

Abstract Abstract
The endothelial monolayer partitioning underlying tissue from blood components in the vessel wall maintains tissue fluid balance and host defense through dynamically opening intercellular junctions. Edemagenic agonists disrupt endothelial barrier function by signaling the opening of the intercellular junctions leading to the formation of protein-rich edema in the interstitial tissue, a hallmark of tissue inflammation that, if left untreated, causes fatal diseases, such as acute respiratory distress syndrome. In this review, we discuss how intercellular junctions are maintained under normal conditions and after stimulation of endothelium with edemagenic agonists. We have focused on reviewing the new concepts dealing with the alteration of adherens junctions after inflammatory stimulus.
Keywords: vascular endothelium, adherens junctions, permeability, signaling mechanisms
The vascular endothelium forming the innermost lining of all blood vessels performs a vital task of providing nutrients to the underlying tissue as well as maintains tissue oncotic pressure.1-3 Maintenance of the size-selective sieving property of endothelium is crucial for several physiological functions, including normal tissue-fluid homeostasis, angiogenesis, vessel tone, and host defense.2-9 Endothelium is permeable to molecules ranging from 0.1 nm (sodium ion) to 11.5 nm (immunoglobulin G [IgG]) in diameter.2,3,9,10 Interestingly, the sieving property of endothelium linearly correlated with molecular radii from 0.1 nm to 3.6 nm but became independent of the molecular radius above 3.6 nm.2,3 On the basis of these findings, two endothelial transport mechanisms were defined for solute/ion influx.2,3,7,11,12 The transport of macromolecules larger than 3 nm, such as albumin, IgG, and other macromolecules, occurs through the transcellular pathway, which is also called transcytosis or vesicular transport.3,12-14 Molecules smaller than 3 nm, such as glucose, water, and ions, can pass through interendothelial junctions (IEJs) through the paracellular pathway.2,3,14 Both transcellular and paracellular pathways work in concert to maintain tissue-oncotic pressure and thereby maintain the endothelial barrier.3,7,12
Caveolae, a flask-shaped vesicle composed of several caveolins-1 along with other proteins, such as dynamin and intersectin, constitutes the transcellular pathway.2,12,15-18 The paracellular pathway is regulated by a complex interaction of various junctional proteins and actomyosin motors.2,12,17 In an unperturbed endothelium, IEJs dynamically open to allow the passage of small molecules and inflammatory cells for tissue homeostasis and immune surveillance.2,8,9,12,14 Proinflammatory agonists such as thrombin, vascular endothelial growth factor (VEGF), and platelet-activating factor,19-22 by binding to their receptors, disorganize IEJs, leading to increase in endothelial permeability. Thus, to understand how tissue-fluid homeostasis is modulated under normal conditions and pathological processes, we must understand the signaling mechanisms that regulate IEJs.
Endothelial barrier permeability
Endothelial cells originate from embryonic precursor cells known as hemangioblasts.4,5,23,24 However, permeability of endothelial cells under basal conditions and in response to edemagenic agents varies remarkably in different vascular beds.2 Vascular beds of coronary, pulmonary, splenchnic, and skeletal muscle are composed of continuous nonfenestrated endothelial cells, which form a restrictive barrier.5,25 Organs such as liver, kidney, and lymphatics are formed from discontinuous and highly permeable endothelial monolayers.2 Also, segmental differences are observed between endothelial barrier permeability at the cell-culture level and intact vessels of the same vascular bed.5,25-27 Measurement of the coefficient of vascular permeability (Kfc) across the endothelium in isolated perfused lung preparations under basal conditions in experimental animals showed a gradation of permeability across the microvascular bed, with the microvascular site constituting approximately 42%, the arterial region approximately 19%, and the venous area approximately 37%,26 indicating that the arterial system was more restrictive than either venous or capillary linings. In pulmonary microvessels, endothelial cells formed a monolayer that was approximately 4 times less permeable to albumin compared with the confluent monolayer of the cells from venous or arterial regions.2,5 Furthermore, transendothelial electrical resistance (TER), which is a measure of interendothelial adhesion in real time, was lower in cells isolated from larger arteries than in microvascular endothelial cells.2
How can these differences in the endothelial permeability of various vascular beds be accounted for? Studies have shown that the constitution of IEJs, extracellular matrix (ECM), and cytoskeletal proteins may contribute toward segmental variability in the endothelium.4,5,25,28 The arterial segment has approximately 18 times more occludin, a component of tight junctions (TJs), than its venular counterpart, making the arterial endothelial barrier tighter than the venular barrier.2 Also, occludin is found to be expressed in brain endothelial cells more than in any other organ.2,29-31 Microvascular endothelial cells express collagen 4α1, collagen 4α2, and laminin in their ECM, whereas collagen 5α1 and collagen 5α2 are expressed more in macrovascular endothelia.2,28 Moreover, there is an abundance of myosin light-chain kinase (MYLK; serine/threonine–specific protein kinase that phosphorylates the regulatory light chain of myosin II),1,2,32 LIM kinase (actin-binding kinases that phosphorylate members of the actin depolymerizing factor/cofilin family of actin-binding proteins),33,34 Rho-GTPases (Vav),2,32 and myosin motor cytoskeleton in microvascular cells. Microvascular endothelial cells also showed a higher density of caveolae. Besides the above-mentioned intrinsic mechanisms, extrinsic mechanisms, such as shear stress caused by linear versus pulsatile blood flow, were also shown to regulate permeability differences between microvascular and macrovasvular endothelial cells. It has been shown that shear stress increases intracellular Ca2+ and inositol triphosphate (IP3) generation and induces activities of small GTPases, such as Rac and RhoA, and reorganization of actin fibers.2,35,36 Thus, well-organized transcellular and paracellular pathways, by balancing extrinsic fluid dynamics, may impact endothelial barrier phenomena.
Starling and Kedem explained the transport of solute and water across the endothelial barrier.2 Briefly, the rate of the transport of fluid across the endothelial barrier is based on the Starling forces,
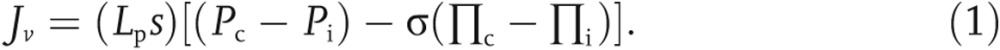 |
Jv denotes volume of flux of fluid, (mL/min); s is the capillary surface area (cm2); Lp is the hydraulic conductivity (cm · min−1 · mmHg−1); Pc and Pi are capillary and interstitial hydrostatic pressures (mmHg), respectively; Πc and Πi are capillary and interstitial colloidal osmotic pressures (mmHg), respectively; and σ is osmotic reflection coefficient of the capillary wall. If the σ = 0, vessel is completely permeable to fluid and solutes, whereas if the σ = 1, capillary is impermeable.2,3,37,38
IEJ proteins
IEJs are composed of adherens junctions (AJs), TJs, and connexins (Fig. 1).2 IEJs are presumed to be covered by an extra layer of fibrous matrix composed of proteoglycans, glycosaminoglycans, and glycolipids known as glycocalyx.2,3,39-41
Figure 1.

Structural organization of endothelial cell intercellular and matrix interactions. Interenthothelial junctions (IEJs) comprised of tight junctions, adherens junctions, and gap junctions interact through actin cytoskeleton with integrin receptors enabling endothelial cell adhesion with contiguous cells and to the underlying matrix. Occludin, claudins, and junctional adhesion molecules (JAMs) are the backbones of tight junctions, whereas vascular endothelial cadherin (VE-cadherin) is required for the formation of adherens junctions. Connexins form gap junctions. Intracellular domains provide junctional stability through their linkages with the actin cytoskeleton via catenins (α, β, γ, and p120) or zona occluden 1 protein (ZO). Gap junctions allow the rapid exchange of information between cells. The cytosolic domains of integrins are linked with actin cytoskeleton through proteins talin and vinculin (Vin), involved in integrin-mediated signaling. Membrane metalloproteases (MMPs) control remodeling of the extracellular matrix (ECM). FN: fibronectin; VN: vitronectin.
Tight junctions
TJs composed of claudin, occludin, and junctional adhesion molecules (JAMs) constitute about 20% of the total junctional proteins in endothelial cells.3,40-43 TJs are well-developed in arterial vessels compared with venular segments. Occludin was the first TJ protein identified in endothelial cells. Occludin interacts with zona occludens (ZO-1) in cytoplasm, which in turn associates with α-catenin and actin cytoskeleton and thus stabilizes endothelial barrier function.3,7,43 Occludin expression in endothelial cells was shown to regulate the tightness of the barrier.2,43 Depletion of occludin using small interference RNA (siRNA) increased endothelial barrier permeability as indicated by the decrease in TER.44 However, occludin-lacking mice showed normal TJs morphology and no apparent change in intestinal epithelial barrier function, indicating compensation in TJs function by TJ proteins.45
Several claudins have been identified to date, but endothelial cells predominantly express claudin 5.46,47 Claudin 5 also binds with ZO-1 protein through its cytoplasmic domain. ZO-1 thus allows TJ proteins to interact with each other and with actin.48 Unlike occludin null mice which survive and grow well, claudin null mice die at 10 h after birth because of the increased permeability of blood brain barrier to small molecules.47
JAM-A, JAM-B, and JAM-C, having 30%–40% sequence homology, belong to immunoglobulin superfamily and participate in formation of TJs in endothelial cells.49-51 Although JAM-B is specifically present in endothelial cells, JAM-A and JAM-C can form TJs in both endothelial cells and epithelial cells. JAM-C is also an important component of lymphatic venular endothelial cells.49,50 JAMs contain PDZ binding motif and may therefore form a platform for mediating the interaction between several proteins containing PDZ domain.51 JAM-A and JAM-C can also be cleaved by membrane-bound metalloproteinase ADAM 10 and ADAM 17 upon activation of endothelial cells by inflammatory agonist, leading to disruption of barrier function and inflammatory signaling.52 Consistently, destabilization of JAM-A protein using anti-JAM-A antibodies prevented the re-annealing of TJs in epithelial cell monolayer.53,54 JAM-A, JAM-B, and JAM-C also participate in transendothelial leukocyte transmigration. However, loss of JAM-A in mice reduced polymorphonuclear leukocyte diapedesis,51,55 indicating JAM-A may play a predominant role in regulating neutrophil transmigration. In line with this idea, it was shown that JAM-A controls the internalization and recycling of β1 integrin after chemotactic stimulation of neutrophils.
ZOs are family members of membrane-associated guanylate kinases.56 Three ZO subtypes have been identified in endothelial cells. ZO-1 interacts with claudins, JAMs, and ocludin. Through PDZ, Src homology 3 and guanylate kinase domains, ZO-1 allow TJ proteins to interact with actin cytoskeleton. Besides TJ protein, ZO-1 is also known to interact with AJ proteins, such as α-catenin, gap junction (GJ) protein conexin-43, actin polymerizing proteins vasodilator-stimulated phosphoprotein (VASP), and spectrin.57 ZO-1 may thereby regulate endothelial permeability by serving as a scaffold for mediating the interaction of TJs with AJs and connexin.56 It has been speculated that ZO-1 stabilizes the endothelial barrier function through the stabilization of F-actin and F-actin itself stabilizes the localization of ZO-1.29,58
Gap junctions
GJs, formed by interaction of two connexins (Cx) from opposite cells, provide a means to facilitate direct cell-to-cell transfer of signaling molecules, ions, current, and transmembrane potential.59 Each connexin is made of six connexin subunits (hexamers). Cx37, Cx40, and Cx43 are expressed in endothelial cells.60 GJs are low-resistance pathways for the flow of current from one cell to another cell.61 Additionally, they allow exchange of information in the form of second messengers, such as Ca2+, IP3, and so on. GJs form a pore size of about 2 nm. Phosphorylation of connexins is shown to regulate GJ function in endothelial cells.34 Serine/threonine and tyrosine phosphorylation of connexins closes the channels, and this prevents intercellular communication. Moreover, phosphorylation of connexins also regulates the assembly and formation of channels. Whether phosphorylation of connexins is casually regulated to alteration in endothelial permeability by edemagenic agonist remains elusive. Recently, Bhattacharya group elegantly demonstrated that bone marrow–derived mesenchymal stromal cells (BMSCs) form GJ channels with the alveolar epithelium through connexin 43, enabling the transfer of mitochondria-containing microvesicles. They demonstrated that BMSC-induced transfer of mitochondria was required for alveolar bioenergetics, thereby preventing endotoxin-induced alveolar leukocytosis and protein leak as well as inhibition of surfactant secretion and mortality.62
Adherent junctions
AJs have a prominent role regulating endothelial barrier function.41 Homotypic adhesion between vascular endothelial (VE) cadherin from contiguous endothelial cells in a calcium-dependent manner forms AJs.2,8,12 VE-cadherin contains 5 extracellular cadherin-like repeats (Fig. 1).2 These cadherin oligomerize to form cis-oligomers or trans-oligomers between adjacent cells. The cytoplasmic tail of the VE-cadherin contain two domains, namely juxtamembrane domain (JMD) and COOH-terminal domain (CTD).2,12,40 These cytoplasmic domains interact with three Armadillo family proteins known as β‐catenin, plakoglobin (γ‐catenin), and p-120 catenin. JMD binds with β‐catenin, whereas CTD domain interacts with plakoglobin and p-120 catenin.40,63 β‐catenin or plakoglobin then binds with α‐catenin and links the whole complex with actin cytoskeleton.64,65
VE-cadherin is required for forming vasculature, because loss of VE-cadherin embryonically induces lethality at E9.5 d.2,12,66 Neutralization of VE-cadherin with anti-VE-cadherin antibody or chelation of extracellular calcium using EDTA induce endothelial barrier leak. Truncation of the β-catenin binding site at the cytosolic domain of VE-cadherin or conditional inactivation of β‐catenin induces lethality in mice because of the disruption of endothelial junctions.67 Expression of VE-cadherin mutant lacking the extracellular domain but with an intact β‐catenin binding site was shown to disrupt endothelial barrier function. Overexpression of JMD of VE-cadherin, depleting endogenous p120-catenin, disrupted AJs, leading to the increase in endothelial permeability.40,68 Interestingly, depletion of p120-catenin in endothelial cells also modulated lung innate immune function by interfering with the association of Toll-like receptor 4 (TLR4) and its adaptor myeloid differentiation primary response gene (MyD88), preventing nuclear factor κ B cell (NF‐κB) activation and thereby the lung inflammatory response to endotoxin.69 These findings suggest that β‐ and p120-catenin bound to VE-cadherin maintains endothelial permeability and innate response.
Signaling mechanisms regulating AJs
As discussed above, alteration of AJs adhesion primarily determines endothelial permeability to plasma proteins. In the following section, we describe the role of the signaling pathways that regulate the opening and resealing of AJs during the phases of increased permeability and of restoration of barrier function (Fig. 2).
Figure 2.

Signaling modulating adherens junctions (AJs). Agonists increase intracellular Ca2+, which, by activating the myosin light-chain kinase (MLCK) and RhoA-Rho kinase pathway, induces actin stress fiber formation, leading to disruption of AJs. Additionally, protein kinase C (PKC), Src, and end-binding protein 3 (EB3) disrupt AJs adhesion by either phosphorylating them or by increasing microtubule dynamics. Several mechanisms are activated in parallel or synergistically to induce re-annealing of AJs. For example, focal adhesion kinase (FAK) phosphorylates neural Wiskott–Aldrich syndrome protein (N-WASP), enabling actin-related protein 2/3 (Arp2/3) to mediate cortical actin formation. Activated N-WASP also links p120-catenin to Arp2/3 and actin to stabilize AJs. FAK also induces Rac1 activity by inhibiting RhoA activity, which may further stabilize AJs. Activated Cdc42 may merge with FAK signaling to restore AJs formation.
Role of actin-myosin motor
Endothelial cells contract in response to permeability-increasing agonists, leading to opening of IEJs. Stress fibers composed of bundles of polymerized actin and myosin filaments form the contractile machinery of endothelial cells.2 These fibers assemble in a characteristic manner in cultured endothelial cells in response to permeability-increasing mediators.12,70 Thrombin, an edemagenic agonist, increased isometric force in endothelial cell monolayers grown on a collagen matrix from a basal value of 70 to 132 dynes.71,72 However, endothelial cells developed comparatively less tension (1.3 × 105 dynes/cm2) than did contracting arterial smooth muscle cells (6.7 × 106 dynes/cm2),71,72 perhaps due to strong interendothelial adhesion between endothelial cells. Actinomyosin-generated contraction requires MYLK-dependent phosphorylation of the regulatory myosin light-chain (MLC) on Ser-19 (monophosphorylation) or Ser-19/Thr-18 (diphosphorylation). On thrombin stimulation of human umbilical vein endothelial cells, isometric contraction increased twofold to 2.5-fold within 5 minutes, whereas MLC phosphorylation increased from a basal value of 0.54 to 1.61 mol PO4/mol MLC within 60 seconds, primarily as the result of diphosphorylation of MLC.73-79 These studies showed diphosphorylation of MLC as an important determinant of endothelial contraction. MLC phosphorylation has also been shown to regulate the permeability of intact isolated postcapillary venules and pulmonary microvessels in response to histamine and thrombin.80-82 Mice lacking only the long isoform of MYLK present in endothelial cells support the in vivo role of MYLK-induced endothelial contraction in the mechanism of increased vascular permeability.82 Mylk−/− mice were protected against increased lung vascular permeability and neutrophil adhesion induced by intraperitoneal injection of the bacterial product lipopolysaccharide (LPS),82 indicating a critical role of endothelial contraction induced by MYLK in mediating the increase in vascular permeability and inflammatory signaling.78
Role of calcium
An increase in cytosolic Ca2+ precedes changes in endothelial cell shape and the opening of IEJs.22,70,83,84 After cell stimulation with inflammatory mediators, an increase in cytosolic Ca2+ concentration is apparent in two phases.22,70,84 There is an initial transient peak as the result of Ca2+ release from endoplasmic reticulum (ER) stores, which is followed by a more sustained response secondary to Ca2+ entry via plasmalemma channels. Ca2+ entry refills ER stores and sustains Ca2+ signaling.84
Transient receptor potential (TRP) family has emerged as a predominant regulator of nonselective Ca2+ entry in endothelial cells.22,70,85,86 Approximately 28 TRP channels have been described so far in mammalian cells, which include TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPP (polycystin), TRPML (mucolipin), and TRPA (ankyrin). TRPC, TRPM, and TRPV all have six transmembrane domains with a pore-forming unit located between transmembrane domains 5 and 6.2,87-89 TRPC and TRPM family members also contain protein-rich sequences in the C-terminal region of the TRP domain designated as TRP box 2, which binds phosphatidyl insositol (PI) phosphates, such as PI (4, 5) P2.2,87,89 TRPC and TRPV family members also contain 3–4 ankyrin repeats at their N-terminus.2,87,90 The functional TRP channel is a tetramer and may be composed of homo- or heterotetramers. Evidence suggests that most of the TRP channels are modulated by Ca2+ itself, which may form positive or negative feedback signaling loops. In addition to their direct involvement in mediating Ca2+ entry, TRP channels are shown to be linked functionally with voltage-gated Ca2+ channels and to thereby amplify Ca2+ entry through them in a voltage-dependent manner.91,92
TRPC channels are by far the most studied in endothelial cells.83,90,93,94 Expression of the various TRPCs differs among human endothelial cells, which may contribute to endothelial heterogeneity. The majority of human endothelial cells highly express TRPC1 and TRPC6, whereas TRPC3, TRPC4, and TRPC7 are expressed only weakly.70,90,95,96 TRPC1, TRPC4, and TRPC5 are activated by depletion of ER stores by IP3 and are therefore called store-operated channels (SOCs), whereas TRPC3 and TRPC6 are activated by diacylglycerol (DAG) independently of store depletion and are referred to as receptor-operated channels (ROCs).70,83,95,96 SOCs and ROCs provide the primary structures by which Ca2+ enters endothelial cells. Multiple heterologous combinations of TRPCs (TRPC1 with TRPC4 or TRPC5 and TRPC3 with TRPC6) combine to produce tetrameric channels with unique properties.83,86,90,97
SOC activity
TRPC1 and/or TRPC4 have been clearly shown to regulate SOC activity and thereby IEJs.22,85,86,98,99 Alterations of TRPC1 function in endothelial cells using TRPC1 antisense or an anti-TRPC1-blocking antibody reduced Ca2+ entry by 50% and also decreased endothelial permeability in response to thrombin.22 Overexpression of TRPC1 in endothelial cells increased Ca2+ entry and, in turn, the formation of actin stress fibers, and it also increased endothelial permeability.100 In addition, the in vivo relevance of TRPC-induced Ca2+ entry in regulating permeability of microvessels has been demonstrated using TRPC4−/− mice.90 The PAR-1 (thrombin receptor) activation-induced increase in lung microvessel permeability in mice was reduced by 50% using lanthanum (La3+), a known Ca2+ channel blocker, indicating Ca2+ entry by this pathway contributed to a portion of the increased vascular permeability response. Trpc4−/− mice also demonstrated a reduction in the thrombin-induced increase in lung microvessel permeability by ∼50%, whereas this response was not affected by La3+.85
Evidence from human endothelial cells indicates that a coupling between the IP3 receptor and TRPC1 could be mediated through small GTPase RhoA, because inhibition of RhoA prevented the thrombin-stimulated association of the IP3 receptor with TRPC1 and influx of Ca2+.2,22 RhoA may facilitate the interaction of the IP3 receptor with TRPC1 by promoting actin polymerization.22 Studies also showed that TRPC1 activity was suppressed by depolymerization of actin filaments consistent with this model.22,101 In addition, SOC-induced Ca2+ entry (SOCE) was shown to require the interaction of spectrin, an actin cross-linking protein, with protein 4.1.102,103 Another signaling possibility is that phosphorylation by protein kinase C (PKC) α is required for TRPC1 activation.98 MYLK may also be involved in the regulation of SOC-induced Ca2+ entry, because inhibition of MYLK with ML7 prevented the SOC-mediated Ca2+ entry in endothelial cells.104 Together, these findings raise several possibilities concerning the mechanism of SOC activation and influx of Ca2+ in endothelial cells.
STIM1, a type 1A single-transmembrane protein originally identified as a tumor-suppressor protein, has been established as a Ca2+ sensor within the ER stores.105,106 STIM1 contains N-terminal EF hand domain, a Ca2+ binding domain, a sterile α motif or SAM domain, a single transmembrane domain (TM), an Ezrin-radixin-moesin (ERM) domain, a serine-proline–rich region (S/P-region), and a lysine-rich region (E-region).105,107,108 The Ca2+-binding EF domain and SAM domain regulate aggregation of STIM1 as well as its translocation to the plasma membrane from ER. When bound with Ca2+ in ER lumen, STIM1 exists as an individual unit or a monomer. Upon store depletion, the EF hand domains and SAM domains from different STIM1 protein aggregate and form multimeric puncta, which leads to its interaction with store-operated Ca2+ channel and activation of SOCE.84,106,108 STIM1 has been shown to regulate TRPC channels, such as TRPC1, TRPC4, TRPC5, and ORAI channels, which are the important constituents of SOC and CRAC channels, respectively.109,110 EF hand mutant STIM1D76A does not bind with Ca2+ and is constitutively active, whereas ΔERM-STIM1 mutant acts as dominant negative STIM1.111 Tiruppathi’s group showed that SOC activates p38 MAP kinase, which, by phosphorylating STIM1, suppresses the endothelial SOCE.84 These findings suggest that a negative feedback loop may be important in terminating SOCE and thereby restoring AJs.
STIM2 shares a sequence homology between the N-terminal domain as well as transmembrane sequences with STIM1 and is also present in endothelial cells as well as other cells. STIM2 differs from STIM1 protein in the C-terminal region. STIM2 deletion seems to alter basal cytosolic Ca2+ but not SOCE.108,112 However, overexpression of STIM2 inhibited SOCE in HEK and smooth muscle cells as well as abolished CRAC activity in jurkat cell line.108,113,114 Thus, unlike STIM1, the expression of STIM2 is inversely linked with SOC and CRAC activities.108,114
ROC activity
TRPC6 is highly expressed in lung and lung endothelial cells.70,83,115 Several studies showed that TRPC6 plays an important role in regulating endothelial permeability.20,70,83,93 Flufenamic acid, a TRPC6 activator, increased water conductivity in frog mesenteric vessels.20,93 Singh et al.70 showed that siRNA-induced suppression of the TRPC6 channel in human pulmonary artery endothelial cells decreased endothelial cell permeability in response to thrombin. Recently, we showed that TRPC6 plays a key role in signaling both LPS-induced lung vascular permeability and inflammation.83 Tauseef et al.83 showed that LPS resulted in DAG production, which activated TRPC6. Activated TRPC6 induced MYLK activity that, by stimulating actomyosin cross-bridging, mediates endothelial cell contraction, leading to increased lung vascular permeability.83 Additionally, activated MYLK promoted the interaction of MyD88 with inteterleukin receptor 1–associated kinase 4, which is involved in triggering NF-kB signaling and pulmonary inflammation downstream of TLR4.83 Thus, TRPC6 appears to be at the center of the signaling pathways mediating acute lung injury (ALI) owing to its dual role in increasing lung vascular permeability and mediating TLR4 signaling (Fig. 3).83
Figure 3.

Ca2+ entry through canonical transient receptor potential (TRPC) mediates loss of endothelial barrier function and inflammatory signaling. Agonists such as thrombin and vascular endothelial growth factor (VEGF), by activating their receptors on the endothelial cell surface, lead to generation of inositol triphosphate (IP3) and diacylglycerol (DAG). IP3 induces Ca2+ entry through store-operated channels (SOCs) such as TRPC1 and TRPC4. DAG directly activates the receptor-operated channel (TRPC6). Endotoxin lipopolysaccharide (LPS), by activating Toll-like receptor 4 (TLR4), induces DAG production, which activates TRPC6. Activated TRPC6 induces myosin light-chain kinase (MYLK) activity that, by stimulating actomyosin cross-bridging, mediates endothelial cell contraction, leading to increased lung vascular permeability. Additionally, activated MYLK promotes myeloid differentiation primary response gene (MyD88) interaction with interleukin-1 receptor-associated kinase 4 (IRAK4), triggering nuclear factor κ B cell (NF‐κB) signaling and pulmonary inflammation. CD14: cluster of differentiation 14; ER: endoplasmic reticulum; PAR-1: protease activating receptor 1.
Role of PKC
PKC forms the family of protein kinase enzymes that are involved in controlling the function of several other proteins through the phosphorylation of hydroxyl groups of serine and threonine amino acid residues on these proteins.64 PKC are activated by signals such as increases in the concentration of DAG or cytosolic calcium ions (Ca2+).2,70 The PKC family consists of conventional (or classical), novel, and atypical isoforms.116 Conventional PKCs (α, βI, βII, and γ) require Ca2+, DAG, and a phospholipid, such as phosphatidylserine, for activation.116-118 Novel PKCs, which include δ, ε, η, and θ isoforms, are activated by DAG independent of Ca2+, whereas atypical PKCs, such as PKCζ, require neither Ca2+ nor DAG for activity.2,116,119 Thus, conventional and novel PKCs are activated through the same signal transduction pathway as phospholipase C.
PKCα has a crucial role in mediating IEJ disassembly,98 although Ca2+-independent isoforms, notably PKCζ, may also be important in IEJ disruption.119 Upon activation by endothelial permeability–increasing agonists, PKCα translocated from the cytosol to the IEJ, where it phosphorylated p120 catenin at serine 879, which destabilized junctions and increased endothelial permeability. Vandenbroucke St Amant et al.64 showed that p120 catenin phosphorylation at serine-879 exposed the VE-cadherin binding site for endocytic protein AP1, leading to internalization of VE-cadherin. Expression of phosphorylation-deficient mutant of p-120 catenin mutant in mouse lung microvessel failed to induce lung vascular permeability in response to thrombin receptor activation, thereby identifying p-120 phoshorylation at serine 879 residue as a crucial regulator of VE-cadherin internalization and endothelial monolayer permeability.64
PKCβ and PKCζ inhibition also decreased endothelial permeability through impairment of Ca2+ levels, MLC contraction, and RhoGTPases.120-123 Studies have shown that PKCδ is required for the permeability increase caused by stimulation with PKC activators or DAG, whereas others suggest that PKCδ has a barrier-protective function.123 Overexpression of PKCδ decreases endothelial permeability by increasing focal adhesion contacts.123 Additional approaches, such as using PKCβ or PKCδ knockout mice models, will help to clarify the contribution of these isofoms in mediating increased endothelial permeability.
Role of Rho-GTPases
Evidence supports the general concept that each of the GTPases, RhoA, Rac, and Cdc42, contribute to the regulation of AJ function, but their relationship with AJs appears to be complex, because AJs themselves can modify RhoGTPase activity.7,8,124 RhoA was found to induce disassembly of endothelial AJs in response to thrombin, histamine, bradykinin, or PAF.125 Inhibition of Rac with Clostridium sordellii toxin (a specific inhibitor of Rac) caused AJ disruption in cultured endothelial cells and increased Lp of rat venular microvessels,126 implicating Rac in promoting barrier function by stabilizing AJs. Schmidt et al.127 recently showed that a fine balance between the activities of RhoA and Rac1 GTPases is required for maintaining AJs in mice lungs. They showed, using a mouse model lacking endothelial focal adhesion kinase (FAK, described below), that, in the absence of FAK, activated RhoA antagonized Rac1 activity, destabilizing AJs. Inhibition of Rho kinase, a downstream effector of RhoA, reinstated normal endothelial barrier function in FAK−/− endothelial cells and lung vascular integrity in EC-FAK−/− mice. The mechanism by which RhoA antagonizes Rac1 remains to be identified. FilGAP, a GTPase, specifically inhibits Rac1 downstream of RhoA. Thus, it is possible that RhoA may induce FilGAP activity to suppress Rac1 and thereby disrupt AJs. Kouklis et al.128 showed that the re-annealing of AJs following the increase in endothelial permeability by thrombin required the activation of Cdc42. Mice or ECs transducing activated form of Cdc42 showed reduced barrier leak, indicating that Cdc42 activity preserved junctional integrity. RhoA activity in V12Cdc42-expressing endothelial monolayers was reduced, compared with untransfected cells, suggesting that activated Cdc42 functions by counteracting the canonical RhoA-mediated mechanism of endothelial hyperpermeability.129
Role of FAK
FAK, a 120-kD nonreceptor tyrosine kinase, regulates turnover of focal adhesion formation by binding to focal adhesion proteins, such as vinculin, talin, and paxillin.2,127 Global as well as endothelial cell–specific deletion of FAK induces lethality in embryos because of improper formation of blood vessels. Several studies have shown that FAK maintains basal endothelial barrier function and induces resealing of endothelial junctions following the increase in endothelial permeability by thrombin or H2O2. For example, FAK was shown to bind with p120-catenin and suppress the activity of the small GTPase RhoA to restrict endothelial contraction and thereby inducing AJs formation.109 Quadri et al.130 also showed that FAK prevents oxidant-induced barrier dysfunction by regulating VE-cadherin phosphorylation. Also, siRNA-induced depletion of FAK or expression of dominant FAK impaired AJ formation and enhancement of barrier function by sphingosine 1 phosphate and lysophosphatidic acid.131 However, expression of dominant negative FAK or kinase-dead FAK mutant in endothelial cells prevented AJ disruption in response to VEGF or oxidants, indicating FAK infact disrupts the barrier function.132 To resolve the role of FAK in regulating lung vascular barrier function, Schmidt et al.127 generated tamoxifen-inducible endothelial-specific FAK knockout mice. These authors showed that loss of EC-FAK induced diffuse lung hemorrhage and increased transvascular albumin influx, edema, and neutrophil accumulation in the lung due to disruption of AJs. Induction of ALI by intraperitoneal administration of LPS or cecal ligation and puncture in wild-type mice markedly decreased FAK expression in lungs, indicating that decreased endothelial FAK expression is therefore a likely factor responsible for the pulmonary vascular hyperpermeability and edema formation seen during ALI.
Role of Src kinase
Src activation occurs downstream of integrin activation that follows integrin clustering. Among various members of Src family kinases, pp60Src kinase has been shown to contribute to increased endothelial permeability in response to production of superoxide anion (O−),133 thrombin,134 and VEGF.135 VEGF, histamine, and thrombin resulted in Src activation and tyrosine phosphorylation of VE-cadherin at Y658 and Y731, which correspond to the binding sites for p120 and β-catenin, respectively.136 Phosphorylation of VE-cadherin resulted in internalization of the protein leading to disruption of barrier. In addition, three other tyrosine residues, namely Y645, Y685, and Y733, in VE-cadherin were identified as phosphorylated. More importantly, point mutations Y658F and Y685F prevented internalization of VE-cadherin and thus blocked vascular permeability.137 The endogenous phosphatase for VE-cadherin, VE-PTP, is frequently associated with the protein and prevents VE-cadherin phosphorylation, promoting an important stabilizing role of endothelial contacts in vivo.138 How VE-PTP and p60Src compete for binding VE-cadherin to regulate AJs needs to be parsed out.
Proline-rich tyrosine kinase 2 (PYK2), a Ca2+-dependent noneceptor tyrosine kinase structurally related to FAK, also binds to integrins and is highly expressed in pulmonary endothelial cells.139 PYK2 is rapidly phosphorylated following exposure of endothelial cells to agonists (e.g., angiotensin) and mechanical stress (e.g., cyclic stretch).140 Evidence indicates that PYK2 can activate RhoA in fibroblasts,141 suggesting that PYK2 may be involved in regulating endothelial barrier function by this mechanism.
Role of end-binding proteins
Microtubules form a lattice of rigid hollow rods spanning the cell in a polarized fashion from nucleus to periphery. Microtubules are presumed to resist compression of cells by opposing actin-myosin contractility.142 Microtubules (typically having a diameter of ˜25 nm) are heterodimers organized by self-assembly of α‐ and β‐tubulins (molecular weight of 55 kDa each).142 Microtubules undergo “dynamic treadmilling” (i.e., alternating addition and removal of tubulin dimers from the microtubule).8 The faster-growing end, the plus end, is made up of β‐tubulin that binds to and hydrolyzes GTP to GDP, whereas α‐tubulin constitutes the minus end and binds to but does not hydrolyse GTP. The plus ends of microtubules are attached to the cell cortex and exhibit “dynamic instability,” because they shift between phases of lengthening and shortening.143 The minus ends are typically joined at a common attachment site in the cell called the microtubule-organizing center.144 Disruption of MT is shown to impair AJs. Likewise, stabilzation of MTs prevented AJs disruption by ermeability-increasing agonists.144
Endbinding protein (EB) family members are evolutionary conserved proteins that bind MT+ ends.145 EB1 and EB3 are ubiquitously expressed, whereas expression levels of EB2 vary between cell lines.146 They can form homo- or heterodimers.147 Komarova et al.148 recently identified a key role of EB3 in regulating AJs adhesion by stabilizing MTs. They showed that phosphorylation of EB3 at S162 induced MT growth in confluent endothelial monolayers, which disassembled AJs. Whether EB3 bound directly with VE-cadherin and thereby orchesterated VE-cadherin homophilic interaction remains unclear.
Role of neural Wiskott-Aldrich syndrome protein
The cortical actin, the actin ring near the plasma membrane, stabilizes AJs. Disruption of preformed actin filaments by C2 toxin or cytochalasin is known to increase endothelial permeability in intact microvessels and cultured cells. Edemagenic agonists, such as thrombin, rapidly induce actin reorganization from cortical actin into actin stress fibers, which induce AJ disruption. In this regard, proteins that maintain cortical actin confirmation play a key role in maintaining AJs.
The actin-related protein 2/3 (Arp2/3) complex generates actin filament-driven protrusive force, which may stabilize AJs. Arp2 was shown to interact with E-cadherin, indicating protein-regulating Arp2 function may contribute to AJs stability. The family of Wiskott–Aldrich syndrome proteins (WASPs) share similar domain structure and are involved in transduction of signals from receptors on the cell surface to the actin cytoskeleton. Neural WASP (N-WASP) induces actin polymerization by activating Arp2.149 In resting cells, N-WASP is auto-inhibited due to binding of C-terminus verprolin central acidic (VCA) domain with its GTPase domain. Several mechanisms have been implicated in facilitating the unfolding of the VCA domain from the GBD domain, which includes WASP phosphorylation by tyrosine kinases, interaction with PIP3, or Cdc42 GTPases.150 Rajput et al.151 showed that the depletion of N-WASP impaired AJ adhesion and promoted actin organization and stress fiber formation, leading to leaky endothelial barrier. These authors showed that N-WASP formed a complex with Arp2/3 and p120 catenin. Knockdown of Arp2 did not inhibit N-WASP interaction with p120 catenin, suggesting that N-WASP binds directly to p120 catenin and induces cortical actin formation, thereby stabilizing AJs.151 However, N-WASP phosphorylation at Y256 residue by FAK was required for reformation of cortical actin and p120-catenin interaction, because expression of phosphodefective N-WASP mutant impaired its interaction with p120-catenin and Arp2, leading to disruption of AJs and loss of endothelial barrier function. Overexpression of phosphomimicking mutant of N-WASP, on the other hand, enhanced basal barrier function and facilitated re-annealing of AJs and barrier restoration. These findings suggest that FAK-mediated N-WASP phosphorylation is a critical event in maintaining AJs and in restoration of endothelial barrier.151
Mediators of increased endothelial permeability
Inflammatory mediators
Histamine, thrombin, bradykinin, VEGF, and Ang2 are shown to disrupt AJs, thereby leading to increase in endothelial permeability. Thrombin disrupts AJs by binding to its receptors, PAR-1, on endothelial cells.95 Thrombin induces proteolysis of the PAR-1 extracellular extension (between Arg 41 and Ser 42), generating PAR1 ligand which, by binding to the second extracellular loop of heptahelical PAR-1, initiates downstream signaling events.152 The synthetic 14–amino acid peptide, SFLLRNPNDKYEPF (TRP-14), can substitute for this tethered ligand.153 Three other isoforms of PARs, PAR2, PAR3, and PAR4 have been identified in endothelial cells, of which only PAR3 and PAR4 are activated by thrombin.154 PAR-2 is activated by tryptase.155 The role of the other PARs in mediating increases in endothelial barrier permeability has not been fully elucidated, but those PARs may work in concert with PAR1 to facilitate changes in barrier function. For example, the transactivation of PAR2 by thrombin ligation of PAR1 has been demonstrated in COS7 cells and in HUVECs, which in turn contributes to thrombin signaling.156 Selective activation and desensitization of PAR2 reduces subsequent responses to thrombin, suggesting that the transactivation of PAR2 by cleaved PAR1 occurs normally. A study showed that PAR2 activity was required for reversing PAR1-induced vascular leak following sepsis.157 Similarly, it has been shown that PAR2 agonism exerts anti-inflammatory effects following LPS-induced lung injury in rodents.158 Therefore, although PAR1 mediates thrombin-induced increase in lung vascular permeability, the role of other PARs in amplifying or suppressing thrombin-induced increase in endothelial permeability has yet to be understood. The role of PAR-3 and PAR-4 in mediating an increase in endothelial permeability has not been established, but evidence indicates that they may cooperate with PAR1 to increase endothelial permeability. PAR-1 induces a transient increase in vascular permeability that is followed by an equally rapid restoration of barrier function. PAR1 couples to the heterotrimeric G proteins of the Gq, G12/13, Gi, and Gs families,159,160 and initial studies predicted all of the G proteins to mediate increases in endothelial barrier permeability. It has been generally accepted that thrombin increases endothelial permeability by activating MYLK and RhoA via the Gαq and Gα12/13 subunits; however, Gαs enhances barrier function through cAMP generation.80 Additionally, dissociated Gβγ subunits have also emerged as barrier protective by inducing FAK activity.161 Studies also suggest that spingosine kinase 1 mantains AJs and reverses the increase in endothelial permeability by thrombin by paracrine mechanism involving S1P generation.162
Histamine increases endothelial permeability through increasing intracellular calcium and MYLK activation but, in addition, mediates Src-dependent phosphorylation of AJ and TJ proteins. Conversely, bradykinin, acting through B1 and B2 receptors, results in an eNOS/iNOS-dependent increase in permeability, although it is unclear whether nitrosylation of junctional proteins leads to barrier destabilization.6
Sphingosine 1 phosphate (S1P) circulates at high levels in the blood and signals through the G-coupled protein receptor S1P receptors (S1PR), including S1PR1, S1PR2, and S1PR3.163 S1PR1 is predominantly studied in endothelial cells and is shown to strengthen endothelial barrier integrity (discussed below). Garcia’s group recently showed a barrier-disruptive role of S1PR3.164 They showed that human lung vascular ECs shed S1PR3 in microparticles following activation with LPS or low–molecular weight hyaluronan. Exposure of normal lung ECs to S1PR3-containing microparticles significantly reduced TER, consistent with increased permeability. These changes were attenuated by RNAi-mediated depletion of S1PR3 in ECs. Intriguingly, elevated total S1PR3 plasma concentrations were linked to sepsis and ALI mortality, indicating S1PR3 as a novel ALI biomarker linked to disease severity and outcome.164
Long-term mediators of permeability, such as endotoxin, LPS, and tumor necrosis factor, result in NF-κB transcriptional expression of cytokines and leukocyte adhesion molecules.2,7 ICAM-1 cell surface activation results in RhoA-directed stress fiber formation as well as increased NO production, which further potentiates increased permeability. Many studies have shown that the ECs are crucial in mediating the lung’s inflammatory response by LPS. LPS binds the endothelial TLR4 via CD14, a membrane-bound glycosylphosphatidyl inositol–anchored protein. TLR4 in turn activates signaling pathways responsible for the generation of proinflammatory cytokines via MyD 88, which in turn activates the NF‐κB signaling and produces the proinflammatory cytokines. We have recently shown that TRPC6 mediates Ca2+ entry into ECs secondary to TLR4-induced DAG generation, which participates in mediating both lung vascular barrier disruption and inflammation induced by endotoxin.83
VEGF induces vascular permeability by several mechanisms, including AJ remodelling, induction of fenestrae, and VVOs formation.165 VEGF concurrently activates multiple signaling pathways downstream of VEGFR2 that have been implicated in vascular permeability. These include PLC-dependent intracellular calcium release, Src kinase–mediated phosphorylation/internalization of junctional proteins, RhoGTPase activation, cytoskeletal rearrangement, and eNOS signalling.166 More recently, in vivo data showed that VEGF mediates these effects by inducing VE-PTP/VE-cadherin dissociation167 and FAK-dependent β-catenin phosphorylation.132 Furthermore, the T cell–specific adapter, TSAd, was found to be essential for Src activation and subsequent phosphorylation of junctional proteins downstream of VEGFR2.168 The contribution of eNOS signaling upon VEGFR2 activation has remained elusive, but recent data suggest that nitrosylation of β-catenin by NO may be an additional mechanism of junctional destabilization by mediating the dissociation of β-catenin from VE-cadherin.169
Ang-2 sensitizes the endothelium to both growth factors and inflammatory mediators, which increases vascular destabilization.170 Although both Ang-1 and Ang-2 mediate Tie-2 clustering at cell-cell contacts, their differential signaling may explain their opposing effects on vascular stability. Several groups have also demonstrated that Ang-2 can act as a partial agonist of Tie-2 signaling through Tie-2 phosphorylation171 and can enhance barrier function following endothelial stress. Generation of mice with endothelial-specific deletion of Ang-2 will help to address its physiological role in vivo and enable a better understanding of the homeostatic functions of Ang-2 in the endothelium. To determine the role of Ang2 in sepsis, Ang2 antibody treatment attenuated LPS-induced hemodynamic alterations and reduced the mortality rate at 36 hours from 95% to 61%. These data clearly indicate that Ang2-mediated microvascular disintegration contributes to septic shock and suggest that inhibition of the Ang2/Tie2 interaction during sepsis is a potential therapeutic target.172
Endothelial barrier–stabilizing mediators
Barrier restoration is critical for maintenance of basal permeability and recovery following exposure to acute inflammatory events, yet our understanding of how this process occurs at the molecular level has remained elusive. Here, we discuss some of the mediators of barrier stability and their known mechanisms of action.
Increase in cAMP levels reduces vascular leakage through activation of protein kinase A (PKA) and the guanine exchange factor, Epac. Epac-mediated Rap1 activation results in increased junctional adhesions and reorganization of actin filaments.173 Emerging evidence on Rap1 suggests it has a cooperative association with VE-cadherin, because they can both modulate each other’s responses. Interestingly, Rap1 can increase Krev interaction trapped gene 1 (KRIT-1) targeting to endothelial cell-cell junctions, suppressing stress fiber formation and stabilizing the barrier (KRIT-1 is known to bind to Rap1, a guanosine triphosphatase that maintains the integrity of endothelial junctions). Thus, defects in Rap1 signaling downstream of mutated KRIT-1 protein may explain the loss of vascular integrity seen in cerebral cavernous malformations.174
More recently, fibroblast growth factor (FGF) has been found to play an important role in AJ integrity. Absence of FGF signaling was found to reduce expression of the phosphatase SHP2, resulting in increased phosphorylation of VE-cadherin, impairing its ability to bind p120 catenin.175 VE-cadherin itself can affect barrier stability by inhibiting growth factor signalling pathways, including VEGF, TGFβ, and PDGF, which promote permeability following angiogenic responses.41
Another emerging and potent barrier-stabilizing factor is sphingosine-1-phosphate (S1P). S1P mediates its effect on endothelial cells by ligating S1PR1 to induce cortical actin organization via a number of downstream targets, including Rac-1, cortactin, FAK, paxillin, and actinin 1 and 4.129 Two recent studies have unequivocally demonstrated the effect of S1P in barrier stability in vivo. Pharmacological or genetic blockade of the S1P signaling axis results in AJs destabilization, permeability, and, in some cases, aberrant angiogenesis.
Tie receptors and their ligands (Ang1–4) are critical regulators of vascular maturation and quiescence.176 One of the main questions in understanding Ang-1 signaling is how it can orchestrate both vascular remodelling and quiescence by signalling through the same receptor. Recent evidence suggests that Ang-1 stimulation leads to differential Tie-2 localization and signaling, depending on whether endothelial cells have engaged cell-cell contacts. Homotypic cell interactions between endothelial cells trigger recruitment of Tie2 to cell-cell contacts upon Ang-1 exposure, leading to enhanced vascular stability following Akt-mediated eNOS phosphorylation.177
Summary
In this review, we have emphasized the role of signaling mechanisms regulating AJs and thereby regulating endothelial permeability. In general, increase in intracellular calcium by TRPC and consequent activation of RhoA and MLCK induce AJ disruption. In parallel, overlapping signals are initiated by kinases such as Src, PKC, and CAMK to induce AJ disruption either directly by mediating the phosphorylation of AJ components or through EB proteins. We have also discussed the roles of Rac1, Cdc42, FAK, SPHK1, N-WASP, and Rap1 in opposing AJ disruption in facilitating barrier formation. We believe underpinning of the commonalities of these complex signaling pathways will define targets for facilitating AJ formation and certainly will pave the way for many new therapeutic and prophylactic interventions.
Source of Support: This work was supported by National Institutes of Health grants HL71794, HL84153, and HL060678 as well as an American Heart Association postdoctoral fellowship to SS (12 Post 12080112).
Conflict of Interest: None declared.
References
- 1.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol (1985) 2001;91:1487–1500. [DOI] [PubMed]
- 2.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev 2006;86:279–367. [DOI] [PubMed]
- 3.Vogel SM, Malik AB. Cytoskeletal dynamics and lung fluid balance. Compr Physiol 2012;2:449–478. [DOI] [PubMed]
- 4.Aird WC. Endothelial cell heterogeneity. Cold Spring Harb Perspect Med 2012;2:a006429. [DOI] [PMC free article] [PubMed]
- 5.Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res 2007;100:158–173. [DOI] [PubMed]
- 6.Chavez A, Smith M, Mehta D. New insights into the regulation of vascular permeability. Int Rev Cell Mol Biol 2011;290:205–248. [DOI] [PubMed]
- 7.Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann N Y Acad Sci 2008;1123:134–145. [DOI] [PubMed]
- 8.Komarova YA, Mehta D, Malik AB. Dual regulation of endothelial junctional permeability. Sci STKE 2007;2007:re8. [DOI] [PubMed]
- 9.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol 2007;7:803–815. [DOI] [PubMed]
- 10.Wang Z, Malik AB. Nanoparticles squeezing across the blood-endothelial barrier via caveolae. Ther Deliv 2013;4:131–133. [DOI] [PMC free article] [PubMed]
- 11.Dvorak AM, Kohn S, Morgan ES, Fox P, Nagy JA, Dvorak HF. The vesiculo-vacuolar organelle (VVO): a distinct endothelial cell structure that provides a transcellular pathway for macromolecular extravasation. J Leukoc Biol 1996;59:100–115. [PubMed]
- 12.Komarova Y, Malik AB. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol 2010;72:463–493. [DOI] [PubMed]
- 13.Predescu D, Palade GE. Plasmalemmal vesicles represent the large pore system of continuous microvascular endothelium. Am J Physiol 1993;265:H725–H733. [DOI] [PubMed]
- 14.Predescu SA, Predescu DN, Palade GE. Endothelial transcytotic machinery involves supramolecular protein-lipid complexes. Mol Biol Cell 2001;12:1019–1033. [DOI] [PMC free article] [PubMed]
- 15.Drab M, Verkade P, Elger M, et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 2001;293:2449–2452. [DOI] [PubMed]
- 16.Miyawaki-Shimizu K, Predescu D, Shimizu J, Broman M, Predescu S, Malik AB. siRNA-induced caveolin-1 knockdown in mice increases lung vascular permeability via the junctional pathway. Am J Physiol Lung Cell Mol Physiol 2006;290:L405–L413. [DOI] [PubMed]
- 17.Machleidt T, Li WP, Liu P, Anderson RG. Multiple domains in caveolin-1 control its intracellular traffic. J Cell Biol 2000;148:17–28. [DOI] [PMC free article] [PubMed]
- 18.Shajahan AN, Tiruppathi C, Smrcka AV, Malik AB, Minshall RD. Gβγ activation of Src induces caveolae-mediated endocytosis in endothelial cells. J Biol Chem 2004;279:48055–48062. [DOI] [PubMed]
- 19.Esser S, Lampugnani MG, Corada M, Dejana E, Risau W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci 1998;111(Pt 13):1853–1865. [DOI] [PubMed]
- 20.Bates DO, Harper SJ. Regulation of vascular permeability by vascular endothelial growth factors. Vascul Pharmacol 2002;39:225–237. [DOI] [PubMed]
- 21.Knezevic N, Roy A, Timblin B, et al. GDI-1 phosphorylation switch at serine 96 induces RhoA activation and increased endothelial permeability. Mol Cell Biol 2007;27:6323–6333. [DOI] [PMC free article] [PubMed]
- 22.Mehta D, Ahmmed GU, Paria BC, et al. RhoA interaction with inositol 1,4,5-trisphosphate receptor and transient receptor potential channel-1 regulates Ca2+ entry. Role in signaling increased endothelial permeability. J Biol Chem 2003;278:33492–33500. [DOI] [PubMed]
- 23.Munoz-Chapuli R, Perez-Pomares JM, Macias D, Garcia-Garrido L, Carmona R, Gonzalez M. Differentiation of hemangioblasts from embryonic mesothelial cells? a model on the origin of the vertebrate cardiovascular system. Differentiation 1999;64:133–141. [DOI] [PubMed]
- 24.Park C, Kim TM, Malik AB. Transcriptional regulation of endothelial cell and vascular development. Circulation Research 2013;112:1380–1400. [DOI] [PMC free article] [PubMed]
- 25.Stevens T, Phan S, Frid MG, Alvarez D, Herzog E, Stenmark KR. Lung vascular cell heterogeneity: endothelium, smooth muscle, and fibroblasts. Proc Am Thorac Soc 2008;5:783–791. [DOI] [PMC free article] [PubMed]
- 26.Parker JC, Stevens T, Randall J, Weber DS, King JA. Hydraulic conductance of pulmonary microvascular and macrovascular endothelial cell monolayers. Am J Physiol Lung Cell Mol Physiol 2006;291:L30–L37. [DOI] [PubMed]
- 27.Lowe K, Alvarez D, King J, Stevens T. Phenotypic heterogeneity in lung capillary and extra-alveolar endothelial cells. Increased extra-alveolar endothelial permeability is sufficient to decrease compliance. J Surg Res 2007;143:70–77. [DOI] [PMC free article] [PubMed]
- 28.Chi JT, Chang HY, Haraldsen G, et al. Endothelial cell diversity revealed by global expression profiling. Proc Natl Acad Sci U S A 2003;100:10623–10628. [DOI] [PMC free article] [PubMed]
- 29.Bolton SJ, Anthony DC, Perry VH. Loss of the tight junction proteins occludin and zonula occludens-1 from cerebral vascular endothelium during neutrophil-induced blood-brain barrier breakdown in vivo. Neuroscience 1998;86:1245–1257. [DOI] [PubMed]
- 30.Mavrakis AG, Havaki S, Marinos E, Chroni E, Konstantinou D. Occludin dislocation in brain capillary endothelium of rats with bile duct ligation induced cholestasis. Neurosci Lett 2012;528:180–184. [DOI] [PubMed]
- 31.Van Itallie CM, Fanning AS, Holmes J, Anderson JM. Occludin is required for cytokine-induced regulation of tight junction barriers. J Cell Sci 2010;123:2844–2852. [DOI] [PMC free article] [PubMed]
- 32.Giorgi D, Ferraz C, Mattei MG, Demaille J, Rouquier S. The myosin light chain kinase gene is not duplicated in mouse: partial structure and chromosomal localization of Mylk. Genomics 2001;75:49–56. [DOI] [PubMed]
- 33.Cheng AK, Robertson EJ. The murine LIM-kinase gene (limk) encodes a novel serine threonine kinase expressed predominantly in trophoblast giant cells and the developing nervous system. Mech Dev 1995;52:187–197. [DOI] [PubMed]
- 34.Zhang J, Wang W, Sun J, et al. Gap junction channel modulates pulmonary vascular permeability through calcium in acute lung injury: an experimental study. Respiration 2010;80:236–245. [DOI] [PubMed]
- 35.Malek AM, Jiang L, Lee I, Sessa WC, Izumo S, Alper SL. Induction of nitric oxide synthase mRNA by shear stress requires intracellular calcium and G-protein signals and is modulated by PI 3 kinase. Biochem Biophys Res Commun 1999;254:231–242. [DOI] [PubMed]
- 36.Tseng H, Peterson TE, Berk BC. Fluid shear stress stimulates mitogen-activated protein kinase in endothelial cells. Circ Res 1995;77:869–878. [DOI] [PubMed]
- 37.Kedem O, Katchalsky A. Thermodynamic analysis of the permeability of biological membranes to non-electrolytes. Biochim Biophys Acta 1958;27:229–246. [DOI] [PubMed]
- 38.Kedem O, Katchalsky A. Thermodynamic analysis of the permeability of biological membranes to non-electrolytes. 1958. Biochim Biophys Acta 1989;1000:413–430. [PubMed]
- 39.Melchior B, Frangos JA. Shear-induced endothelial cell-cell junction inclination. Am J Physiol Cell Physiol 2010;299:C621–C629. [DOI] [PMC free article] [PubMed]
- 40.Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell 2013;26:441–454. [DOI] [PubMed]
- 41.Dejana E, Giampietro C. Vascular endothelial-cadherin and vascular stability. Curr Opin Hematol 2012;19:218–223. [DOI] [PubMed]
- 42.Taddei A, Giampietro C, Conti A, et al. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol 2008;10:923–934. [DOI] [PubMed]
- 43.Runkle EA, Mu D. Tight junction proteins: from barrier to tumorigenesis. Cancer Lett 2013;337:41–48. [DOI] [PMC free article] [PubMed]
- 44.Kevil CG, Okayama N, Trocha SD, et al. Expression of zonula occludens and adherens junctional proteins in human venous and arterial endothelial cells: role of occludin in endothelial solute barriers. Microcirculation 1998;5:197–210. [PubMed]
- 45.Saitou M, Furuse M, Sasaki H, et al. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell 2000;11:4131–4142. [DOI] [PMC free article] [PubMed]
- 46.Morita K, Sasaki H, Furuse M, Tsukita S. Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol 1999;147:185–194. [DOI] [PMC free article] [PubMed]
- 47.Nitta T, Hata M, Gotoh S, et al. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol 2003;161:653–660. [DOI] [PMC free article] [PubMed]
- 48.Tsukita S, Furuse M. The structure and function of claudins, cell adhesion molecules at tight junctions. Ann N Y Acad Sci 2000;915:129–135. [DOI] [PubMed]
- 49.Aurrand-Lions M, Johnson-Leger C, Wong C, Du Pasquier L, Imhof BA. Heterogeneity of endothelial junctions is reflected by differential expression and specific subcellular localization of the three JAM family members. Blood 2001;98:3699–3707. [DOI] [PubMed]
- 50.Aurrand-Lions MA, Duncan L, Du Pasquier L, Imhof BA. Cloning of JAM-2 and JAM-3: an emerging junctional adhesion molecular family? Curr Top Microbiol Immunol 2000;251:91–98. [DOI] [PubMed]
- 51.Arcangeli ML, Frontera V, Aurrand-Lions M. Function of junctional adhesion molecules (JAMs) in leukocyte migration and homeostasis. Arch Immunol Ther Exp (Warsz) 2013;61:15–23. [DOI] [PubMed]
- 52.Koenen RR, Pruessmeyer J, Soehnlein O, et al. Regulated release and functional modulation of junctional adhesion molecule A by disintegrin metalloproteinases. Blood 2009;113:4799–4809. [DOI] [PubMed]
- 53.Liang TW, DeMarco RA, Mrsny RJ, et al. Characterization of huJAM: evidence for involvement in cell-cell contact and tight junction regulation. Am J Physiol Cell Physiol 2000;279:C1733–C1743. [DOI] [PubMed]
- 54.Lippoldt A, Liebner S, Andbjer B, et al. Organization of choroid plexus epithelial and endothelial cell tight junctions and regulation of claudin-1, -2 and -5 expression by protein kinase C. Neuroreport 2000;11:1427–1431. [DOI] [PubMed]
- 55.Lakshmi SP, Reddy AT, Naik MU, Naik UP, Reddy RC. Effects of JAM-A deficiency or blocking antibodies on neutrophil migration and lung injury in a murine model of ALI. Am J Physiol Lung Cell Mol Physiol 2012;303:L758–L766. [DOI] [PMC free article] [PubMed]
- 56.Gonzalez-Mariscal L, Betanzos A, Avila-Flores A. MAGUK proteins: structure and role in the tight junction. Semin Cell Dev Biol 2000;11:315–324. [DOI] [PubMed]
- 57.Fanning AS, Jameson BJ, Jesaitis LA, Anderson JM. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J Biol Chem 1998;273:29745–29753. [DOI] [PubMed]
- 58.Tsukita S, Katsuno T, Yamazaki Y, Umeda K, Tamura A. Roles of ZO-1 and ZO-2 in establishment of the belt-like adherens and tight junctions with paracellular permselective barrier function. Ann N Y Acad Sci 2009;1165:44–52. [DOI] [PubMed]
- 59.Goodenough DA, Paul DL. Beyond the gap: functions of unpaired connexon channels. Nat Rev Mol Cell Biol 2003;4:285–294. [DOI] [PubMed]
- 60.Van Rijen H, van Kempen MJ, Analbers LJ, et al. Gap junctions in human umbilical cord endothelial cells contain multiple connexins. Am J Physiol 1997;272:C117–C130. [DOI] [PubMed]
- 61.Carter TD, Ogden D. Acetylcholine-stimulated changes of membrane potential and intracellular Ca2+ concentration recorded in endothelial cells in situ in the isolated rat aorta. Pflugers Arch 1994;428:476–484. [DOI] [PubMed]
- 62.Islam MN, Das SR, Emin MT, et al. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med 2012;18:759–765. [DOI] [PMC free article] [PubMed]
- 63.Dejana E, Tournier-Lasserve E, Weinstein BM. The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell 2009;16:209–221. [DOI] [PubMed]
- 64.Vandenbroucke St Amant E, Tauseef M, Vogel SM, et al. PKCalpha activation of p120-catenin serine 879 phospho-switch disassembles VE-cadherin junctions and disrupts vascular integrity. Circ Res 2012;111:739–749. [DOI] [PMC free article] [PubMed]
- 65.Zebda N, Tian Y, Tian X, et al. Interaction of p190RhoGAP with C-terminal domain of p120-catenin modulates endothelial cytoskeleton and permeability. J Biol Chem 2013;288:18290–18299. [DOI] [PMC free article] [PubMed]
- 66.Carmeliet P, Lampugnani MG, Moons L, et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 1999;98:147–157. [DOI] [PubMed]
- 67.Tian Z, Dong B, Blackwell JW, Stewart PW, Egan TM. Effect of a vascular endothelial cadherin antagonist in a rat lung transplant model. Ann Thorac Surg 2013;95:1028–1033. [DOI] [PubMed]
- 68.Broman MT, Kouklis P, Gao X, et al. Cdc42 regulates adherens junction stability and endothelial permeability by inducing alpha-catenin interaction with the vascular endothelial cadherin complex. Circ Res 2006;98:73–80. [DOI] [PubMed]
- 69.Wang YL, Malik AB, Sun Y, et al. Innate immune function of the adherens junction protein p120-catenin in endothelial response to endotoxin. J Immunol 2011;186:3180–3187. [DOI] [PMC free article] [PubMed]
- 70.Singh I, Knezevic N, Ahmmed GU, Kini V, Malik AB, Mehta D. Galphaq-TRPC6-mediated Ca2+ entry induces RhoA activation and resultant endothelial cell shape change in response to thrombin. J Biol Chem 2007;282:7833–7843. [DOI] [PubMed]
- 71.Kolodney MS, Wysolmerski RB. Isometric contraction by fibroblasts and endothelial cells in tissue culture: a quantitative study. J Cell Biol 1992;117:73–82. [DOI] [PMC free article] [PubMed]
- 72.Morel NM, Petruzzo PP, Hechtman HB, Shepro D. Inflammatory agonists that increase microvascular permeability in vivo stimulate cultured pulmonary microvessel endothelial cell contraction. Inflammation 1990;14:571–583. [DOI] [PubMed]
- 73.Katoh K, Kano Y, Amano M, Kaibuchi K, Fujiwara K. Stress fiber organization regulated by MLCK and Rho-kinase in cultured human fibroblasts. Am J Physiol Cell Physiol 2001;280:C1669–C1679. [DOI] [PubMed]
- 74.Kaye AD, Nossaman BD, Ibrahim IN, Kadowitz PJ. Influence of PLC and MLCK inhibitors and the role of L-calcium channels in the cat pulmonary vascular bed. Am J Physiol 1995;269:L507–L513. [DOI] [PubMed]
- 75.Lazar V, Garcia JG. A single human myosin light chain kinase gene (MLCK; MYLK). Genomics 1999;57:256–267. [DOI] [PubMed]
- 76.Rao K, He JA, Halayko AJ, Pan N, Kepron W, Stephens NL. Increased ATPase activity and myosin light chain kinase (MLCK) content in airway smooth muscle from sensitized dogs. Adv Exp Med Biol 1991;304:369–376. [DOI] [PubMed]
- 77.Totsukawa G, Yamakita Y, Yamashiro S, Hartshorne DJ, Sasaki Y, Matsumura F. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J Cell Biol 2000;150:797–806. [DOI] [PMC free article] [PubMed]
- 78.Yanase M, Ikeda H, Ogata I, et al. Functional diversity between Rho-kinase- and MLCK-mediated cytoskeletal actions in a myofibroblast-like hepatic stellate cell line. Biochem Biophys Res Commun 2003;305:223–228. [DOI] [PubMed]
- 79.Goeckeler ZM, Wysolmerski RB. Myosin light chain kinase-regulated endothelial cell contraction: the relationship between isometric tension, actin polymerization, and myosin phosphorylation. J Cell Biol 1995;130:613–627. [DOI] [PMC free article] [PubMed]
- 80.Vogel SM, Gao X, Mehta D, et al. Abrogation of thrombin-induced increase in pulmonary microvascular permeability in PAR-1 knockout mice. Physiol Genomics 2000;4:137–145. [DOI] [PubMed]
- 81.Yuan Y, Huang Q, Wu HM. Myosin light chain phosphorylation: modulation of basal and agonist-stimulated venular permeability. Am J Physiol 1997;272:H1437–H1443. [DOI] [PubMed]
- 82.Wainwright MS, Rossi J, Schavocky J, et al. Protein kinase involved in lung injury susceptibility: evidence from enzyme isoform genetic knockout and in vivo inhibitor treatment. Proc Natl Acad Sci U S A 2003;100:6233–6238. [DOI] [PMC free article] [PubMed]
- 83.Tauseef M, Knezevic N, Chava KR, et al. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J Exp Med 2012;209:1953–1968. [DOI] [PMC free article] [PubMed]
- 84.Sundivakkam PC, Natarajan V, Malik AB, Tiruppathi C. Store-operated Ca2+ entry (SOCE) induced by protease-activated receptor-1 mediates STIM1 protein phosphorylation to inhibit SOCE in endothelial cells through AMP-activated protein kinase and p38beta mitogen-activated protein kinase. J Biol Chem 2013;288:17030–17041. [DOI] [PMC free article] [PubMed]
- 85.Tiruppathi C, Freichel M, Vogel SM, et al. Impairment of store-operated Ca2+ entry in TRPC4(-/-) mice interferes with increase in lung microvascular permeability. Circ Res 2002;91:70–76. [DOI] [PubMed]
- 86.Sundivakkam PC, Freichel M, Singh V, et al. The Ca(2+) sensor stromal interaction molecule 1 (STIM1) is necessary and sufficient for the store-operated Ca(2+) entry function of transient receptor potential canonical (TRPC) 1 and 4 channels in endothelial cells. Mol Pharmacol 2012;81:510–526. [DOI] [PMC free article] [PubMed]
- 87.Birnbaumer L. The TRPC class of ion channels: a critical review of their roles in slow, sustained increases in intracellular Ca(2+) concentrations. Annu Rev Pharmacol Toxicol 2009;49:395–426. [DOI] [PubMed]
- 88.Montell C, Birnbaumer L, Flockerzi V. The TRP channels, a remarkably functional family. Cell 2002;108:595–598. [DOI] [PubMed]
- 89.Abramowitz J, Birnbaumer L. Physiology and pathophysiology of canonical transient receptor potential channels. FASEB J 2009;23:297–328. [DOI] [PMC free article] [PubMed]
- 90.Tiruppathi C, Ahmmed GU, Vogel SM, Malik AB. Ca2+ signaling, TRP channels, and endothelial permeability. Microcirculation 2006;13:693–708. [DOI] [PubMed]
- 91.Curry FR, Glass CA. TRP channels and the regulation of vascular permeability: new insights from the lung microvasculature. Circ Res 2006;99:915–917. [DOI] [PubMed]
- 92.Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol Rev 2001;81:1415–1459. [DOI] [PubMed]
- 93.Pocock TM, Foster RR, Bates DO. Evidence of a role for TRPC channels in VEGF-mediated increased vascular permeability in vivo. Am J Physiol Heart Circ Physiol 2004;286:H1015–H1026. [DOI] [PubMed]
- 94.Dietrich A, Kalwa H, Gudermann T. TRPC channels in vascular cell function. Thromb Haemost 2010;103:262–270. [DOI] [PubMed]
- 95.Tiruppathi C, Minshall RD, Paria BC, Vogel SM, Malik AB. Role of Ca2+ signaling in the regulation of endothelial permeability. Vascul Pharmacol 2002;39:173–185. [DOI] [PubMed]
- 96.Ahmmed GU, Malik AB. Functional role of TRPC channels in the regulation of endothelial permeability. Pflugers Arch 2005;451:131–142. [DOI] [PubMed]
- 97.Obukhov AG, Schultz G, Luckhoff A. Regulation of heterologously expressed transient receptor potential-like channels by calcium ions. Neuroscience 1998;85:487–495. [DOI] [PubMed]
- 98.Ahmmed GU, Mehta D, Vogel S, et al. Protein kinase Calpha phosphorylates the TRPC1 channel and regulates store-operated Ca2+ entry in endothelial cells. J Biol Chem 2004;279:20941–20949. [DOI] [PubMed]
- 99.Jho D, Mehta D, Ahmmed G, et al. Angiopoietin-1 opposes VEGF-induced increase in endothelial permeability by inhibiting TRPC1-dependent Ca2 influx. Circ Res 2005;96:1282–1290. [DOI] [PubMed]
- 100.Paria BC, Vogel SM, Ahmmed GU, et al. Tumor necrosis factor-alpha-induced TRPC1 expression amplifies store-operated Ca2+ influx and endothelial permeability. Am J Physiol Lung Cell Mol Physiol 2004;287:L1303–L1313. [DOI] [PubMed]
- 101.Holda JR, Blatter LA. Capacitative calcium entry is inhibited in vascular endothelial cells by disruption of cytoskeletal microfilaments. FEBS Lett 1997;403:191–196. [DOI] [PubMed]
- 102.Wu S, Sangerman J, Li M, Brough GH, Goodman SR, Stevens T. Essential control of an endothelial cell ISOC by the spectrin membrane skeleton. J Cell Biol 2001;154:1225–1233. [DOI] [PMC free article] [PubMed]
- 103.Hartwig JH. Actin-binding proteins. 1: Spectrin super family. Protein Profile 1995;2:703–800. [PubMed]
- 104.Watanabe H, Tran QK, Takeuchi K, et al. Myosin light-chain kinase regulates endothelial calcium entry and endothelium-dependent vasodilation. FASEB J 2001;15:282–284. [DOI] [PubMed]
- 105.Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol 2012;13:549–565. [DOI] [PMC free article] [PubMed]
- 106.Putney JW. Pharmacology of store-operated calcium channels. Mol Interv 2010;10:209–218. [DOI] [PMC free article] [PubMed]
- 107.Bird GS, Hwang SY, Smyth JT, Fukushima M, Boyles RR, Putney JW Jr. STIM1 is a calcium sensor specialized for digital signaling. Curr Biol 2009;19:1724–1729. [DOI] [PMC free article] [PubMed]
- 108.Cahalan MD. STIMulating store-operated Ca(2+) entry. Nat Cell Biol 2009;11:669–677. [DOI] [PMC free article] [PubMed]
- 109.Yuan JP, Kim MS, Zeng W, et al. TRPC channels as STIM1-regulated SOCs. Channels (Austin) 2009;3:221–225. [DOI] [PubMed]
- 110.Huang GN, Zeng W, Kim JY, et al. STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat Cell Biol 2006;8:1003–1010. [DOI] [PubMed]
- 111.Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat Cell Biol 2007;9:636–645. [DOI] [PMC free article] [PubMed]
- 112.Brandman O, Liou J, Park WS, Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007;131:1327–1339. [DOI] [PMC free article] [PubMed]
- 113.Smyth JT, Hwang SY, Tomita T, DeHaven WI, Mercer JC, Putney JW. Activation and regulation of store-operated calcium entry. J Cell Mol Med 2010;14:2337–2349. [DOI] [PMC free article] [PubMed]
- 114.Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol 2010;28:491–533. [DOI] [PMC free article] [PubMed]
- 115.Dietrich A, Mederos YSM, Gollasch M, et al. Increased vascular smooth muscle contractility in TRPC6−/− mice. Mol Cell Biol 2005;25:6980–6989. [DOI] [PMC free article] [PubMed]
- 116.Asaoka Y, Nakamura S, Yoshida K, Nishizuka Y. Protein kinase C, calcium and phospholipid degradation. Trends Biochem Sci 1992;17:414–417. [DOI] [PubMed]
- 117.Smart EJ, Ying YS, Anderson RG. Hormonal regulation of caveolae internalization. J Cell Biol 1995;131:929–938. [DOI] [PMC free article] [PubMed]
- 118.Tang ZL, Scherer PE, Lisanti MP. The primary sequence of murine caveolin reveals a conserved consensus site for phosphorylation by protein kinase C. Gene 1994;147:299–300. [DOI] [PubMed]
- 119.Minshall RD, Vandenbroucke EE, Holinstat M, et al. Role of protein kinase Czeta in thrombin-induced RhoA activation and inter-endothelial gap formation of human dermal microvessel endothelial cell monolayers. Microvasc Res 2010;80:240–249. [DOI] [PMC free article] [PubMed]
- 120.Rigor RR, Shen Q, Pivetti CD, Wu MH, Yuan SY. Myosin light chain kinase signaling in endothelial barrier dysfunction. Med Res Rev 2013;33:911–933. [DOI] [PMC free article] [PubMed]
- 121.Grinnell KL, Harrington EO. Interplay between FAK, PKCdelta, and p190RhoGAP in the regulation of endothelial barrier function. Microvasc Res 2012;83:12–21. [DOI] [PMC free article] [PubMed]
- 122.Huang F, Subbaiah PV, Holian O, et al. Lysophosphatidylcholine increases endothelial permeability: role of PKCalpha and RhoA cross talk. Am J Physiol Lung Cell Mol Physiol 2005;289:L176–L185. [DOI] [PubMed]
- 123.Harrington EO, Brunelle JL, Shannon CJ, Kim ES, Mennella K, Rounds S. Role of protein kinase C isoforms in rat epididymal microvascular endothelial barrier function. Am J Respir Cell Mol Biol 2003;28:626–636. [DOI] [PubMed]
- 124.Chava KR, Tauseef M, Sharma T, Mehta D. Cyclic AMP response element-binding protein prevents endothelial permeability increase through transcriptional controlling p190RhoGAP expression. Blood 2012;119:308–319. [DOI] [PMC free article] [PubMed]
- 125.Wojciak-Stothard B, Potempa S, Eichholtz T, Ridley AJ. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J Cell Sci 2001;114:1343–1355. [DOI] [PubMed]
- 126.Schlegel N, Burger S, Golenhofen N, Walter U, Drenckhahn D, Waschke J. The role of VASP in regulation of cAMP- and Rac 1-mediated endothelial barrier stabilization. Am J Physiol Cell Physiol 2008;294:C178–C188. [DOI] [PubMed]
- 127.Schmidt TT, Tauseef M, Yue L, et al. Conditional deletion of FAK in mice endothelium disrupts lung vascular barrier function due to destabilization of RhoA and Rac1 activities. Am J Physiol Lung Cell Mol Physiol 2013;305:L291–L300. [DOI] [PMC free article] [PubMed]
- 128.Kouklis P, Konstantoulaki M, Vogel S, Broman M, Malik AB. Cdc42 regulates the restoration of endothelial barrier function. Circ Res 2004;94:159–166. [DOI] [PubMed]
- 129.Ramchandran R, Mehta D, Vogel SM, Mirza MK, Kouklis P, Malik AB. Critical role of Cdc42 in mediating endothelial barrier protection in vivo. Am J Physiol Lung Cell Mol Physiol 2008;295:L363–L369. [DOI] [PMC free article] [PubMed]
- 130.Quadri SK, Bhattacharya J. Resealing of endothelial junctions by focal adhesion kinase. Am J Physiol Lung Cell Mol Physiol 2007;292:L334–L342. [DOI] [PubMed]
- 131.Sun X, Shikata Y, Wang L, et al. Enhanced interaction between focal adhesion and adherens junction proteins: involvement in sphingosine 1-phosphate-induced endothelial barrier enhancement. Microvasc Res 2009;77:304–313. [DOI] [PMC free article] [PubMed]
- 132.Chen XL, Nam JO, Jean C, et al. VEGF-induced vascular permeability is mediated by FAK. Dev Cell 2012;22:146–157. [DOI] [PMC free article] [PubMed]
- 133.Chen Z, Bakhshi FR, Shajahan AN, et al. Nitric oxide-dependent Src activation and resultant caveolin-1 phosphorylation promote eNOS/caveolin-1 binding and eNOS inhibition. Mol Biol Cell 2012;23:1388–1398. [DOI] [PMC free article] [PubMed]
- 134.Harper MT, Sage SO. Src family tyrosine kinases activate thrombin-induced non-capacitative cation entry in human platelets. Platelets 2010;21:445–450. [DOI] [PubMed]
- 135.Wu T, Zhang B, Ye F, Xiao Z. A potential role for caveolin-1 in VEGF-induced fibronectin upregulation in mesangial cells: involvement of VEGFR2 and Src. Am J Physiol Renal Physiol 2013;304:F820–F830. [DOI] [PubMed]
- 136.Potter MD, Barbero S, Cheresh DA. Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J Biol Chem 2005;280:31906–31912. [DOI] [PubMed]
- 137.Orsenigo F, Giampietro C, Ferrari A, et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun 2012;3:1208. [DOI] [PMC free article] [PubMed]
- 138.Vestweber D, Winderlich M, Cagna G, Nottebaum AF. Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends Cell Biol 2009;19:8–15. [DOI] [PubMed]
- 139.Arnold KM, Goeckeler ZM, Wysolmerski RB. Loss of focal adhesion kinase enhances endothelial barrier function and increases focal adhesions. Microcirculation 2013;20:637–649. [DOI] [PubMed]
- 140.Loot AE, Schreiber JG, Fisslthaler B, Fleming I. Angiotensin II impairs endothelial function via tyrosine phosphorylation of the endothelial nitric oxide synthase. J Exp Med 2009;206:2889–2896. [DOI] [PMC free article] [PubMed]
- 141.Ying Z, Giachini FR, Tostes RC, Webb RC. PYK2/PDZ-RhoGEF links Ca2+ signaling to RhoA. Arterioscler Thromb Vasc Biol 2009;29:1657–1663. [DOI] [PMC free article] [PubMed]
- 142.Field JJ, Diaz JF, Miller JH. The binding sites of microtubule-stabilizing agents. Chem Biol 2013;20:301–315. [DOI] [PubMed]
- 143.Stec-Martyna E, Ponassi M, Miele M, Parodi S, Felli L, Rosano C. Structural comparison of the interaction of tubulin with various ligands affecting microtubule dynamics. Curr Cancer Drug Targets 2012;12:658–666. [DOI] [PubMed]
- 144.Vignaud T, Blanchoin L, Thery M. Directed cytoskeleton self-organization. Trends Cell Biol 2012;22:671–682. [DOI] [PubMed]
- 145.Akhmanova A, Hoogenraad CC. Microtubule plus-end-tracking proteins: mechanisms and functions. Curr Opin Cell Biol 2005;17:47–54. [DOI] [PubMed]
- 146.Bajer S, Kasprzak AA. [Plus end tracking proteins and their role in mitotic spindle organization]. Postepy Biochem 2009;55:223–231. [PubMed]
- 147.Li W, Miki T, Watanabe T, et al. EB1 promotes microtubule dynamics by recruiting Sentin in Drosophila cells. J Cell Biol 2011;193:973–983. [DOI] [PMC free article] [PubMed]
- 148.Komarova YA, Huang F, Geyer M, et al. VE-cadherin signaling induces EB3 phosphorylation to suppress microtubule growth and assemble adherens junctions. Mol Cell 2012;48:914–925. [DOI] [PMC free article] [PubMed]
- 149.Padrick SB, Rosen MK. Physical mechanisms of signal integration by WASP family proteins. Annu Rev Biochem 2010;79:707–735. [DOI] [PMC free article] [PubMed]
- 150.Takenawa T, Suetsugu S. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol 2007;8:37–48. [DOI] [PubMed]
- 151.Rajput C, Kini V, Smith M, et al. Neural Wiskott-Aldrich syndrome protein (N-WASP)-mediated p120-catenin interaction with Arp2-Actin complex stabilizes endothelial adherens junctions. J Biol Chem 2013;288:4241–4250. [DOI] [PMC free article] [PubMed]
- 152.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature 2000;407:258–264. [DOI] [PubMed]
- 153.Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991;64:1057–1068. [DOI] [PubMed]
- 154.Ishihara H, Connolly AJ, Zeng D, et al. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature 1997;386:502–506. [DOI] [PubMed]
- 155.Mirza H, Yatsula V, Bahou WF. The proteinase activated receptor-2 (PAR-2) mediates mitogenic responses in human vascular endothelial cells. J Clin Invest 1996;97:1705–1714. [DOI] [PMC free article] [PubMed]
- 156.Wells CD, Liu MY, Jackson M, et al. Mechanisms for reversible regulation between G13 and Rho exchange factors. J Biol Chem 2002;277:1174–1181. [DOI] [PubMed]
- 157.Kaneider NC, Leger AJ, Agarwal A, et al. ‘Role reversal’ for the receptor PAR1 in sepsis-induced vascular damage. Nat Immunol 2007;8:1303–1312. [DOI] [PMC free article] [PubMed]
- 158.Klarenbach SW, Chipiuk A, Nelson RC, Hollenberg MD, Murray AG. Differential actions of PAR2 and PAR1 in stimulating human endothelial cell exocytosis and permeability: the role of Rho-GTPases. Circ Res 2003;92:272–278. [DOI] [PubMed]
- 159.Barr AJ, Brass LF, Manning DR. Reconstitution of receptors and GTP-binding regulatory proteins (G proteins) in Sf9 cells. A direct evaluation of selectivity in receptor.G protein coupling. J Biol Chem 1997;272:2223–2229. [DOI] [PubMed]
- 160.Hung DT, Wong YH, Vu TK, Coughlin SR. The cloned platelet thrombin receptor couples to at least two distinct effectors to stimulate phosphoinositide hydrolysis and inhibit adenylyl cyclase. J Biol Chem 1992;267:20831–20834. [PubMed]
- 161.Knezevic N, Tauseef M, Thennes T, Mehta D. The G protein betagamma subunit mediates reannealing of adherens junctions to reverse endothelial permeability increase by thrombin. J Exp Med 2009;206:2761–2777. [DOI] [PMC free article] [PubMed]
- 162.Tauseef M, Kini V, Knezevic N, et al. Activation of sphingosine kinase-1 reverses the increase in lung vascular permeability through sphingosine-1-phosphate receptor signaling in endothelial cells. Circ Res 2008;103:1164–1172. [DOI] [PMC free article] [PubMed]
- 163.Thennes T, Mehta D. Heterotrimeric G proteins, focal adhesion kinase, and endothelial barrier function. Microvasc Res 2012;83:31–44. [DOI] [PMC free article] [PubMed]
- 164.Sun X, Ma SF, Wade MS, et al. Functional promoter variants in sphingosine 1-phosphate receptor 3 associate with susceptibility to sepsis-associated acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol 2013;305:L467–L477. [DOI] [PMC free article] [PubMed]
- 165.Weis SM, Cheresh DA. Pathophysiological consequences of VEGF-induced vascular permeability. Nature 2005;437:497–504. [DOI] [PubMed]
- 166.Bates DO. Vascular endothelial growth factors and vascular permeability. Cardiovasc Res 2010;87:262–271. [DOI] [PMC free article] [PubMed]
- 167.Nottebaum AF, Cagna G, Winderlich M, et al. VE-PTP maintains the endothelial barrier via plakoglobin and becomes dissociated from VE-cadherin by leukocytes and by VEGF. J Exp Med 2008;205:2929–2945. [DOI] [PMC free article] [PubMed]
- 168.Sun Z, Li X, Massena S, et al. VEGFR2 induces c-Src signaling and vascular permeability in vivo via the adaptor protein TSAd. J Exp Med 2012;209:1363–1377. [DOI] [PMC free article] [PubMed]
- 169.Thibeault S, Rautureau Y, Oubaha M, et al. S-nitrosylation of beta-catenin by eNOS-derived NO promotes VEGF-induced endothelial cell permeability. Mol Cell 2010;39:468–476. [DOI] [PubMed]
- 170.Scharpfenecker M, Fiedler U, Reiss Y, Augustin HG. The Tie-2 ligand angiopoietin-2 destabilizes quiescent endothelium through an internal autocrine loop mechanism. J Cell Sci 2005;118:771–780. [DOI] [PubMed]
- 171.Yuan HT, Khankin EV, Karumanchi SA, Parikh SM. Angiopoietin 2 is a partial agonist/antagonist of Tie2 signaling in the endothelium. Mol Cell Biol 2009;29:2011–2022. [DOI] [PMC free article] [PubMed]
- 172.Ziegler T, Horstkotte J, Schwab C, et al. Angiopoietin 2 mediates microvascular and hemodynamic alterations in sepsis. J Clin Invest 2013;123(8):3436–3445. [DOI] [PMC free article] [PubMed]
- 173.Cullere X, Shaw SK, Andersson L, Hirahashi J, Luscinskas FW, Mayadas TN. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood 2005;105:1950–1955. [DOI] [PubMed]
- 174.Glading A, Han J, Stockton RA, Ginsberg MH. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J Cell Biol 2007;179:247–254. [DOI] [PMC free article] [PubMed]
- 175.Hatanaka K, Simons M, Murakami M. Phosphorylation of VE-cadherin controls endothelial phenotypes via p120-catenin coupling and Rac1 activation. Am J Physiol Heart Circ Physiol 2011;300:H162–H172. [DOI] [PMC free article] [PubMed]
- 176.Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol 2009;10:165–177. [DOI] [PubMed]
- 177.Fukuhara S, Sako K, Minami T, et al. Differential function of Tie2 at cell-cell contacts and cell-substratum contacts regulated by angiopoietin-1. Nat Cell Biol 2008;10:513–526. [DOI] [PubMed]