

Abstract Abstract
Pulmonary arterial hypertension (PAH) is a potentially life-threatening complication of thalassemia. A sexagenarian with β-thalassemia intermedia presented with new-onset dyspnea and syncope. Right heart catheterization confirmed severe PAH. Her functional class IV symptoms and severely elevated mean pulmonary artery pressure prompted the initiation of continuous epoprostenol therapy. Clinical follow-up documented significant improvement in functional class, 6-minute walk distance, and right ventricular size and function as well as pulmonary arterial pressure on echocardiogram. At the patient’s request, epoprostenol was down-titrated and eventually discontinued. The patient was then safely transitioned to nifedipine therapy after verification of vasoresponsiveness.
Keywords: β-thalassemia intermedia, pulmonary arterial hypertension, pulmonary hypertension, epoprostenol treatment
The association of pulmonary arterial hypertension (PAH) with chronic hemolytic anemias is well documented; however, published prevalences vary with study method and sample size. In thalassemia major, the prevalence of PAH ranges between 2% and 79%; for thalassemia intermedia, the range is 40%–50%.1-8 Most of the studies used echocardiogram to determine the prevalence, but a recently published multicenter study confirmed the PAH diagnosis by right heart catheterization and reported a prevalence of 2.1%.8
PAH is a life-threatening complication of thalassemia; however, published guidelines for the treatment of PAH only minimally mention the association and offer no specific recommendations for treatment. In addition, the newly published classification guidelines from the World Symposium on Pulmonary Hypertension9 have relegated PAH due to chronic hemolytic anemia to diagnostic group 5, for which PAH-specific therapy is not approved by the United States Food and Drug Administration (FDA). We describe a patient with 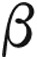 thalassemia intermedia who developed severe PAH and was successfully treated with continuous epoprostenol infusion and later transitioned to oral calcium blocker therapy.
thalassemia intermedia who developed severe PAH and was successfully treated with continuous epoprostenol infusion and later transitioned to oral calcium blocker therapy.
Case Description
A sexagenarian with history of 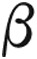 thalassemia intermedia presented with new-onset dyspnea and syncope. She had a complicated medical history, including insulin-dependent diabetes mellitus from iron overload in the pancreas, paralyzed hemidiaphragm, extramedullary hematopoiesis along lumbar and thoracic spine, prothrombin gene mutation, and hyperhomocysteinemia complicated by multiple episodes of deep venous thrombosis in her lower extremity and pulmonary embolism. Earlier medical treatment had included anticoagulation, multiple transfusions, and splenectomy (at 16 years of age). The patient was referred for evaluation after echocardiogram revealed severely elevated right heart pressures.
thalassemia intermedia presented with new-onset dyspnea and syncope. She had a complicated medical history, including insulin-dependent diabetes mellitus from iron overload in the pancreas, paralyzed hemidiaphragm, extramedullary hematopoiesis along lumbar and thoracic spine, prothrombin gene mutation, and hyperhomocysteinemia complicated by multiple episodes of deep venous thrombosis in her lower extremity and pulmonary embolism. Earlier medical treatment had included anticoagulation, multiple transfusions, and splenectomy (at 16 years of age). The patient was referred for evaluation after echocardiogram revealed severely elevated right heart pressures.
A second echocardiogram with contrast demonstrated normal left ventricle (LV) function with hyperdynamic global systolic function; however, the LV was D-shaped, which was indicative of severe right ventricular (RV) pressure overload. The LV ejection fraction was noted to be normal at 73%. There was mild left atrial enlargement and mild-to-moderate tricuspid regurgitation (TR). The peak TR jet velocity was significantly elevated at 4.8 m/s. The right atrial pressure (RAP) was estimated to be 10 mmHg on the basis of the inferior vena cava diameter and only partial collapse with inspiration. The estimated right ventricular systolic pressure (RVSP) was 102 mmHg using the Bernoulli formula technique. The findings were consistent with severe pulmonary hypertension (PH).
Blood work screening for connective tissue disease and human immunodeficiency virus (HIV) had results that were negative or normal, including antinuclear antibody, anti-DNA, rheumatoid factor, SCL-70 antibody, and HIV enzyme-linked immunosorbent assays. Arterial blood gas showed a pH of 7.49, PaO2 of 65 mmHg, PaCO2 of 35 mmHg on room air, and the patient’s chest radiograph was remarkable only for prominent pulmonary arteries. Pulmonary function test results were within normal limits except for mildly depressed oxygen saturation by pulse oximetry, 91% at rest. Nuclear medicine ventilation-perfusion lung scan showed multiple small-to-subsegmental mismatched perfusion defects consistent with high probability for pulmonary embolism.
Additional testing was ordered in pursuit of the abnormal lung scan findings. Doppler ultrasound of the lower extremities had findings that were negative for deep venous thrombosis. Chest computed tomography with intravenous contrast (pulmonary embolism protocol) showed no acute or chronic pulmonary embolus. The right heart and the main and proximal pulmonary arteries were enlarged. No pulmonary parenchymal disease was present. Evidence of earlier splenectomy was noted. Pulmonary angiogram was negative for pulmonary embolism. Significant vascular pruning, consistent with PAH, provided explanation for the abnormal ventilation perfusion scan findings.
Hematologic and hepatic evaluation included confirmation of the patient’s thalassemia by review of outside test results. In addition, iron studies showed an iron level of 222 μg/dL with a total iron binding capacity of 233 μg/dL and 95% saturation. Ferritin level was 2,809 μg/L. The patient’s liver function test results showed only a mildly elevated aspartate aminotransferase level of 42 u/L and alanine transaminase level of 53 u/L with normal total and direct bilirubin. Liver ultrasound findings were within normal limits. Of note, the patient received treatment with deferoxamine with her red blood cell transfusions and was receiving deferasirox for transfusion-related hemosiderosis.
Right heart catheterization (RHC) revealed an RAP of 8 and pulmonary artery systolic pressure (PASP), pulmonary artery diastolic pressure (PADP), and mean pulmonary artery pressure (MPAP) of 92, 37, and 55 mmHg, respectively. The pulmonary capillary wedge pressure (PCWP) was 10 mmHg with a transpulmonary gradient of 45 mmHg. Cardiac output (CO) was 6.5 L/min with a cardiac index (CI) of 3.9 L/min/m2. The calculated pulmonary vascular resistance (PVR) was 681 dyn·s·cm−5. The patient’s systemic blood pressure was 98/61/73 mmHg with a heart rate of 80 beats per minute.
Epoprostenol was initiated intravenously at 2 ng/kg/min and was titrated upward to 6 ng/kg/min with a reduction in MPAP and PVR to 35 mmHg and PVR to 290 dyn·s·cm−5 and an associated increase in CO to 6.9 L/min. The acute hemodynamic improvement with epoprostenol was considered a positive vasoreactive response; therefore, the patient received a trial of oral calcium channel blockade while the pulmonary artery catheter remained in place to monitor hemodynamics using a published protocol.10 After 8 successive hourly doses of oral nifedipine 20 mg (total dose, 160 mg), she sustained a significant reduction in her systemic blood pressure. The CO decreased to 4.4 L/min. Subsequently, diltiazem was administered after a washout period for the nifedipine with essentially the same result. Diltiazem 60 mg was administered every hour for 3 doses (total dose, 180 mg) but was discontinued because of systemic hypotension. The CO decreased to 5.6 L/min. Although the patient was acutely vasoresponsive, she was intolerant of oral calcium channel blockers.
The patient’s diagnosis was confirmed to be severe PAH, idiopathic versus in association with chronic hemolytic anemia. Although she was acutely vasoresponsive to intravenous epoprostenol, she demonstrated intolerance to oral calcium channel blockers with systemic hypotension. The most appropriate treatment was continuous intravenous epoprostenol. A permanent central venous catheter was inserted, and the patient was educated on epoprostenol preparation, administration, and sterile technique. She was discharged from the hospital with receipt of continuous infusion epoprostenol with upward titration as an outpatient.
Ultimately, the patient’s peak epoprostenol dosage was 18 ng/kg/min. She had regular follow-up with us to insure improvement of her condition and tolerability of therapy. She had gradual improvement in her dyspnea and no additional episodes of syncope. After 10 months of intravenous epoprostenol, an echocardiogram showed RAP of 5 mmHg, RVSP of 46 mmHg with a peak TR velocity of 3.2 m/s, and normal RV size and function. Regular and frequent clinical follow-up documented significant improvement in functional class, 6-minute walk distance (6MWD), and RV size and function as well as pulmonary arterial pressure on echocardiogram.
After approximately 12 months of continuous infusion epoprostenol, the patient was back to her baseline function with normalization of RV size and function. She was functional class II, had dyspnea only with extreme exertion, and experienced no associated chest pain, near-syncope, syncope, or fluid retention. Her brain natriuretic peptide (BNP) was 29 pg/mL (normal range, 0–82 pg/mL). Echocardiogram showed a peak TR velocity of 2.4 m/s with RAP, MPAP, and RVSP of 5, 23, and 28 mmHg, respectively. At the request of the patient, epoprostenol was down-titrated to 12 ng/kg/min to reduce the systemic adverse effects. After approximately 3.5 years of epoprostenol therapy, the patient stated that she would like to discontinue the medication and convert to oral therapy if possible. At that time, she was functional class II with a BNP of 73 pg/mL and 6MWD in excess of 380 m. Echocardiogram continued to demonstrate a normal RV. After significant discussion, and with the patient’s consent, she was admitted to the intensive care unit for placement of a pulmonary artery catheter and potential conversion from epoprostenol to oral therapy. Right heart catheterization showed PASP/PADP at 34/14 mmHg, MPAP of 20 mmHg, and PCWP of 12 mmHg. The patient was titrated slowly off of epoprostenol under careful observation and did not have any elevations in her pulmonary artery pressure. Her baseline MPAP, PVR, and CO after discontinuing epoprostenol therapy were 38 mmHg, 284 dyn·s·cm−5, and 6.5 L/min. A trial of nifedipine decreased MPAP and PVR to 29 mmHg and 178 dyn·s·cm−5 with preserved CO at 8.7 L/min. The patient was then safely transitioned to nifedipine therapy after verification of vasoresponsiveness by RHC. Vasoresponsiveness at that time was defined as at least a 20% decrease in both the MPAP and PVR on the basis of the protocol published by Rich et al.,10 which is different from the current definition of vasodilator response as a decrease in MPAP by at least 10 mmHg to an absolute level of less than 40 mmHg without a decrease in cardiac output. An additional echocardiogram obtained 2 weeks after the patient was discharged from the hospital showed normal LV and RV size and function with estimated RAP and RVSP of 5 and 41 mmHg, respectively. Over the subsequent 3 years, the patient tolerated oral nifedipine well, with only intermittent fluid retention that required upward adjustment of her diuretic dosing. In 2007, the patient had worsening dyspnea consistent with functional class III symptoms. Her BNP was 56 pg/mL, and her 6WMD was 301 m. An echocardiogram obtained at that time revealed that the RV was of normal size and contracting normally. RAP, RVSP, and MPAP were 5, 49, and 34 mmHg, respectively, which was slightly more elevated than in the patient’s earlier study. We discussed an additional RHC, but the patient preferred to try empirical diuresis; however, the findings of an echocardiogram obtained after diuresis were not significantly improved. Again, the patient preferred a trial of medical therapy to repeated catheterization; therefore, the nifedipine dosage was increased to 40 mg administered 3 times daily. After tolerability of the higher dosage was confirmed, the patient’s treatment was converted to a long-acting preparation, nifedipine extended release 90 mg/day. The patient’s functional class improved to class II after this intervention. Subsequent follow-up demonstrated sustained improvement and stability with the same dosage of nifedipine. At her last outpatient assessment, in June 2012, the patient’s functional class was II, her BNP was 41 pg/mL, and her 6MWD was 333 m. An echocardiogram obtained at that time showed normal LV size and function, LV ejection fraction of 63%, borderline RV enlargement with preserved contractility, indeterminate diastolic dysfunction, ratio of early transmitral flow velocity to early diastolic mitral annulus velocity (E/e’) of 11.7, and tricuspid annular plane systolic excursion of 21 mm. RAP, RVSP, and MPAP by echocardiogram were 5, 32, and 23 mmHg, respectively. The clinical data are summarized in Figure 1.
Figure 1.

Time line of patient’s clinical course. E/e’: ratio of early transmitral flow velocity to early diastolic mitral annulus velocity; LV: left ventricle; LVEF: left ventricular ejection fraction; MPAP: mean pulmonary artery pressure; NYHA: New York Heart Association; RHC: right heart catheterization; RV: right ventricle; RVSP: right ventricular systolic pressure; TAPSE: tricuspid annular plane systolic excursion; TID: 3 times per day; XL: extended release.
Discussion
PAH is a life-threatening complication and increasingly recognized in patients with thalassemia. The pathophysiology of PAH in patients with thalassemia is believed to be multifactorial and complex. Chronic hemolysis leads to nitric oxide depletion due to nitric oxide scavenging, arginine catabolism, and endogenous nitric oxide synthesis inhibition; enhanced platelet activation and increased endothelin-1 release lead to vasculopathy characterized by endothelial dysfunction, increased vascular tone inflammation, and vascular remodeling and destruction of pulmonary vasculature, which eventually result in hemolytic anemia–associated PAH. Hypercoagulable state is another contributing factor that resulted from the native erythrocyte precoagulant surface, the previous splenectomy, some coexistent genetic coagulation defects, and endothelial dysfunction and vasculopathy. Patients with thalassemia also have an increased occurrence of left-sided heart disease either from systolic or diastolic dysfunction from high output state, iron overload, and other immunologic factors, which can cause some degree of PAH. Chronic anemia and chronic tissue hypoxia induce a compensatory high output and volume overload that damages the pulmonary vasculature and contributes to the pathogenesis of PAH.1-7
There is no specific treatment guideline established for patients with thalassemia and PAH. In addition, the recently revised classification from the 2013 World Symposium on Pulmonary Hypertension has relocated PAH in association with chronic hemolytic anemia from diagnostic group 1 to group 5.9 None of the currently FDA-approved PAH-specific therapies have a group 5 indication. The clinician must rely on clinical judgment and available published data, which is limited to individual case reports, a case series, and small, open-label clinical trials with phosphodiesterase-5 inhibitors and endothelin antagonists, described below (Table 1). Littera et al.11 reported a case with 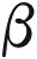 -thalassemia intermedia and PH treated with sildenafil that resulted in normalization of the echocardiogram after 15 months. Similarly, Correale et al.12 reported improvement in both the New York Heart Association (NYHA) functional class and echocardiogram with long-term sildenafil treatment in a patient with β‐thalassemia major. A case series of patients (2 with β‐thalassemia major, 4 with β‐thalassemia intermedia, and 1 with sickle cell anemia) with severe PAH defined by echocardiogram findings received long-term sildenafil treatment that resulted in improved NYHA functional class, 6MWD, and pulmonary pressure by echocardiogram.13 A 12-week, prospective, open-label pilot study of sildenafil involving 10 patients with β‐thalassemia with increased TR velocity greater than 2.5 m/s showed a 13% reduction in TR velocity and improved NYHA functional class but no significant change in 6MWD.14 Treatment with bosentan for 1 year in a patient with β‐thalassemia intermedia and PH confirmed by RHC reported an improvement in pulmonary hemodynamics.15 El-Beshlawy et al.16 found that oral L-carnitine can reduce mean pulmonary pressure in 12 children with β‐thalassemia major from 34 to 24 mmHg at 3 months. Treatment was based on the hypothesis that L-carnitine plays a role in long-chain fatty acid mitochondrial oxidation for energy production and can be attributable to the effect on myocardial energy production, stabilizing red blood cell membranes, and improving the anemic state.16
-thalassemia intermedia and PH treated with sildenafil that resulted in normalization of the echocardiogram after 15 months. Similarly, Correale et al.12 reported improvement in both the New York Heart Association (NYHA) functional class and echocardiogram with long-term sildenafil treatment in a patient with β‐thalassemia major. A case series of patients (2 with β‐thalassemia major, 4 with β‐thalassemia intermedia, and 1 with sickle cell anemia) with severe PAH defined by echocardiogram findings received long-term sildenafil treatment that resulted in improved NYHA functional class, 6MWD, and pulmonary pressure by echocardiogram.13 A 12-week, prospective, open-label pilot study of sildenafil involving 10 patients with β‐thalassemia with increased TR velocity greater than 2.5 m/s showed a 13% reduction in TR velocity and improved NYHA functional class but no significant change in 6MWD.14 Treatment with bosentan for 1 year in a patient with β‐thalassemia intermedia and PH confirmed by RHC reported an improvement in pulmonary hemodynamics.15 El-Beshlawy et al.16 found that oral L-carnitine can reduce mean pulmonary pressure in 12 children with β‐thalassemia major from 34 to 24 mmHg at 3 months. Treatment was based on the hypothesis that L-carnitine plays a role in long-chain fatty acid mitochondrial oxidation for energy production and can be attributable to the effect on myocardial energy production, stabilizing red blood cell membranes, and improving the anemic state.16
Table 1.
Review of pulmonary hypertension treatment in patients with thalassemia
| Source | No. of patients | Diagnosis | Medication | Follow-up | Outcome |
|---|---|---|---|---|---|
| Present report | 1 | β‐thalassemia intermedia | Epoprostenol | 13 years, by RHC | Symptoms, NYHA, 6MWD, hemodynamic improvement |
| Littera et al. 200211 | 1 | β‐thalassemia intermedia | Sildenafil 50–100 mg/day | 15 months, by echo | Symptoms, echo (RV dimension, RVSP, and MPAP) improvement |
| Correale et al. 201212 | 1 | β‐thalassemia major | Sildenafil 120 mg/day | 2 years, by echo | NYHA, echo (PASP) improvement |
| Derchi et al. 200513 | 7 | Thalassemia intermedia (n = 4); thalassemia major (n = 2); sickle thalassemia (n = 1) | Sildenafil 50 mg BID | 4–48 months, by echo | Symptoms, NYHA, 6MWD, echo (TG gradient) improvement |
| Morris et al. 201314 | 10 | β‐thalassemia major (n = 5); β‐thalassemia intermedia (n = 5) | Sildenafil 50 mg TID to 100 mg TID | 12 weeks, by echo | 6MWD no change; NYHA, echo (TR jet velocity) improvement |
| Anthi et al. 201215 | 1 | β‐thalassemia intermedia | Bosentan 125 mg BID | 1 year, by RHC | NYHA, hemodynamic improvement |
| El-Beshlawy et al. 200916 | 32 | β‐thalassemia major | L-carnitine 50 mg/kg/day | 3 months, by echo | Echo (PASP) improvement |
| Tam et al. 200617 | 1 | β‐thalassemia major | Epoprostenol | 5 years, by RHC | Symptoms,hemodynamic improvement |
BID: twice per day; echo∶ echocardiogram; MPAP: mean pulmonary arterial pressure; NYHA: New York Heart Association; PASP: pulmonary artery systolic pressure; RHC: right heart catheterization; RV: right ventricle; RVSP: right ventricular systolic pressure; TG: tricuspid gradient; TR: tricuspid regurgitation; 6MWD: 6-minute walk distance.
Most analogous to our case, Tam and Farber17 reported the first case of β‐thalassemia treated with epoprostenol infusion. After 5 years of therapy, their patient experienced clinical improvement and repeated RHC demonstrated a lower MPAP of 33 mmHg, decreased from 47 mmHg. The dosage of epoprostenol had been increased to 22 ng/kg/min. There is no information regarding subsequent transition to oral therapy in this report.
Our patient presented with severe functional class IV PAH that promptly needed epoprostenol therapy, and she clearly had a dramatic response to the treatment. She had complete compensation of right heart size and function as well as acceptable right-sided heart pressure after epoprostenol was slowly weaned that made her a candidate for potential conversion from intravenous therapy to oral therapy. Her therapy was successfully transitioned from intravenous epoprostenol to oral nifedipine without a rebound in MPAP or PVR. Recent (January 2014) communication with the patient confirmed that she continues to do well with stable functional class II symptoms while receiving her current nifedipine dosage.
To date, there is no randomized, controlled trial demonstrating a benefit of any specific PAH medications in this patient group. The review of earlier studies involving PAH therapy for patients with thalassemia and our case are summarized in Table 1.
Epoprostenol is a synthetic prostacyclin, a member of the prostanoids group normally produced by vascular epithelium. It is a potent vasodilator and inhibitor of platelet aggregation in conjunction with its anti-proliferative effect. Long-term therapy for PAH improves symptoms, exercise capacity, and hemodynamics; is approved as the gold standard for advanced PAH; and can improve survival.
Long-term treatment is usually initiated at a dosage ranging from 2–4 ng/kg/min and increased at a rate limited by adverse effects (e.g., flushing, headache, diarrhea, and leg pain). Target dosage for the first 2–4 weeks is usually 10–15 ng/kg/min, and periodic dosage increases are required to maximize efficacy and to maintain the results because of possible tolerance of the drug. Abrupt interruption of the epoprostenol infusion can lead to a rebound worsening of PAH with symptomatic deterioration and even death in some patients.18,19
PAH is often considered an irreversible pulmonary vasculopathy that results in a fixed elevation of PVR; therefore, once intravenous therapy is initiated, it is unlikely to be discontinued. Nonetheless, some patients respond sufficiently to permit transition to oral therapies.20,21 Extended exposure to epoprostenol may reverse the pulmonary vasculopathy or enhance the vasoresponsiveness to oral agents. To that end, Ziesche et al.22 followed up 7 patients with idiopathic PAH who were nonresponsive to inhaled nitric oxide at diagnosis. Treatment with epoprostenol infusion resulted in improved pulmonary hemodynamics and reversal of the initial refractoriness to nitric oxide. Although the long-term effect of epoprostenol presumably has the potential to reverse the pulmonary vasculopathy, this has not been histologically confirmed in humans. Indeed, Rich et al.23 reported a case of a patient with idiopathic PAH treated with epoprostenol for 18 years who did well without right-sided heart failure but unfortunately died from metastatic colon cancer. A postmortem examination revealed severe muscular hypertrophy and intimal fibrosis with occasional complete occlusion of the pulmonary arterial vascular lumen and persistent evidence of cellular proliferation. The RV had severe concentric hypertrophy, but it did not have the characteristic dilatation and interventricular septal distortion that is typically seen in chronic PAH, indicating that epoprostenol did not reverse the patient’s vascular disease. The case illustrated excellent survival in PAH despite an advanced proliferative vasculopathy in support of the hypothesis that preserved RV function is the primary determinant of survival.23
The most significant limitation is that this report represents only a single case experience. In addition, our patient continued to have low 6MWD that did not reach our typical goal of 380 m; however, she was chronically anemic, had a paralyzed hemidiaphragm, and had other comorbidities that may have contributed to her short 6MWD. We also cannot exclude the component of diastolic dysfunction, but PCWP was normal at RHC and the E/e’ ratios by echocardiography were generally less than 12. The strengths of the report include the available hemodynamic data by RHC at the time of epoprostenol to nifedipine transition and the long-term clinical stability represented by functional class II status, normal BNP levels, and normal RV function determined by echocardiogram.
We described a patient with β-thalassemia intermedia complicated by severe PAH confirmed by RHC treated with continuous infusion epoprostenol therapy for over 3 years whose clinical improvement permitted transition to oral nifedipine. Long-term stability since the transition to oral therapy has been documented by subsequent outpatient assessment over the last 9 years. This case would indicate that epoprostenol therapy can be considered as the treatment option in thalassemia patients with advanced PAH with the expectation of clinical improvement and the possibility of long-term survival.
Furthermore, epoprostenol therapy may potentially reverse the pathogenic process to such a degree that transition to oral therapies may be possible. This may be particularly true with the advent of multiple FDA-approved, oral PAH-specific therapies that are now currently available.24,25 Lastly, the report provides additional support for use of PAH-specific therapies in severe PAH associated with thalassemia, now relegated to diagnostic group 5 under the most recent published guidelines.9
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Morris CR, Vichinsky EP. Pulmonary hypertension in thalassemia. Ann N Y Acad Sci 2010;1202:205–213. [DOI] [PubMed]
- 2.Aessopos A, Farmakis D, Deftereos S, Tsironi M, Tassiopoulos S, Moyssakis I, et al. Thalassemia heart disease: a comparative evaluation of thalassemia major and thalassemia intermedia. Chest 2005;127(5):1523–530. [DOI] [PubMed]
- 3.Karimi M, Musallam KM, Cappellini MD, Daar S, El-Beshlawy A, Belhoul K, et al. Risk factors for pulmonary hypertension in patients with β thalassemia intermedia. Eur J Intern Med 2011;22(6):607–610. [DOI] [PubMed]
- 4.Hagar RW, Morris CR, Vichinsky EP. Pulmonary hypertension in thalassaemia major patients with normal left ventricular systolic function. Br J Haematol 2006;133(4):433–435. [DOI] [PubMed]
- 5.Farmakis D, Aessopos A. Pulmonary hypertension associated with hemoglobinopathies: prevalent but overlooked. Circulation 2011;123(11):1227–1232. [DOI] [PubMed]
- 6.Vij R, Machado RF. Pulmonary complications of hemoglobinopathies. Chest 2010;138(4):973–983. [DOI] [PubMed]
- 7.Anthi A, Orfanos SE, Armaganidis A. Pulmonary hypertension in β thalassaemia. Lancet Respir Med 2013;1(6):488–496. [DOI] [PubMed]
- 8.Derchi G, Galanello R, Bina P, Cappellini MD, Piga A, Lai ME, et al. Prevalence and risk factors for pulmonary arterial hypertension in a large group of β-thalassemia patients using right heart catheterization: a Webthal study. Circulation 2014;129(3):338–345. [DOI] [PubMed]
- 9.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013;62(25 suppl):D34–D41. [DOI] [PubMed]
- 10.Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med 1992;327(2):76–81. [DOI] [PubMed]
- 11.Littera R, La Nasa G, Derchi G, Cappellini MD, Chang CY, Contu L. Long-term treatment with sildenafil in a thalassemic patient with pulmonary hypertension. Blood 2002;100(4):1516–1517. [DOI] [PubMed]
- 12.Correale M, De Rosa F, Ieva R, Di Biase M, Brunetti ND. Long-term treatment with high-dose of sildenafil in a thalassemic patient with pulmonary hypertension. Monaldi Arch Chest Dis 2012;78(2):105–106. [DOI] [PubMed]
- 13.Derchi G, Forni GL, Formisano F, Cappellini MD, Galanello R, D’Ascola G, et al. Efficacy and safety of sildenafil in the treatment of severe pulmonary hypertension in patients with hemoglobinopathies. Haematologica 2005;90(4):452–458. [PubMed]
- 14.Morris CR, Kim HY, Wood J, Porter JB, Klings ES, Trachtenberg FL, et al. Sildenafil therapy in thalassemia patients with Doppler-defined risk of pulmonary hypertension. Haematologica 2013;98(9):1359–1367. [DOI] [PMC free article] [PubMed]
- 15.Anthi A, Tsangaris I, Hamodraka ES, Lekakis J, Armaganidis A, Orfanos SE. Treatment with bosentan in a patient with thalassemia intermedia and pulmonary arterial hypertension. Blood 2012;120(7):1531–1532. [DOI] [PubMed]
- 16.El-Beshlawy A, Youssry I, El-Saidi S, El Accaoui R, Mansi Y, Makhlouf A, et al. Pulmonary hypertension in β-thalassemia major and the role of L-carnitine therapy. Pediatr Hematol Oncol 2008;25(8):734–743. [DOI] [PubMed]
- 17.Tam DH, Farber HW. Pulmonary hypertension and β-thalassemia major: report of a case, its treatment, and a review of the literature. Am J Hematol 2006;81(6):443–447. [DOI] [PubMed]
- 18.McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation 2002;106(12):1477–1482. [DOI] [PubMed]
- 19.Galiè N, Torbicki A, Barst R, Dartevelle P, Haworth S, Higenbottam T, et al. Guidelines on diagnosis and treatment of pulmonary arterial hypertension. The Task Force on Diagnosis and Treatment of Pulmonary Arterial Hypertension of the European Society of Cardiology. Eur Heart J 2004;25(24):2243–2278. [DOI] [PubMed]
- 20.Demerouti EA, Manginas AN, Athanassopoulos GD, Karatasakis GT, Leontiadis ED, Pavlides GS. Successful epoprostenol withdrawal in pulmonary arterial hypertension: case report and literature review. Respir Care 2013;58(2):e1–e5. [DOI] [PubMed]
- 21.Kim NH, Channick RN, Rubin LJ. Successful withdrawal of long-term epoprostenol therapy for pulmonary arterial hypertension. Chest 2003;124(4):1612–1615. [PubMed]
- 22.Ziesche R, Petkov V, Wittmann K, Kopatschka J, Stiebellehner L, Schenk P, et al. Treatment with epoprostenol reverts nitric oxide non-responsiveness in patients with primary pulmonary hypertension. Heart 2000;83(4):406–409. [DOI] [PMC free article] [PubMed]
- 23.Rich S, Pogoriler J, Husain AN, Toth PT, Gomberg-Maitland M, Archer SL. Long-term effects of epoprostenol on the pulmonary vasculature in idiopathic pulmonary arterial hypertension. Chest 2010;138(5):1234–1239. [DOI] [PMC free article] [PubMed]
- 24.Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med 2004;351(14):1425–1436. [DOI] [PubMed]
- 25.Galiè N, Corris PA, Frost A, Girgis RE, Granton J, Jing ZC, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol 2013;62(25 suppl):D60–D72. [DOI] [PubMed]