitro with normal (HbAA) or sickle cell (HbSS) peptides. Adult rat lungs were prepared for studies as described in “Methods.” Proteins were prepared, and PAFR was measured by Western blotting. HbSS peptide increased expression of PAFR. Data are means + SEM. An asterisk indicates significant (P < 0.05) difference from Krebs buffer control; a pound sign (#) indicates significant (P < 0.05) difference from effects of both Krebs buffer control and HbAA peptide.
Figures 10 and 11 show expression of COX1 and COX2 proteins, respectively, by lungs stimulated with HbSS and HbAA peptides, compared to expression by Krebs control lungs. There was no difference in COX1 protein expression by the 3 groups of lungs (Figure 10). On the other hand, whereas COX2 protein expression by lungs stimulated with HbAA peptide was not different from that by Krebs control lungs, HbSS peptide stimulated COX2 protein expression 20% greater than that by either Krebs control or HbAA lungs (Figure 11).
Figure 10.
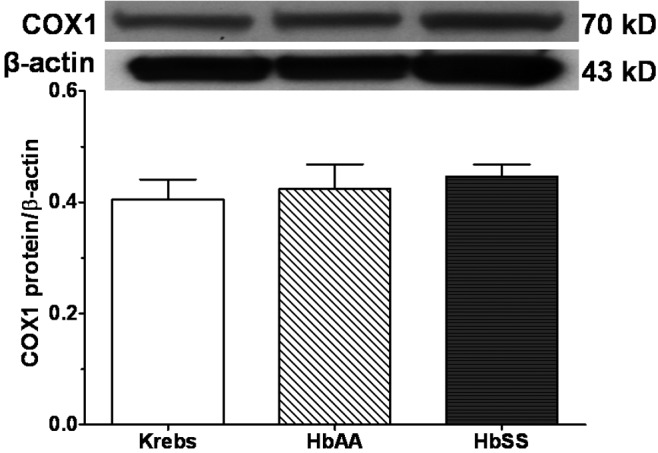
Expression of cyclooxygenase 1 (COX1) protein by adult rat lungs stimulated in vitro with normal (HbAA) or sickle cell (HbSS) peptides. Adult rat lungs were prepared for studies as described in “Methods.” Proteins were prepared, and COX1 expression was measured by Western blotting. There was no significant difference in COX1 expression among the 3 groups of lung explants. “Krebs” denotes the Krebs buffer control. Data are means + SEM.
Figure 11.
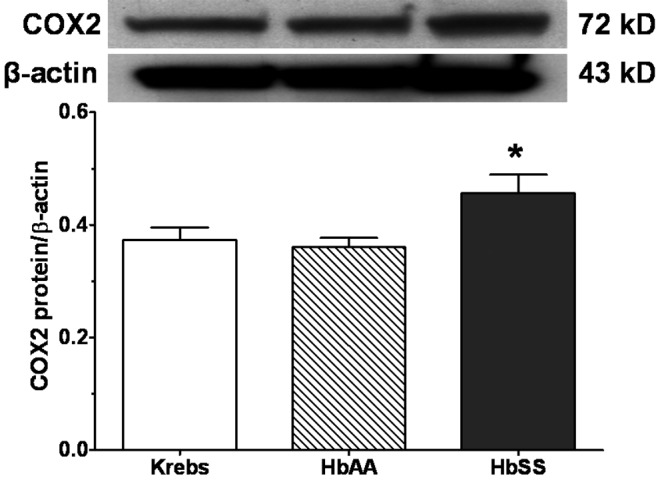
Expression of cyclooxygenase 2 (COX2) protein by adult rat lungs stimulated in vitro with normal (HbAA) or sickle cell (HbSS) peptides. Adult rat lungs were prepared for studies as described in “Methods.” Proteins were prepared, and COX2 expression was measured by Western blotting. HbSS peptide increased expression of COX2 protein. Data are means + SEM. An asterisk indicates significant (P < 0.05) difference from effects of both Krebs buffer control and HbAA peptide.
HbSS peptides stimulate greater production of prostacyclin (6-keto-PGF1α) and LTs
The amount of prostacyclin, as 6-keto-PGF1α (6-keto), produced by lungs stimulated for 4 hours with the HbAA and HbSS peptides is shown in Figure 12. Compared to Krebs control, the calcium ionophore A23187 caused greater release of 6-keto, and HbAA peptide did not stimulate release of 6-keto (Figure 12). On the other hand, 6-keto release by lungs stimulated with HbSS peptide was comparable to the effect of A23187, suggesting, perhaps, tissue injury to the lung comparable to that caused by A23187. A23187 also stimulated greater leukotriene production than Krebs control and HbAA-treated lungs (Figure 13). However, leukotriene production by lungs stimulated with HbSS peptide was greater than that by Krebs control and HbAA-peptide lungs. Of note, leukotriene production by HbAA-peptide lungs was less than that by Krebs control lungs.
Figure 12.
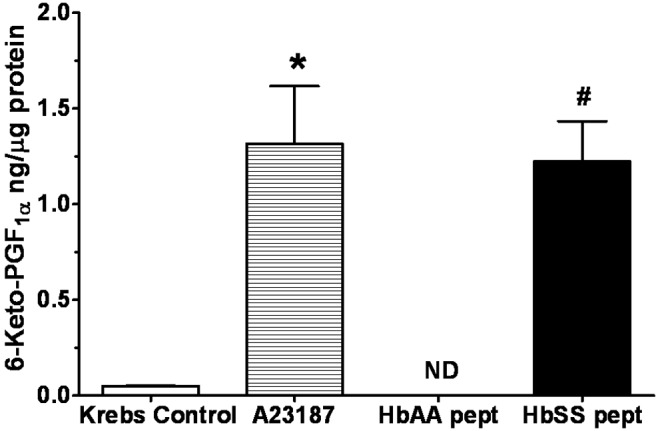
Amount of 6-keto-PGF1α measured from lung explants cultured with normal (HbAA) or sickle cell (HbSS) peptides. HbSS peptide stimulated release of 6-keto-PGF1α at a level comparable to effect of A23187 and higher than the effect of HbAA peptide or Krebs buffer control. Data are means + SEM. ND: none detected. An asterisk indicates release significantly (P < 0.05) greater than that for Krebs control; a pound sign (#) indicates release significantly (P < 0.05) greater than that for both HbAA peptide and Krebs control.
Figure 13.
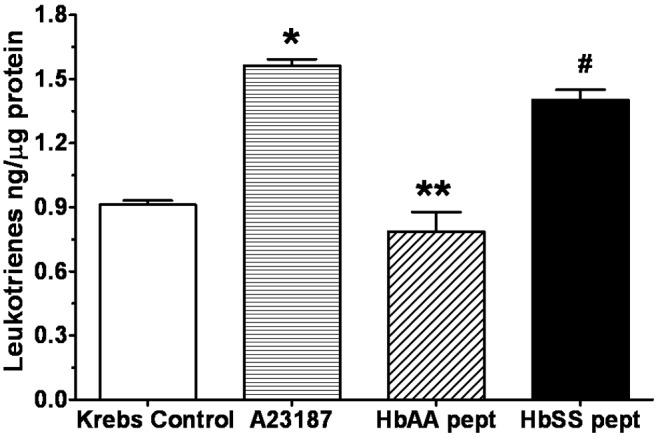
Amount of leukotrienes measured from lung explants cultured with normal (HbAA) or sickle cell (HbSS) peptides. HbSS peptide stimulated release of leukotrienes at a level comparable to effect of A23187 and higher than the effect of HbAA peptide or Krebs buffer control. Data are means + SEM. A single asterisk indicates release significantly (P < 0.05) greater than that for Krebs control; double asterisks indicate release significantly (P < 0.05) different from that under all other treatments; a pound sign (#) indicates release significantly (P < 0.05) greater than that for both HbAA peptide and Krebs control.
Discussion
In this study, we show that rat lungs perfused with HbSS erythrocytes or HbSS erythrocytes together with autologous PRP produced significantly greater amounts of LTC4 and PGE2, compared to lungs perfused with HbAA erythrocytes or perfusion buffer under the same conditions. Weight gain by the 3 groups of lungs after perfusion was not different. Also, the acid-base status, pco2, and po2 of the perfusates of the 3 groups of lungs were not different. There were some differences in vital characteristics between the SCD patients and the normal controls (Table 1).
SCD is characterized by recurrent hypoxemia, chronic hemolysis, recurring vaso-occlusive pain episodes, and tissue damage.22,23 It has been shown that tissue ischemia results in proinflammatory conditions in SCD, as a high level of platelet-activating factor (PAF) is measured in the circulation of SCD patients in a steady state,24 and indeed the initiation, progression, and resolution of vaso-occlusive pain episodes due to red blood cell sickling and adhesion to endothelium in the affected tissues have been characterized as “reperfusion injury.”17 Reperfusion of tissues after interruption of blood flow causes free-radical generation,25 provokes an inflammatory response, and exposes tissues to inflammatory mediators such as PAF, leukotrienes, and other cytokines.24-26
Sickle cell lung disease is also characterized by the inflammatory state of the tissues, occlusion of the lung microvessels by sickle erythrocytes, and lung segments that exhibit either a paucity or the absence of visible arterioles and venules, suggesting vessel collapse as a result of microthrombi within their lumen or upstream occlusion of segmental arterioles.10-12,22,23 In a previous report, Haynes et al.27 showed that ventilation of the lungs with a hypoxic gas increased pulmonary capillary pressure only when the lungs were perfused with HbSS red blood cells. This suggests that the vasoconstriction that occurred in the pulmonary microvasculature in response to alveolar hypoxia28,29 probably augments hemoglobin S polymerization and diminishes the deformability of sickle erythrocytes. In the present study, the perfusion rate was doubled in an attempt to determine the possible effect of diminished deformability of sickle erythrocytes during low-flow conditions. Production of metabolites increased even when the flow rate was increased, suggesting that diminished erythrocyte deformability may not be the factor causing increased metabolite release, but perhaps free radicals generated in situ by the sickle cell erythrocytes may be contributing to injury of the endothelium and the inflammatory response.
Leukotrienes are potent mediators of inflammation as well as vasoconstrictors in the pulmonary circulation. LTB4, another eicosanoid from the 5-lox pathway, promotes leukocyte chemotaxis and adhesion to the endothelium of postcapillary venule.30 The CystLTs LTC4, LTD4, and LTE4 elicit macromolecular leakage from this vessel segment. Leukocyte adhesion to the endothelium and macromolecular leakage from postcapillary venules are characteristic of the microcirculatory failure after ischemia-reperfusion. This suggests a role of leukotrienes as mediators of ischemia-reperfusion injury in sickle cell lung disease.17,23 Our observation that HbSS erythrocytes augment production of LTC4, compared to control HbAA erythrocytes, is supported by the report of LTC4 synthase expression by human lung membranes and human platelet homogenates, as determined by Western blotting.8 Davidson and Drafta31 also suggested that pulmonary hypertension in the rat lung induced by PAF, a proinflammatory mediator, is caused by prolonged activation of 5-lox leading to LTC4 production. Rat lung explants incubated with HbSS and HbAA peptides expressed 5-lox protein, and HbSS peptides caused greater expression of 5-lox. This suggests that increased LTC4 production by HbSS erythrocyte–perfused rat lungs may result from activation of the 5-lox enzyme, as observed in this tissue culture study, and perhaps activation of LTC4 synthase downstream from 5-lox. This contention is supported by findings by Daak and associates,32 who found no difference in the levels of arachidonic acid in cell membranes between SCD patients and normal individuals. Therefore, the underlying difference is not the level of arachidonic acid but the activities of leukotriene-metabolizing enzymes in the lung endothelium. In our study with rat lung explants, leukotrienes were measured after all treatments, but HbSS peptide stimulated greater production of leukotrienes than did HbAA peptide, and release was comparable to effect of the phospholipase A2 (PLA2) activator A23187. We can infer from this result that HbAA peptide is less effective in stimulating arachidonic acid metabolism following PLA2 activation. In this in vitro study with explants, the lung vasculature was washed devoid of rat blood components to ensure that protein expression or metabolite release was induced by the peptide treatments. These results also suggest that in some patients, systemic hypoxia may produce a rapid microvascular inflammatory response characterized by release of reactive oxygen species (ROS), leukocyte-endothelial adherence and emigration, increased vascular permeability, and consequently elevated production of inflammatory mediators such as leukotrienes and PAF.15,24 PAF is a potent inflammatory mediator and pulmonary vasoconstrictor that acts through its receptor to evoke responses in target cells or organs. We found that HbSS peptides stimulated greater PAFR expression than does HbAA peptide, suggesting that in vivo, PAF released in the lung can bind to its receptors to induce the inflammatory response directly or stimulate the vascular endothelium and white cells to release leukotrienes. Furthermore, LTB4 is involved in early hypoxia-induced responses, such as ROS generation and leukocyte adherence,33 and has been measured in plasma of SCD patients. Pulmonary hypoxia associated with SCD may induce production of increased ROS levels by actions of LTB4, LTC4, and PAF.18,24,30,33,34 Leukotrienes, in particular LTB4, are receiving significant attention in their role in lung diseases such as chronic obstructive pulmonary disease, where LTB4 binds to its cell surface receptors to evoke inflammatory response and airway remodeling.35
Contribution of platelets to the transcellular metabolism of arachidonic acid represents an important pathway of leukotriene production. We demonstrate here that rat lungs perfused with HbSS erythrocytes and autologous PRP synthesized and produced significantly greater amounts of LTC4 and PGE2 than lungs perfused with the GPBS buffer alone, GPBS+Plts, or control HbAA erythrocytes. Other studies have also demonstrated that intact erythrocytes enhanced platelet activation and recruitment.36-38 An important consequence of platelet activation is release of intracellular bioactive compounds, which in turn activate additional platelets that can interact with other cells in the microvasculature via receptor activation, i.e., the “recruitment phase” of homeostasis or thrombosis.38,39 Of note in this report is the observation that inclusion of platelets, whether from HbAA or HbSS blood, in GPBS perfusion buffer did not result in augmentation of measured metabolites. This suggests that the SCD erythrocytes are the primary stimuli for platelet activation and subsequent induction of metabolite production and release by the lung vascular endothelium. Prostaglandins, COX metabolites of arachidonic acid, can serve as indicators of inflammatory conditions,40,41 for example, during ischemia and inflammatory conditions.17,29,42 Lungs perfused with HbSS erythrocytes produced more PGE2, suggesting that HbSS erythrocytes can also induce acute inflammatory condition in the pulmonary microvasculature, perhaps because of reperfusion injury. Sustained production of PGE2 also suggests tissue injury compounded with inflammatory conditions.43-45 We found no difference in COX1 protein expression between HbAA peptide– and HbSS peptide–stimulated lungs. However, lungs stimulated with HbSS peptide expressed more COX2 protein. COX2 protein is the inducible form of cyclooxygenases, and so we can surmise that HbSS peptide is able to induce COX2 expression in culture. Furthermore, HbSS peptide–stimulated release of prostacyclin is comparable to effect of the PLA2 activator A23187, whereas prostacyclin is not detected in lungs stimulated with HbAA peptides. This suggests that HbSS peptide is able to stimulate arachidonic acid release, which is an indication of cell membrane injury, presented by COX2 activation and then prostacyclin production. It is likely that the stimulation of prostacyclin production by lungs incubated with HbSS peptide may be for protection of pulmonary vascular integrity, as expression of COX2 enzyme was also activated by the HbSS peptide.
SCD is complicated by the coexistence of vascular occlusion, the acute chest syndrome, and pulmonary hypertension, among others.46,47 These factors combine to cause increased morbidity and mortality of SCD patients. However, the mechanisms by which these processes occur or affect the general physiological well-being of the SCD patients are poorly understood. Our data from isolated rat lungs perfused with HbSS erythrocytes and those from lung explants cultured with purified HbSS peptide suggest that HbSS erythrocytes can stimulate release of inflammatory molecules, most likely from the pulmonary vascular endothelium, as well as induce expression of inflammatory proteins. These properties of HbSS hemoglobin may cause injury to the pulmonary vascular endothelium, leading to vaso-occlusive processes and subsequently the acute chest syndrome.
Source of Support: This study was supported in part by Seed Grant 520522 from the Los Angeles Biomedical Research Institute at Harbor-UCLA Medical Center, Torrance, California, and grant HL-38521 from the National Heart, Lung, and Blood Institute (National Institutes of Health) to JK-M.
Conflict of Interest: None declared.
References
- 1.Samuelsson B, Dahlen SE, Lindgren JA, Rouzer CA, Serhan CN. Leukotrienes and lipoxins: structure, biosynthesis, and biological effects. Science 1987;237(4819):1171–1176. [DOI] [PubMed]
- 2.Voelkel NF. Species variation in the pulmonary responses to arachidonic acid metabolites. Protaglandins 1985;29(5):867–889. [DOI] [PubMed]
- 3.Morganroth ML, Stenmark KR, Zirrolli JA, Mauldin R, Mathias M, Reeves JT, Murphy RC, Voelkel NF. Leukotriene C4 production during hypoxic vasoconstriction in the isolated rat lung. Protaglandins 1984;28(6):867–875. [DOI] [PubMed]
- 4.Hallstrand TS, Lai Y, Henderson WR Jr., Altemeier WA, Gelb MH. Epithelial regulation of eicosanoid production in asthma. Pulm Pharmacol Ther 2012;25(6):432–437. [DOI] [PMC free article] [PubMed]
- 5.Knight-Perry J, DeBaun MR, Strunk RC, Field JJ. Leukotriene pathway in sickle cell disease: a potential target for directed therapy. Expert Rev Hematol 2009;2(1):57–68. [DOI] [PubMed]
- 6.Foster HR, Fuerst E, Lee TK, Cousins DJ, Woszczek G. Characterization of P2Y12 receptor responsiveness to cysteinyl leukotrienes. PLoS ONE 2013;8(3):e58305. doi:10.1371/journal.pone.0058305. [DOI] [PMC free article] [PubMed]
- 7.Barrrett NA, Fernandez JM, Maekawa A, Xing W, Li L, Parsons MW, Austen KF, Kanaoka Y. Cysteinyl leukotriene 2 receptor on dendritic cells negatively regulates ligand-dependent allergic pulmonary inflammation. J Immunol 2012;189(9):4556–4565. [DOI] [PMC free article] [PubMed]
- 8.Scoggan KA, Jakobsson PJ, Ford-Hutchinson AW. Production of leukotriene C4 in different human tissues is attributable to distinct membrane bound biosynthetic enzymes. J Biol Chem 1997;272(15):10182–10187. [DOI] [PubMed]
- 9.Nicholson DW, Ali A, Vaillancourt JP, Calaycay JR, Mumford RA, Zamboni RJ, Ford-Hutchinson AW. Purification to homogeneity and the N-terminal sequence of human leukotriene C4 synthase: a homodimeric glutathione S-transferase composed of 18-kDa subunits. Proc Natl Acad Sci USA 1993;90(5):2015–2019. [DOI] [PMC free article] [PubMed]
- 10.Bhalla M, Abboud MR, McLoud TC, Shepard JO, Munden MM, Jackson SM, Beaty JR, Laver JH. Acute chest syndrome in sickle cell disease: CT evidence of microvascular occlusion. Radiology 1993;187(1):45–49. [DOI] [PubMed]
- 11.Gray A, Anionwu EN, Davies SC, Brozovic M. Patterns of mortality in sickle cell disease in the United Kingdom. J Clin Pathol 1991;44(6):459–463. [DOI] [PMC free article] [PubMed]
- 12.Powars D, Weidman JA, Odom-Maryon T, Niland JC, Johnson C. Sickle cell chronic lung disease: prior morbidity and the risk of pulmonary failure. Medicine 1988;67(1):66–76. [PubMed]
- 13.Zennadi R, Chien A, Xu K, Batchvarova M, Telen MJ. Sickle red cells induce adhesion of lymphocytes and monocytes to endothelium. Blood 2008;112(8):3474–3483. [DOI] [PMC free article] [PubMed]
- 14.Kaul DK, Finnegan E, Barabino GA. Sickle red cell–endothelium interactions. Microcirculation 2009;16(1):97–111. [DOI] [PMC free article] [PubMed]
- 15.Ibe BO, Kurantsin-Mills J, Raj JU, Lessin LS. Plasma and urinary leukotrienes in sickle cell disease: possible role in the inflammatory process. Eur J Clin Invest 1994;24(1):57–64. [DOI] [PubMed]
- 16.Kurantsin-Mills J, Ibe BO, Natta CL, Raj JU, Siegel RS, Lessin LS. Elevated urinary levels of thromboxane and prostacyclin metabolites in sickle cell disease reflects activated platelets in the circulation. Br J Haematol 1994;87(3):580–585. [DOI] [PubMed]
- 17.Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J Clin Invest 2000;106(3):411–420. [DOI] [PMC free article] [PubMed]
- 18.Haynes J. Jr., Obiako B. Activated polymorphonuclear cells increase sickle red blood cell retention in lung: role of phospholipids. Am J Physiol Heart Circ Physiol 2002;282(1):H122–H130. [DOI] [PubMed]
- 19.Ibe BO, Morris J, Kurantsin-Mills J, Raj JU. Sickle erythrocytes induce prostacyclin and thromboxane synthesis by isolated perfused lung. Am J Physiol Lung Cell Mol Physiol 1997;272(4):L597–L602. [DOI] [PubMed]
- 20.Ibe BO, Portugal AM, Raj JU. Metabolism of platelet activating factor by intrapulmonary vascular smooth muscle cells: effect of oxygen on phospholipase A2 protein expression and activities of acetyl–CoA acetyltransferase and cholinephosphotransferase. Mol Genet Metab 2002;77(3):237–248. [DOI] [PubMed]
- 21.Ibe BO, Abdallah MF, Portugal AM, Raj JU. Platelet-activating factor stimulates ovine foetal pulmonary vascular smooth muscle cell proliferation: role of nuclear factor-kappa B and cyclin-dependent kinases. Cell Prolif 2008;41(2):208–229. [DOI] [PMC free article] [PubMed]
- 22.Aquino SL, Gamsu G, Fahy JV, Claster S, Embury SH, Mentzer WC, Vichinsky EP. Chronic pulmonary disorders in sickle cell disease: findings at thin-section CT. Radiology 1994;193(3):807–811. [DOI] [PubMed]
- 23.Kaul DK, Fabry ME, Costantini F, Rubin EM, Nagel RL. In vivo demonstration of red cell–endothelial interaction, sickling and altered microvascular response to oxygen in the sickle transgenic mouse. J Clin Invest 1995;96(6):2845–2853. [DOI] [PMC free article] [PubMed]
- 24.Oh SO, Ibe BO, Johnson C, Kurantsin-Mills J, Raj JU. Platelet-activating factor in plasma of patients with sickle cell disease in steady state. J Lab Clin Med 1997;130(2):191–196. [DOI] [PubMed]
- 25.McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med 1985;312(3):159–163. [DOI] [PubMed]
- 26.Matsui NM, Borsig L, Rosen SD, Yaghmai M, Varki A, Embury SH. P-selectin mediates the adhesion of sickle erythrocytes to endothelium. Blood 2001;98(6):1955–1962. [DOI] [PubMed]
- 27.Haynes J Jr., Taylor AE, Dixon D, Voelkel N. Microvascular hemodynamics in the sickle red blood cell perfused isolated rat lung. Am J Physiol Heart Circ Physiol 1993;264(2):H484–H489. [DOI] [PubMed]
- 28.Raj JU, Toga H, Ibe BO, Anderson J. Effects of endothelin, platelet activating factor and thromboxane A2 in ferret lungs. Respir Physiol 1992;88(1–2):129–140. [DOI] [PubMed]
- 29.Pritchard KA Jr., Ou J, Ou Z, Shi Y, Franciosi JP, Signorino, P, Kaul S, et al. Hypoxia-induced acute lung injury in murine models of sickle cell disease. Am J Physiol Lung Cell Mol Physiol 2004;286(3):L705–L714. [DOI] [PubMed]
- 30.Setty BNY, Stuart MJ. Eicosanoids in sickle cell disease: potential relevance of neutrophil leukotriene B4 to disease pathophysiology. J Lab Clin Med 2002;139(2):80–89. [DOI] [PubMed]
- 31.Davidson D, Drafta D. Prolonged pulmonary hypertension caused by platelet-activating factor and leukotriene C4 in the rat lung. J Appl Physiol 1992;73(3):955–961. [DOI] [PubMed]
- 32.Daak AA, Ghebremeskel K, Elbashir MI, Bakhita A, Hassan Z, Crawford MA. Hydroxyurea therapy mobilises arachidonic acid from inner cell membrane aminophospholipids in patients with homozygous sickle cell disease. J Lipids 2011;2011:718014. doi:10.1155/2011/718014. [DOI] [PMC free article] [PubMed]
- 33.Lehr HA, Guhlmann A, Nolte D, Keppler D, Messmer K. Leukotrienes as mediators in ischemia-reperfusion injury in a microcirculation model in the hamster. J Clin Invest 1991;87(6):2036–2041. [DOI] [PMC free article] [PubMed]
- 34.Steiner DRS, Gonzalez NC, Wood JG. Leukotriene B4 promotes reactive oxidant generation and leukocyte adherence during acute hypoxia. J Appl Physiol 2001;91(3):1160–1167. [DOI] [PubMed]
- 35.Kilfeather S. 5-lipoxygenase inhibitors for the treatment of COPD. Chest 2002;121(5 suppl.):197S–200S. [DOI] [PubMed]
- 36.Brittain HA, Eckman JR, Swerlick RA, Howard RJ, Wick TM. Thrombospondin from activated platelets promotes sickle erythrocyte adherence to human microvascular endothelium under physiological flow: a potential role for platelet activation in sickle cell disease. Blood 1993;81(8):2137–2143. [PubMed]
- 37.Silvain J, Pena A, Cayla G, Brieger D, Bellemain-Appaix A, Chastre T, Vignalou J-B, et al. Impact of red blood cell transfusion on platelet aggregation in healthy volunteers: results of the TRANSFUSION study. Eur Heart J 2010;31(22):2816–2821. [DOI] [PubMed]
- 38.Vallés J, Santos MT, Aznar J, Martínez M, Moscardó A, Piñón M, Broekman MJ, Marcus AJ. Platelet-erythrocyte interactions enhance αIIbβ3 integrin receptor activation and P-selectin expression during platelet recruitment: down-regulation by aspirin ex vivo. Blood 2002;99(11):3978–3984. [DOI] [PubMed]
- 39.Nobili E, Salvado MD, Folkersen L, Castiglione L, Kastrup J, Watterholm A, Tremoli E, et al. Cysteinyl leukotriene signaling aggravates myocardial hypoxia in experimental atherosclerotic heart disease. PLoS ONE 2012;7(7):e41786. doi:10.1371/journal.pone.0041786. [DOI] [PMC free article] [PubMed]
- 40.Dassé Séry R, N’guessan K, Akré Dagra P, Yao R, Sombo Mambo F. Pulmonary events induced by non-steroidal anti-inflammatory drugs in patients with sickle cell disease [in French]. Sante 2011;21(4):187–191. [DOI] [PubMed]
- 41.Ong HT, Ong LM, Tan TE, Chean KY. Cardiovascular effects of common analgesics. Med J Malays 2013;68(2):189–194. [PubMed]
- 42.Lang PA, Kasinathan RS, Brand VB, Duranton C, Lang C, Koka S, Shumilina E, et al. Accelerated clearance of Plasmodium-infected erythrocytes in sickle cell trait and annexin-A7 deficiency. Cell Physiol Biochem 2009;24(5–6):415–428. [DOI] [PubMed]
- 43.Kaul DK, Zhang X, Dasgupta T, Fabry ME. Arginine therapy of transgenic-knockout sickle mice improves microvascular function by reducing non-nitric oxide vasodilators, hemolysis, and oxidative stress. Am J Physiol Heart Circ Physiol 2008;295(1):H39–H47. [DOI] [PMC free article] [PubMed]
- 44.Kaul DK, Liu X-d, Chang H-Y, Nagel RL, Fabry ME. Effect of fetal hemoglobin on microvascular regulation in sickle transgenic-knockout mice. J Clin Invest 2004;114(8):1136–1143. [DOI] [PMC free article] [PubMed]
- 45.Field JJ, DeBaun MR. Asthma and sickle cell disease: two distinct diseases or part of the same process? Hematology 2009;2009:45–53. [DOI] [PubMed]
- 46.Newaskar M, Hardy KA, Morris CR. Asthma in sickle cell disease. Sci World J 2011;11:1138–1152. [DOI] [PMC free article] [PubMed]
- 47.Machado RF, Gladwin MT. Chronic sickle cell lung disease: new insights into the diagnosis pathogenesis and treatment of pulmonary hypertension. Brit J Haematol 2005;129(4):449–464. [DOI] [PubMed]