

Abstract Abstract
Obesity is causally linked to a number of comorbidities, including cardiovascular disease, diabetes, renal dysfunction, and cancer. Obesity has also been linked to pulmonary disorders, including pulmonary arterial hypertension (PAH). It was long believed that obesity-related PAH was the result of hypoventilation and hypoxia due to the increased mechanical load of excess body fat. However, in recent years it has been proposed that the metabolic and inflammatory disturbances of obesity may also play a role in the development of PAH. To determine whether PAH develops in obese rats in the absence of hypoxia, we assessed pulmonary hemodynamics and pulmonary artery (PA) structure in the diet-resistant/diet-induced obesity (DR/DIO) and Zucker lean/fatty rat models. We found that high-fat feeding (DR/DIO) or overfeeding (Zucker) elicited PA remodeling, neomuscularization of distal arterioles, and elevated PA pressure, accompanied by right ventricular (RV) hypertrophy. PA thickening and distal neomuscularization were also observed in DIO rats on a low-fat diet. No evidence of hypoventilation or chronic hypoxia was detected in either model, nor was there a correlation between blood glucose or insulin levels and PAH. However, circulating inflammatory cytokine levels were increased with high-fat feeding or calorie overload, and hyperlipidemia and oxidant damage in the PA wall correlated with PAH in the DR/DIO model. We conclude that hyperlipidemia and peripheral inflammation correlate with the development of PAH in obese subjects. Obesity-related inflammation may predispose to PAH even in the absence of hypoxia.
Keywords: obesity, pulmonary hypertension, hypoxia, hyperlipidemia, oxidant damage, inflammation, insulin resistance
Introduction
It is currently estimated that two-thirds of Americans are overweight (body mass index from 25 to 30) or obese (body mass index greater than 30), with similar levels reported in other developed nations.1,2 Excess body fat has been linked to a number of comorbidities, including cardiovascular disease, diabetes, renal dysfunction, and certain forms of cancer.3-5 The prevalence of obesity and associated chronic diseases makes it one of the most pressing medical and financial problems facing our society.
Excess body fat has also been linked to several pulmonary disorders, including chronic obstructive pulmonary disease, pneumonia, sleep apnea, pulmonary embolism (as a result of deep-vein thrombosis), and asthma.6 Pulmonary arterial hypertension (PAH), a disease characterized by sustained vasoconstriction and progressive remodeling of the pulmonary artery (PA), is also observed in obese and overweight individuals, although prevalence remains uncertain.6-8 Taraseviciute and Voelkel9 reported that almost 50% of PAH patients they reviewed were clinically obese, while in another study of 20 ambulatory patients presenting with echocardiographic evidence of PAH, all were obese.10 Retrospective review of autopsy records also showed that a significant proportion (44%) of obese individuals display medial thickening of the PAs, indicative of subclinical PAH.10,11 These studies highlight an important link between excess body fat and pathological changes in the pulmonary circulation.
Conventional wisdom has held that obesity-associated PAH is primarily due to transient or chronic hypoventilation resulting from the increased mechanical load imposed by excess adiposity.6,8 Obesity-related obstructive sleep apnea and obesity-related hypoventilation syndrome are clinically recognized conditions characterized by increased upper-airway resistance, decreased inspiratory and expiratory pressure, and decreased chest wall compliance.12,13 These factors lead to lung hypoxia and hypoxemia, which alone are sufficient to induce remodeling and vasoconstriction of the pulmonary arterial tree in animal models.14
While hypoxemia and lung hypoxia undoubtedly contribute to the development of PAH in overweight and obese patients, excess body fat can elicit numerous metabolic and physiologic changes that may also promote pulmonary vascular pathology. For example, blood insulin levels are often elevated with obesity because of global insulin resistance.15 Lack of insulin responsiveness in adipose tissue results in elevated circulating free fatty acids,16 while insulin resistance in the liver promotes hepatic glucose production and release of triglycerides into the blood.17 Adipose production of inflammatory cytokines (e.g., interleukin-6 [IL-6] and tumor necrosis factor α [TNF-α]) and adipokines (e.g., leptin, resistin) is likely a primary cause of the peripheral insulin resistance, diminished energy expenditure, and systemic inflammation associated with obesity.18 Finally, obesity-induced changes in the cardiovascular system, including elevated cardiac output and systemic hypertension, also likely undermine normal pulmonary function.19 With this broad spectrum of metabolic and physiologic disturbances, it is unlikely that hypoventilation and hypoxia alone explain obesity-related PAH. Indeed, several investigators have recently proposed that insulin resistance may promote pulmonary vascular disturbances in obese individuals.20,21
Here we investigated whether obesity-related PAH could be recapitulated in rat models of obesity. We initially characterized PA pressure, right ventricular (RV) hypertrophy, and PA remodeling in the polygenetic diet-induced-obesity rat model developed by Levin and colleagues.22 These investigators originally noted that approximately half of outbred Sprague-Dawley rats develop obesity when fed a high-fat diet, while the remaining half are resistant to obesity (diet-resistant, or DR). Through selective breeding of each type for multiple generations (now more than 50), they produced sublines that are either resistant or prone to diet-induced obesity. When fed a high-fat diet, the obesity-prone animals develop visceral obesity, fasting hyperglycemia, hyperinsulinemia, and hyperlipidemia. This model reflects the polygenetic features of human obesity and permits investigation of both diet and genetic affects on disease initiation and progression.
We also examined the development of PAH in Zucker lean and fatty rats. Zucker fatty rats homozygous for a spontaneous mutation in the leptin receptor exhibit early-onset obesity, hyperphagia, hyperlipidemia, peripheral insulin resistance, and systemic hypertension.23 Interestingly, the elevated blood pressure in this model cannot be reduced by exercise and/or weight loss, indicating that it is associated with the obese genotype rather than with diet or excess body fat.
We report physiological and structural evidence of PAH in obesity-prone (DIO) rats with and without high-fat feeding and in Zucker fatty rats. Hypoventilation and hypoxia were not detected in either model, suggesting that diet and metabolic and/or genetic factors were responsible for the observed physiological and structural changes. Circulating inflammatory cytokine levels correlated with PAH in high-fat-fed DIO rats, and hyperlipidemia and increased oxidant production in the PA wall correlated with PAH in both DR/DIO and Zucker models.
Methods
Material
Male DR and DIO rats and Zucker lean and fatty rats were purchased from Charles River Laboratories (Wilmington, MA). Research diets with 10 kcal% fat (product no. D12450Bi) or 60 kcal% fat (product no. D12492i) were obtained from Research Diets (New Brunswick, NJ). Polyclonal antibodies to 8-OH-guanosine and 4-OH-nonenal were from Abcam (Cambridge, MA). Polyclonal antibodies to α-smooth muscle (α-SM) actin, cardiogreen polymethine dye, and fasudil (HA-1077) were purchased from Sigma (St. Louis, MO). Rabbit polyclonal antibody to CREB was from Cell Signaling Technology (Beverly, MA). Picrosirius red stain was obtained from Electron Microscopy Sciences (Hatfield, PA). Alexa Fluor 594– and Alexa Fluor 488–conjugated secondary antibodies were purchased from Molecular Probes (Eugene, OR). VectaShield mounting medium with 4,6-diamidino-2-phenylindole (DAPI) was purchased from Vector Laboratories (Burlingame, CA). Rat serum adipokines kits (MILLIPLEXMAP) and rat insulin enzyme-linked immunosorbent assay (ELISA) kits were ordered from Millipore (St. Charles, MO), rat adiponectin ELISA kits were from Invitrogen (Carlsbad, CA), and mouse/rat erythropoietin (EPO) ELISA kits were from R&D Systems (Minneapolis, MN). Reagents for measuring nonesterified fatty acids were purchased from Wako Diagnostics (Richmond, VA) and those for triglycerides from Thermo Scientific (Middletown, VA).
Animal care and treatment
All animal procedures were performed with the approval and in accordance with the guidelines of the University of Colorado Anschutz Medical Campus Institutional Animal Care and Use Committee. Male DR and DIO rats and Zucker lean and fatty rats (8 weeks old, 6 per cohort) were maintained under isobaric normoxia (1,600-m altitude, 630 mmHg) for 5 months. DR/DIO rats were fed either a low-fat (10 kcal% fat) or a high-fat (60 kcal% fat) diet, ad lib., as indicated. Zucker rats were fed standard chow ad lib. Fresh water and clean cages with fresh bedding were provided every other day. Light was maintained on a 12-hour cycle, and humidity was 40%–45%, with a temperature of 25°–27°C. The animals were monitored daily, and weight was checked once a week.
Catheterization of anesthetized animals
Rats received a combination of ketamine (75 mg/kg) and xylazine (6 mg/kg) intraperitoneally as anesthetic. The ventral neck area and the area dorsally between the scapulae were shaved, and the areas were scrubbed with betadine solution. A 2-cm incision was made over the right ventral neck area to expose the internal jugular vein. A polyvinyl (PV-1) catheter was threaded into the PA via the right jugular vein. A catheter pressure tracing was transduced and monitored with an oscilloscope, and the location of the tip was identified by the characteristic shapes of the PA pressure waveforms. The catheter was then securely sutured into place. An additional catheter was inserted into the jugular vein for drug infusion and venous return. A polyethylene (PE-50) catheter was placed into the right carotid artery to obtain cardiac output values. All catheters were exteriorized caudal to the scapulae by blunt dissection and attached to pressure transducers for hemodynamic measurements.
Catheterization of awake animals
Catheters were placed as described above 48 hours before measurements. The exteriorized catheters were threaded through a specially designed plastic hub sutured to the back of the neck. Catheters were coiled inside the hub, over which a snug-fitting rubber cap was placed to prevent animals tampering with the catheters. On the day of measurements, rats were placed in a 3-inch-high, 3-inch-wide, and 7-inch-long Plexiglas box with gas delivery ports for the delivery of air. Sampling of the gas inside the box over time showed complete gas exchange within 2 minutes. Exteriorized catheters were attached to pressure transducers for hemodynamic measurements.
Hemodynamic measurements and tissue procurement in anesthetized animals
Pulmonary and systemic arterial pressures were captured from fluid-filled PE-50 catheters (Becton Dickinson, Franklin Lakes, NJ) fitted to TransPac IV (Hospira, Lake Forest, IL) pressure transducers connected to a BIOPAC MP150 acquisition system (BioPac Systems, Camino Goleta, CA). Cardiac output was measured by infusion of cardiogreen polymethine dye, and a specially designed densitometer and software (Deterministic Systems, Boulder, CO) were used to detect and calculate cardiac output from the dye dilution. Total pulmonary and systemic resistance were calculated by dividing PA pressure or systemic pressure by cardiac output.
After hemodynamic measurements, the anesthetized animals were treated with the RhoA/Rho kinase inhibitor fasudil (10 mg/kg intravenously) to dilate the vasculature before fixation. The rats were then euthanized by exsanguination. Lungs were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) containing 5 mM EDTA (ethylenediaminetetraacetic acid) by airway inflation at 30–cm H2O pressure. PAs were perfused with 4% paraformaldehyde–PBS–5 mm EDTA (to maintain vasodilation) at a pressure similar to that measured in vivo. The heart and lungs were then removed in bloc. Wet RV weights were measured and used to calculate the ratio of RV weight to left ventricle plus septum weight. Lung tissue was fixed overnight in 4% paraformaldehyde and embedded in paraffin.
Evaluation of potential hypoxia in lean and obese animals
Hematocrit and circulating EPO levels were measured at the end of each study to determine whether the obese rats experienced hypoxia. Hematocrit was measured in capillary tubes as the ratio of packed red blood cell volume to total blood sample volume. EPO was measured by ELISA. To make sure that the assays were sufficiently sensitive to detect changes in red blood cell volume and EPO in response to hypoxia, these end points were also assessed in cohorts of 6 Zucker lean and fatty rats exposed to hypobaric hypoxia (5,500 m altitude, 410 mmHg) for the last 3 weeks of the 5-month treatment regimen.
Blood oxygen and carbon dioxide concentrations and oxygen saturation (po2, pco2, and Sao2, respectively) were measured as indices of potential hypoventilation. Awake rats were placed in a 3-inch-wide, 3-inch-high, and 7-inch-long Plexiglas box with gas delivery ports for the delivery of air, through which catheters could also be passed. Sampling of the gas inside the box over time showed that complete gas exchange within the box occurred within 2 minutes. Initially, the box was flushed with a normoxic gas mix (21% O2, 79% N2), and an arterial blood sample (0.2 mL) was collected into a blood gas syringe after the animals were resting comfortably. The gas mixture was then gradually changed to a hypoxic mixture (10% O2, 90% N2) over a period of 30 minutes, at which time another blood sample was withdrawn. The chamber was flushed with the hypoxic mixture for another 30 minutes, and another blood sample was taken. Finally, the box was returned to the normoxic mixture over 30 minutes, at which time a final blood sample was recovered. Blood samples were analyzed immediately upon withdrawal (ABL5, Radiometer, Copenhagen; co-oximetry via OSM3, Radiometer).
Lung morphometric analysis
Sections (5 μm) of fixed lung or heart tissue were subjected to hematoxylin-and-eosin staining by the University of Colorado Denver Histology Core Laboratory. Picrosirius red staining of lung sections was performed as directed by the supplier’s instructions. Muscularized distal arterioles were visualized by staining sections with a primary antibody to SM-actin (1∶600 dilution), followed by staining with an Alexa 488–conjugated secondary antibody (1∶250 dilution).
Distal pulmonary vessels (outside diameter: 10–50 m) were assessed by a blinded observer for degrees of circumferential α-SM actin–positive staining indicative of muscularization. Vessels smaller than 10 m were considered capillaries and excluded from further considerations. Proximal vessels (outside diameter: 50–250 m) were analyzed for medial wall thickness by measuring the area between the inner and outer elastic lamellae and correcting for lumen area via computer-assisted image analysis (MetaMorph 6.1 software).
Immunostaining procedures
Oxidative stress in the arterial wall contributes to the development of PAH,24,25 and antioxidants attenuate PA remodeling and vasoconstriction.26-28 The increased availability and inappropriate metabolism of lipids often result in mitochondrial dysfunction and oxidant production in the arterial wall.29,30 The high circulating lipid levels in our obese-rat models led us to assess oxidant production in the PA wall by immunohistochemistry for 8-OH-guanosine or 4-OH-nonenal. Sections (5 μm) of paraformaldehyde-fixed, paraffin-embedded lung tissue were deparaffinized with Hemo-D and rehydrated in a graded ethanol-water series. Sections were subjected to antigen retrieval in citrate buffer in a microwaveable pressure cooker for 20 minutes. Sections were blocked with PBS containing 5% horse serum for 30 minutes at room temperature. The sections were incubated overnight in PBS–5% FBS (fetal bovine serum) at 4°C with 7.5 μg/mL anti-8-OH-guanosine or 10 μg/mL anti-4-OH-nonenal antibodies. The sections were then washed and incubated with the indicated Alexa Fluor–conjugated secondary antibodies (1∶250 dilution) for 1 hour at room temperature. Coverslips were affixed with mounting medium containing DAPI.
Microscopy
Microscopy was performed on a Nikon TE2000-U inverted epifluorescent microscope. Bright-field, phase-contrast, and fluorescent images were captured to a personal computer with either a Spot RT/KE monochrome camera or a Spot Insight color camera (Diagnostic Imaging, Sterling Heights, MI). Images were analyzed and processed with MetaMorph 6.1 software (Molecular Devices, Sunnyvale, CA). The nuclear fluorescence intensity of 8-OH-guanosine (colocalized with DAPI) or total medial (between inner and outer elastic lamellae, colocalized with SM-actin immunofluorescence) 4-OH-nonenal staining was determined by inclusive thresholding in each micrograph, followed by computer-assisted fluorescence signal integration. This number was divided by the lumen radius of the vessel to account for artery size.
Measurement of metabolic end points and cytokines
Blood samples (1 mL) were recovered from anesthetized rats immediately before euthanasia. Blood glucose levels were determined with a OneTouch Ultra glucometer. Plasma was prepared from each blood sample for subsequent analyses. Circulating insulin levels were measured by ELISA according to the manufacturer’s instructions. Blood nonesterified fatty acids and triglycerides were determined with standard clinical reagents. Plasma adiponectin, IL-6, TNF-α, IL-1β, tissue plasminogen activator inhibitor-1 (tPAI-1), leptin, and macrophage chemoattractant protein-1 (MCP-1) levels were measured by multiplex bead assay.
Statistics
Statistical analysis was performed with the Super ANOVA software program (Abacus Concepts, Berkeley, CA). Comparisons were performed via 2-way ANOVA followed by the Scheffé multiple-comparison test for individual comparisons within and between groups of data points. DIO/DR or Zucker lean/fatty data presented in graphs represent averages of values obtained from at least 3 experiments with 6 animals per group. Data were considered statistically significant at P < .05. In all figures, statistically significant differences between control groups (DR low-fat-fed or Zucker lean) and other cohorts are indicated by an asterisk, while significant differences between the DIO low-fat-fed and DIO high-fat-fed groups are indicated by a pound sign (#).
Results
Weight gain in DR/DIO and Zucker rat models
At 5 months of age, low-fat-fed DR rats weighed approximately 420 g, while the high-fat-fed animals were an average of 90 g heavier (Fig. 1A, 1B). While the DIO rats were only slightly heavier than the DR rats at 1 month, by 5 months the low-fat-fed DIO rats were approximately 225 g heavier than the DR rats on the same diet. This was apparently due to increased food intake by the DIO group. DIO rats on the high-fat diet were, on average, 320 g heavier than low-fat-fed DR animals. Zucker lean and fatty rats were also obtained at 1 month of age, but both were fed the same low-fat chow for an additional 4 months. At this time, the Zucker lean average body weight was 340 g, while the Zucker fatty rats were approximately 240 g heavier (Fig. 1C, 1D). Retrospective analysis of magnetic resonance imaging data shows that weight gain in the DR/DIO models is primarily due to increases in body fat content, with only minor increases in muscle and bone weight, which is required to carry the burden of excess adipose tissue (Fig. S1).
Figure 1.
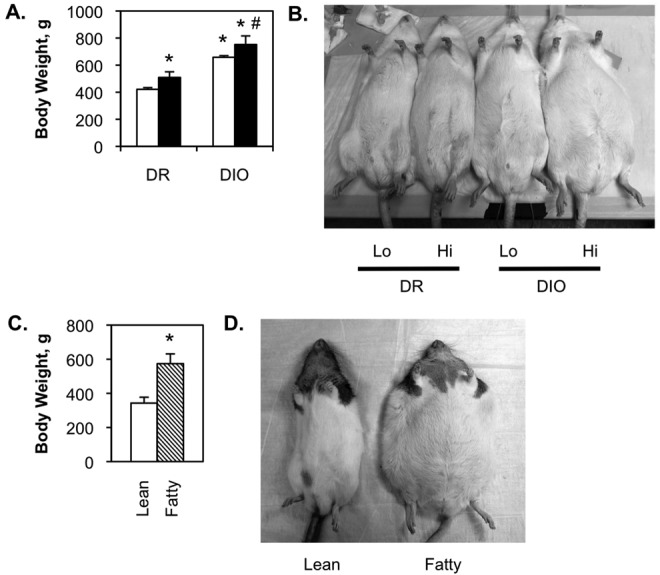
Body weight of diet-resistant/diet-induced obesity (DR/DIO) and Zucker lean/fatty rats. A, B, Eight-week-old male DR and DIO rats were fed either a low-fat (white bars in A; Lo in B) or a high-fat (black bars in A; Hi in B) diet ad lib. for 5 months. C, D, Eight-week-old male Zucker lean (white bar) and fatty (hatched bar) rats were fed standard chow ad lib. for 5 months. Values in A and C are averages from 3 experiments with 6 animals per group.
Evidence of PAH in DIO and Zucker fatty rats
DR rats exhibited a small but statistically significant increase in mean PA pressure when fed a high-fat diet (Fig. 2A), indicating that energy overload or diet composition is capable of altering PA pressure. A similar significant increase was noted between DIO rats fed a low-fat diet and the DR low-fat-fed group, suggesting that the genetic/metabolic environment alone can also elicit changes in pulmonary hemodynamics. A high-fat diet produced a reproducible, but not statistically significant, increase in mean PA pressure in the DIO cohort.
Figure 2.
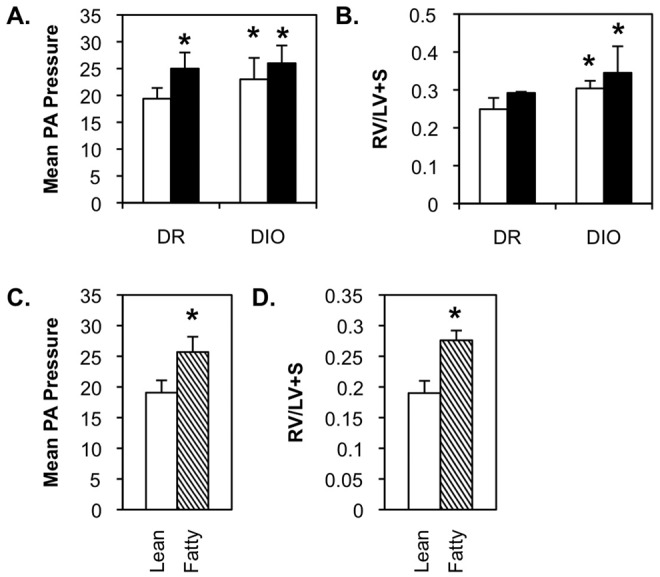
Spontaneous pulmonary arterial hypertension in diet-resistant/diet-induced obesity (DR/DIO) and Zucker lean/fatty rat models. A, B, Eight-week-old male DR and DIO rats were fed either a low-fat (white bars) or a high-fat (black bars) diet ad lib. for 5 months. C, D, Eight-week-old male Zucker lean and fatty rats were fed standard chow ad lib. for 5 months. Mean pulmonary artery (PA) pressure (mmHg; A, C) and right ventricular hypertrophy (right ventricle weight/weight of left ventricle and septum [RV/LV+S]; B, D) were measured as described in “Methods.” Values are averages from 3 experiments with 6 animals per group.
Right ventricle weight was elevated in high-fat-fed DR rats, but this did not achieve statistical significance over the course of our studies (Fig. 2B). Right ventricle weight was significantly higher in low- and high-fat-fed DIO rats than in low-fat-fed DR animals, but there was no significant difference between these groups. Mean PA pressure (Fig. 2C) and right ventricle weight (Fig. 2D) were higher in Zucker fatty rats than in Zucker lean animals.
No differences were noted in cardiac output or mean systemic pressure in either the DR/DIO or the Zucker model (Fig. S2). Likewise, no significant differences were noted in ventricular collagen deposition in the DR/DIO model (Fig. S3). This suggests that the elevated PA pressure observed in obese rats is not related to changes in cardiac function and structure.
Evidence of PA remodeling in DR/DIO and Zucker fatty rats
Thickening of the PA wall was observed in high-fat-fed DR rats, compared to low-fat-fed animals (representative images in Fig. 3A, morphometrics in Fig. 3C). Similar results were observed in high- versus low-fat-fed DIO rats. Thus, a high-fat diet and/or calorie overload alone stimulated remodeling of the PA wall. Low-fat-fed DIO rats also exhibited a significant increase in PA wall thickness, compared to low-fat-fed DR rats, suggesting that the genetic/metabolic environment may also affect vascular structure. Wall area was also significantly higher in high-fat-fed DIO rats than in either low-fat-fed DIO or high-fat-fed DR rats, indicating that diet and genetic environment have an additive affect on PA wall morphology. Significant remodeling of the PA wall was also observed in Zucker fatty rats relative to lean rats (Fig. 3E, 3G).
Figure 3.

PA remodeling and muscularization of distal arterioles in diet-resistant/diet-induced obesity (DR/DIO) and Zucker lean/fatty rat models. A–D, Eight-week-old male DR and DIO rats were fed either a low-fat (white bars in C, D) or a high-fat (black bars in C, D) diet ad lib. for 5 months. E–H, Eight-week-old male Zucker lean and fatty rats were fed standard chow ad lib. for 5 months. A, E, Representative bright-field micrographs of hematoxylin-and-eosin-stained lung sections show medial thickening in high-fat-fed DR and low- and high-fat-fed DIO rats (A) and in Zucker fatty rats (E). Scale bars = 100 μm. C, G, Remodeling was confirmed by morphometric analysis of 10 pulmonary arteries per animal. B, D, Representative fluorescence micrographs of lung parenchyma stained with an anti-rat smooth muscle (SM)-actin antibody, followed by an Alexa Fluor 488–conjugated secondary antibody. Arrows indicate scored actin-positive microvessels. Scale bars = 50 μm. D, H, Morphometric analysis of the number of actin-positive vessels per field in 10 fields per animal. Values in C, D, G, and H are averages from 3 experiments with 6 animals per group.
High-fat-fed DR rats had significantly more actin-positive arterioles than low-fat-fed DR animals (Fig. 3B, 3D). Similarly, low-fat-fed DIO rats also had a greater number of muscularized arterioles than low-fat-fed DR animals. High-fat feeding, in combination with an obesity-prone phenotype, did not appear to have an additive impact on neomuscularization. Zucker fatty rats also had a greater number of actin-positive microvessels, compared to lean controls (Fig. 3F, 3H).
No evidence of hypoxemia or hypoventilation in rat models of obesity
Hematocrit was the same for DR and DIO rats, regardless of low- or high-fat diet (Fig. 4A). Likewise, hematocrit was the same for Zucker lean and fatty rats maintained under isobaric normoxia. As anticipated, exposure to 3 weeks of hypobaric hypoxia elicited polycythemia in both lean and fatty rats.
Figure 4.
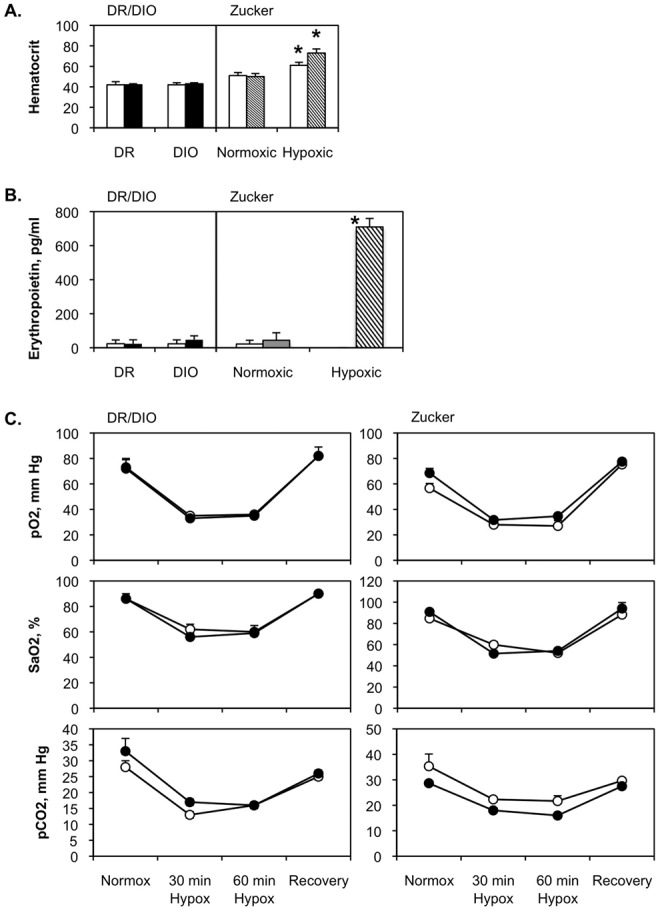
No evidence for hypoventilation or hypoxia in diet-resistant/diet-induced obesity (DR/DIO) and Zucker lean/fatty rat models. Eight-week-old male DR and DIO rats were fed either a low-fat (white bars on left side of A and B) or a high-fat (black bars on left side of A and B) diet ad lib. for 5 months. Eight-week-old male Zucker lean (white bars on right side of A and B) and fatty (hatched bars on right side of A and B) rats were fed standard chow ad lib. for 5 months. In A, separate cohorts of lean and fatty Zucker rats were maintained under hypoxic conditions for the last 3 weeks of the 5-month period. In B, a separate cohort of Zucker fatty rats was held under hypoxic conditions for 48 hours at the end of the 5-month period. C, After 5 months, po2, pco2, and Sao2 were measured in blood samples retrieved from resting low-fat-fed DR, high-fat-fed DIO rats (left; open and filled circles, respectively), or lean and fatty Zucker rats (right; open and filled circles, respectively) in Plexiglas containers continuously flushed with room air (normox). Measurements were also made 30 and 60 minutes after the atmosphere in the containers was switched to hypoxic gas mixture (hypox), and then again 30 minutes after the containers were flushed with room air (recovery). Values in all panels are averages from 3 experiments with 6 animals per group.
EPO was barely detectable in DR and DIO rats fed with low- or high-fat diets (DR low fat: 23.5 ± 22.0 pg/mL; DR high fat: 23.5 ± 23.0 pg/mL; DIO low fat: 23.0 ± 23.0 pg/mL; DIO high fat: 47.0 ± 23.0 pg/mL) or in Zucker lean and fatty rats (lean: 22.0 ± 21.0 pg/mL; fatty: 44 ± 42 pg/mL; Fig. 4B). We also measured EPO in Zucker fatty rats after 48 hours of exposure to hypobaric hypoxia, to test the sensitivity of the assay and demonstrate the robust response of EPO production to brief hypoxic exposure. EPO levels were more than 35-fold higher in the hypoxic rats (710 ± 50 pg/mL) than in those housed at ambient Denver conditions.
No differences in po2, pco2, or Sao2 were noted between the experimental groups. The data from these studies indicate that obese rats do not experience significant hypoxemia or exhibit evidence of hypoventilation (Fig. 4C).
Basic metabolic parameters of obese rat models
There was no significant difference in fasting blood glucose between genetic backgrounds (DR vs. DIO, lean vs. fatty) or diets (Fig. 5A, 5E). Fasting blood insulin was elevated in the DR and DIO rats, with lower levels in the high-fat-fed animals (Fig. 5B). These differences never achieved statistical significance. Blood insulin was normal in Zucker lean rats but highly elevated in Zucker fatty animals (Fig. 5F).
Figure 5.
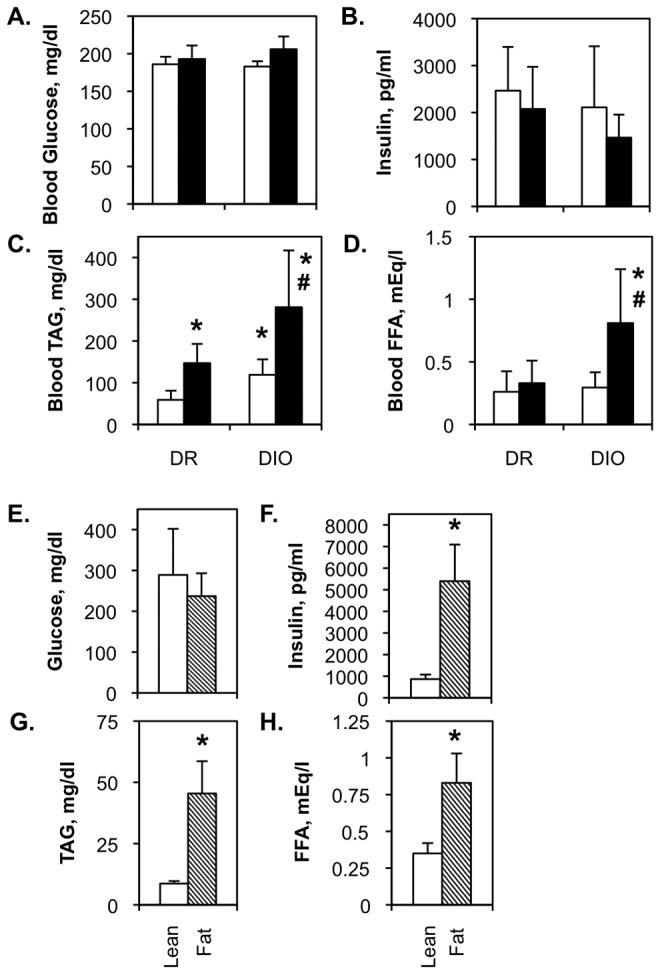
Metabolic parameters in diet-resistant/diet-induced obesity (DR/DIO) and Zucker lean/fatty rat models. A–D, Eight-week-old male DR and DIO rats were fed either a low-fat (white bars) or a high-fat (black bars) diet ad lib. for 5 months. E–H, Eight-week-old male Zucker lean and fatty rats were fed standard chow ad lib. for 5 months. Blood glucose and insulin levels and circulating triacylglycerol (TAG) and free fatty acid (FFA) concentrations were measured as detailed in “Methods.” Values in all panels are averages from 3 experiments with 6 animals per group.
Circulating triglyceride was elevated by high-fat diet in both DR and DIO rats (Fig. 5C) and was higher in low-fat-fed DIO rats than in low-fat-fed DR animals. Triglyceride levels were higher in high-fat-fed DIO rats than in either high-fat-fed DR or low-fat-fed DIO animals, suggesting that diet and genetic background have an additive impact on blood triglyceride levels in this model. Circulating free fatty acids were significantly higher only in the high-fat-fed DIO group (Fig. 5D). However, both triglyceride (Fig. 5G) and fatty acids (Fig. 5H) were increased in Zucker fatty rats.
Oxidative stress in the PA wall of obese rats
Immunofluorescent staining revealed increased levels of 8-OH-guanosine and 4-OH-nonenal with high-fat feeding or obesity proneness in the DR/DIO model (Fig. 6). Fluorescence intensities of individual nuclei in images of multiple arteries were measured and averaged as a quantitative index of 8-OH-guanosine levels (Fig. 7A), and total fluorescence of the endothelial layer and arterial media was measured as an index of 4-OH-nonenal levels (Fig. 7B). Minimal staining for either adduct was observed in endothelial cells or the arterial media in low-fat-fed DR rats. However, discrete foci of intense 4-OH-nonenal staining were present in the perivascular region in all samples/conditions. High-fat feeding of DR rats produced a small but insignificant increase in 8-OH-guanosine but an 8–9-fold increase in 4-OH-nonenal staining. This staining was particularly intense in the media adjacent to the adventitia. Both 8-OH-guanosine and 4-OH-nonenal levels in the artery wall were significantly elevated in low- or high-fat-fed DIO rats, but the impact of high-fat diet and obesity proneness was not additive for either adduct.
Figure 6.

Oxidant damage in the pulmonary artery (PA) wall of diet-induced-obesity-resistant/obesity-prone (DR/DIO) rats. Eight-week-old male DR and DIO rats were fed either a low-fat or a high-fat diet ad lib. for 5 months. Figure shows representative bright-field and fluorescence images of proximal PAs adjacent to airways. Sections were stained with 4,6-diamidino-2-phenylindole (DAPI) to locate nuclei, smooth muscle (SM)-actin to visualize the arterial media, and antibodies to either 8-OH-guanosine (A) or 4-OH-nonenal (B). Sections were subsequently stained with Alexa Fluor 555–conjugated secondary antibodies. Scale bar = 100 μm.
Figure 7.
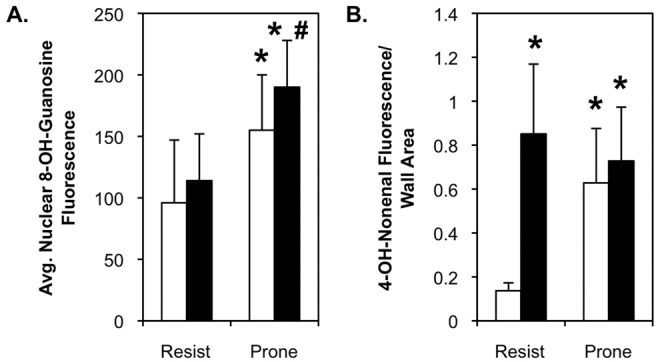
Morphometric analysis confirms oxidant damage in the pulmonary artery (PA) wall of diet-induced-obesity-resistant (Resist) and diet-induced-obesity-prone rats. Nuclear 8-OH-guanosine (A) and total medial 4-OH-nonenal (B) levels were quantified by morphometric analysis of fluorescent images like those in Figure 6. The relative 8-OH-guanosine fluorescence was measured in 6–8 individual nuclei (identified by 4,6-diamidino-2-phenylindole [DAPI] fluorescence) in 10 PAs in lung sections from 6 rats in each group from 2 separate experiments. Relative 4-OH-nonenal fluorescence was measured over the entire region defined by smooth muscle (SM)-actin staining in 10 PAs in lung sections from 6 rats in each group from 2 separate experiments. Fluorescence values represent single-channel luminance. White bars: rats fed low-fat diet; black bars: rats fed high-fat diet.
Inflammatory cytokine levels in obese rat models
Levels of IL-6, TNF-α, IL-1β and tPAI-1 were barely detectable in lean or fatty Zucker rats (Fig. 8). Interestingly, plasma levels of these cytokines were highly elevated in DR and DIO rats with high-fat feeding but were not increased in low-fat-fed DIO rats. Thus, expression of these factors is linked to diet composition but not to genetic background. MCP-1 was elevated in high-fat-fed DR and DIO rats, but it was also increased in Zucker fatty rats, even though they were maintained on low-fat chow. As expected, leptin was increased in Zucker fatty rats because of increased adiposity and global leptin resistance. In the DR/DIO model, leptin was increased by high-fat diet and the DIO genetic background and, in general, correlated with adiposity. Adiponectin was elevated in Zucker fatty rats and in DIO rats compared to DR controls. High-fat diet had no significant impact on adiponectin values.
Figure 8.
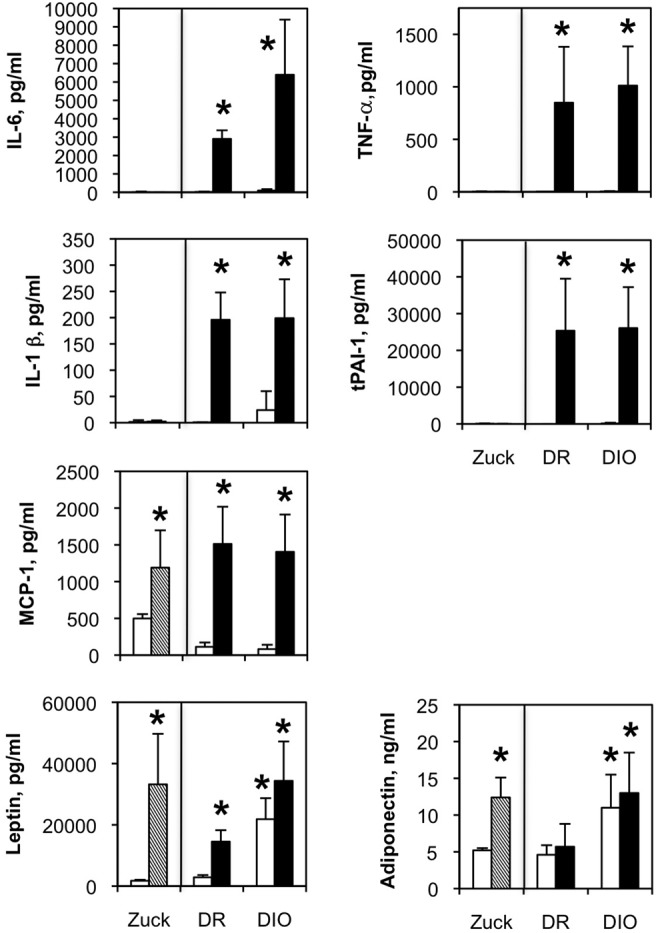
Circulating leptin, adiponectin, and inflammatory cytokine levels in Zucker (Zuck) lean/fatty and diet-resistant/diet-induced obesity (DR/DIO) rat models. Eight-week-old male Zucker lean (white bars) and fatty (hatched bars) rats were fed standard chow ad lib. for 5 months. Eight-week-old male DR and DIO rats were fed either a low-fat (white bars) or a high-fat (black bars) diet ad lib. for 5 months. Cytokine levels were measured in blood plasma by multiplex bead assay. Values are averages from 3 experiments with 6 animals per group. IL: interleukin; MCP-1: macrophage chemoattractant protein-1; TNF-a: tumor necrosis factor α; tPAI-1: tissue plasminogen activator inhibitor-1.
Discussion
Obesity remains one of the foremost medical issues to confront society and medicine. The physical changes and metabolic abnormalities elicited by excess body fat educe numerous comorbidities, including PAH.6-8 Obesity-related PAH has, until recently, been believed to result primarily from lung hypoxia due to hypoventilation. However, many of the comorbidities associated with excess adiposity are directly linked to metabolic and inflammatory disturbances, suggesting that these processes may also play a role in PAH. Here we examined rat models of obesity with two goals. The first was to determine whether measurable changes in pulmonary hemodynamics and pulmonary arterial structure occur in these models, and the second was to assess the relative impact of ventilatory versus metabolic and inflammatory processes on PAH.
Our results show detrimental changes in both pulmonary hemodynamics and PA and RV thickness in the DR/DIO and Zucker rat models. The pulmonary pathology observed in these rats occurred in the absence of the increased hematocrit and EPO levels that occur in response to chronic hypoventilation and hypoxia or of the decreased resting ventilatory efficiency that is reported in obese humans.13 The magnitude of hemodynamic and structural changes was generally less than that reported for other PAH models driven by chronic hypoxia alone,31 hypoxia plus vascular endothelial growth factor receptor antagonists,32 monocrotaline,31,33 or PA banding.34 This suggests that additional factors, including hypoxia, may be necessary to elicit the full magnitude of responses seen in traditional models of PAH. Mild disease is consistent with the subclinical disease found in obese patients at autopsy.10,11 Indeed, preliminary studies (Fig. S4) demonstrate that chronic hypoxia, in combination with high-fat feeding/obesity, exerts additive negative effects on the pulmonary vasculature. We predict that both hypoventilation and metabolic abnormalities contribute to the progressive deterioration in cardiopulmonary function in obese humans.
We also noted that the changes in pulmonary structure and function we observed in obese rats occurred in the absence of a significant increase in systemic vascular pressure or changes in left ventricular size and structure. Although systemic vascular pathology has been reported in DR/DIO35-37 and Zucker fatty38 rats, these changes typically take longer than 20 weeks to manifest. The observation is significant because it indicates that pulmonary pathology can develop in the absence of detectable systemic cardiovascular deterioration. Of note, there is evidence in humans that left-heart pathology is linked to detrimental changes in right-heart function and abnormal pulmonary function, suggesting that clinical presentation occurs with more progressive disease.39 It is possible that the early changes observed here herald later disease progression.
Scrutiny of the DR/DIO data indicates that a high-fat diet alone is sufficient to promote PA remodeling and muscularization of distal arterioles and to elicit elevated PA pressure and RV hypertrophy (Table 1). A high-fat diet also resulted in hypertriglyceridemia, increased circulating inflammatory cytokines, and oxidant damage in the PA wall. The “genetic background” of the DIO rats also stimulated detrimental changes in arterial structure and arterial oxidant damage but had no effect on PA pressure, RV hypertrophy, or inflammatory cytokine production. We surmise that the obesity-favorable genotype of these animals, in the absence of calorie or fat overload, fails to elicit persistent vasoconstriction, which would promote RV hypertrophy. We are examining this idea in ongoing experiments. The DIO rats are also mildly hyperphagic and were fed ad lib. in these studies, which is evident in the higher body weights of low-fat-fed DIO rats relative to the low-fat-fed DR group. The increased calories consumed by the DIO rats may explain the PA thickening, distal neomuscularization, and oxidant damage in this cohort more than genetic changes in metabolism or physiology. We are currently examining this possibility in pair-feeding experiments. Finally, high-fat diet and genetic background had a synergistic impact on PA remodeling, RV hypertrophy, and circulating free fatty acid levels but not on other end points. The reason for this is unclear, but it is interesting that both PA remodeling and RV hypertrophy require hyperplasia and hypertrophy of resident cell populations and changes in extracellular matrix composition and structure. Perhaps overabundance of free fatty acids and metabolic damage produced by their catabolism alter the growth characteristics of smooth muscle cells (SMCs), cardiomyocytes, and other cells, leading to exaggerated arterial and cardiac remodeling. We are exploring this hypothesis, using cells isolated from each of the treatment groups.
Table 1.
Correlation of end points with diet and/or genetic background in DR/DIO and Zucker lean/fatty rats
| DR/DIO model | Zucker model | |||
|---|---|---|---|---|
| Parameter | High-fat diet | Genetic backgrounda | Diet-genetics synergyb | Fatty vs. lean |
| PAH end points | ||||
| Medial thickening | + | + | + | + |
| Mean PAP | + | + | − | + |
| Neomuscularization | + | + | − | + |
| RV hypertrophy | − | + | − | + |
| Metabolic end points | ||||
| Blood glucose | − | − | − | − |
| Plasma insulin | − | − | − | + |
| Plasma TAG | + | + | + | + |
| Plasma FFA | − | − | + | + |
| Oxidant end points | ||||
| 8-OH-guanosine | − | + | + | ND |
| 4-OH-nonenal | + | + | − | ND |
| Inflammatory cytokines | + | − | − | − |
Except as noted, “correlation” is defined as a statistically significant (P < .05) change versus the control cohort. A plus sign indicates a positive correlation and a minus sign indicates that there is no correlation. DIO: diet-induced obesity–prone; DR: diet-induced obesity–resistant; FFA: free fatty acid; ND: not determined; PAH: pulmonary arterial hypertension; PAP: pulmonary artery pressure; RV: right ventricle; TAG: triacylglycerol.
Statistically significant difference between the DR low-fat and DIO low-fat cohorts.
Statistically significant difference between the DIO high-fat cohort and either the DR high-fat or the DIO low-fat cohort. This represents evidence of putative synergism between diet and genetic background.
Another significant observation in our studies is that changes in pulmonary hemodynamics and PA structure did not correlate with blood glucose or insulin levels but rather correlated with circulating levels of triglycerides and, to a lesser extent, free fatty acids (Table 1). In high-fat-fed animals, some of these lipids undoubtedly arise from the diet itself, which is likely a factor in human obesity-related PAH, given the high fat content of the Western diet. However, manifestations of PAH were also observed in low-fat-fed (DR/DIO model) or chow-fed (Zucker model) rats, highlighting adipose tissue and the liver as sources for circulating free fatty acids and triglycerides, respectively. Insulin resistance accompanying obesity increases adipose tissue lipolysis and excretion of free fatty acids into the circulation. Likewise, de novo lipogenesis and triglyceride synthesis are elevated in the insulin-resistant liver, as is production of low-density lipoproteins that carry triglycerides and cholesterol to the peripheral tissues. Clearance of high-density lipoproteins by the liver is also diminished in obesity. The net increase in fat delivery to the PA wall may negatively affect vascular structure and function in several ways. Inefficient oxidation of fatty acids may result in the production of reactive oxygen species (ROSs),40-42 and by-products of incomplete fatty acid oxidation.43-45 In addition, increased delivery of fatty acids could elevate production of lipid-based second messengers, such as sphingolipids, ceramides, and gangliosides, and oxidized arachadonate derivatives within vessel wall cells.46-48 Our data suggest that inefficient oxidation of lipids may be involved in obesity-related PAH, as ROSs were observed in the PA wall in obese DR/DIO rats. The generally low oxidative capacity of SMCs, endothelial cells, and other arterial-wall populations probably contributes to the production of ROSs in the PA wall, while excess delivery of fats undoubtedly shifts catabolic processes in favor of their oxidation, as in other systems.
Inflammatory cytokines were also elevated in obese rats, especially with high-fat feeding. These cytokines were examined on the basis of their expression by adipose tissue in obese subjects. While recognized for their role in promoting insulin resistance and inflammation in adipose and peripheral tissues, these factors also drive pathological remodeling of the PA wall. For example, IL-6-overexpressing mice spontaneously manifest PAH, with muscularization of distal arterioles and formation of plexiform lesions composed of endothelial cells and T lymphocytes.49 In contrast, depletion of IL-6 attenuates hypoxia-induced PAH by blocking arterial remodeling and accumulation of lung macrophage.50 Thus, IL-6 appears to influence SMC migration, endothelial cell proliferation, and recruitment of immune cells during the development of PAH. The ability of IL-6 to induce PA SMC migration has been confirmed in cell culture experiments.51 MCP-1 may also be a crucial factor modulating recruitment of monocytes to the arterial adventitia, which, along with fibroblasts, participate in fibrotic remodeling of the vessel wall.52,53 MCP-1 levels are elevated in lung and plasma from PAH patients, and MCP-1 expression is increased in endothelial cells and SMCs from these individuals.54 Migration of monocytes toward endothelial cells from PAH patients is effectively blocked by anti-MCP-1 antibodies.
Production of inflammatory cytokines and chemokines in PAH is normally attributed to resident PA cells, including endothelial cells and SMCs. The cytokines thus act in a paracrine or autocrine manner in eliciting pathological phenotypic switching of resident arterial cells, and their localized production forms a gradient for recruitment of immune cells. In obesity, it is likely that adipose tissue is the initial (and a continuous) source of inflammatory agents that impinge on peripheral tissues, such as the lung. Although not localized to the lung, these adipose-derived cytokines produce a body-wide state of low-grade inflammation that may be important in eliciting progressive arterial pathology.55 Moreover, IL-6 and TNF-α produced in adipose tissue are believed to promote insulin resistance in peripheral tissues via blockade of the insulin signaling pathway.56,57 In conjunction with elevated circulating lipids, loss of insulin responsiveness may have a potent impact on arterial cell phenotype via changes in fuel metabolism and oxidant production.
Insulin resistance has been suggested to play a causal role in the development of PAH in obese individuals.20 Loss of insulin sensitivity manifests early in many obese individuals, before overt hyperglycemia is observed. We found that blood insulin levels were highly elevated in Zucker fatty rats. Circulating insulin levels were elevated in DR and DIO rats, but there was no significant difference between treatment groups. The results suggest that insulin resistance alone is insufficient to drive changes in pulmonary arterial structure or vascular tone. Efforts to modulate blood insulin levels and insulin sensitivity almost always modify blood lipids, glucose, and inflammation, making it difficult to separate insulin responsiveness from other factors.
Loss of adiponectin in obesity has also been implicated in PAH.58,59 Adiponectin is produced by adipocytes and exerts potent insulin-sensitizing and anti-inflammatory effects throughout the body. Adiponectin expression generally correlates with adiposity, but it is paradoxically diminished in obese human subjects, suggesting impaired adiponectin production or secretion in chronic obesity. Adiponectin-null mice spontaneously exhibit signs of pulmonary hypertension at 1 year of age60 and show exaggerated PA remodeling in response to chronic hypoxia.61 Conversely, adiponectin overexpression in mice prevents PA remodeling in response to either hypoxia or high-dose ovalbumin.62 Circulating adiponectin levels in both Zucker fatty rats and obese DIO rats were elevated in our experiments. These results were not unexpected, as high-fat feeding over periods of 6–20 weeks increases adiposity and adiponectin expression in rat63,64 and mouse65,66 models. In some studies, adiponectin levels are reduced after correction for white adipose tissue mass,64 but the impact on total circulating levels is less robust. The loss of appropriate coupling between fat mass and adiponectin is believed to reflect adipocyte insulin resistance. The time course over which adiponectin production is suppressed in humans has not been evaluated in detail. Our studies suggest that other obesity-related factors, including hyperlipidemia, oxidant stress, and inflammation, may play a more important role than reduced adiponectin production and, in fact, may override elevated adiponectin expression in promoting PAH.
Pulmonary hypertension was noted in an early characterization of the Zucker fatty-rat model, albeit in rats more than 1 year old.38 A more recent report by Moral-Sanz and colleagues67 failed to detect manifestations of PAH in Zucker fatty rats. The reasons underlying the different results reported by the Moral-Sanz group and our laboratory remain unclear. However, they may reflect differences in the source and terminal age of the rats or dietary and housing differences. They may also be due to differences in ambient altitude, as the studies by Moral-Sanz et al.67 were conducted in Madrid, Spain (approximate altitude: 660 m), whereas ours were performed in Denver, Colorado (approximate altitude: 1,650 m). Perhaps the modest hypoxia experienced at Denver altitude was sufficient to unmask the otherwise minimal impact of the obese genetic background. If so, this would further highlight the synergistic impact of genetic background and hypoventilation on the development of PAH.
To our knowledge, this is the first report of pulmonary abnormalities in the DR/DIO model. Data obtained with this model implicate genetic background in the development of PAH in obese subjects, as did those from the Zucker rat model. More importantly, this model also indicates that dietary composition, or at least calorie overload, also promotes detrimental changes in the pulmonary circulation. We believe that this model affords the opportunity to further dissect the roles of genetics, epigenetics, diet, and exercise in regulating pulmonary structure and function. We are currently exploring pair-feeding studies to further define the impact of genetics versus diet and dietary composition on the development of PAH in this model. DR/DIO rats may also be useful for exploring aspects of maternal/fetal obesity, a syndrome characterized by exaggerated postnatal growth in infants of over- or underfed mothers. It would be interesting to examine the impact of calorie excess during gestation on pulmonary hemodynamics and structure in offspring over time. Such studies might reveal genetic and, more importantly, epigenetic phenomena underlying the familial propensity for developing PAH.
In closing, we conclude that obesity-related PAH can be modeled in rat models of obesity. Our data indicate that both high-fat feeding and genetic background contribute to pulmonary arterial pathology. Insulin resistance alone and changes in adiponectin levels do not appear to be major drivers of early PA pathology. However, changes in arterial structure and vascular tone correlated with circulating lipid and inflammatory cytokine levels. We also found evidence of elevated ROS production related to calorie overload and genetic background. The typically low oxidative capacity of cells in the arterial wall may lead to inappropriate or inefficient oxidation of circulating lipids, resulting in increased ROS production. We are currently exploring the impact of agents that alter fuel selection and mitochondrial activity as potential therapeutic strategies to intervene in the development of PAH in obese individuals.
Appendix.
Figure S1.
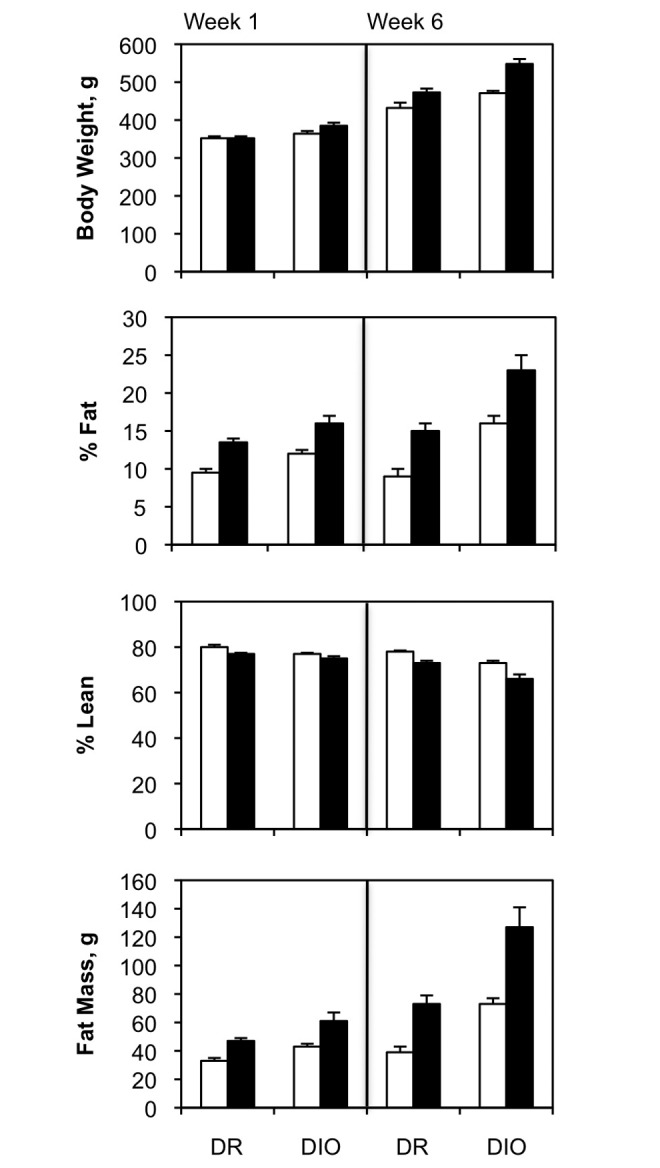
Retrospective analysis of diet-resistant/diet-induced obesity (DR/DIO) rat body composition. DR and DIO rats were fed a low-fat (white bars) or a high-fat (black bars) diet. The various parameters of body composition were measured by magnetic resonance imaging after 1 week and of feeding and again after 6 weeks. The cohorts of rats from which these data were derived were not the same as those used in the experiments for this article. The data were kindly provided by Drs. MacLean and Jackman from the Metabolic Core Facility at the University of Colorado Anschutz Medical Campus. The data show that weight gain in the DR/DIO model is primarily due to expansion of adipose tissue rather than expansion of lean mass.
Figure S2.
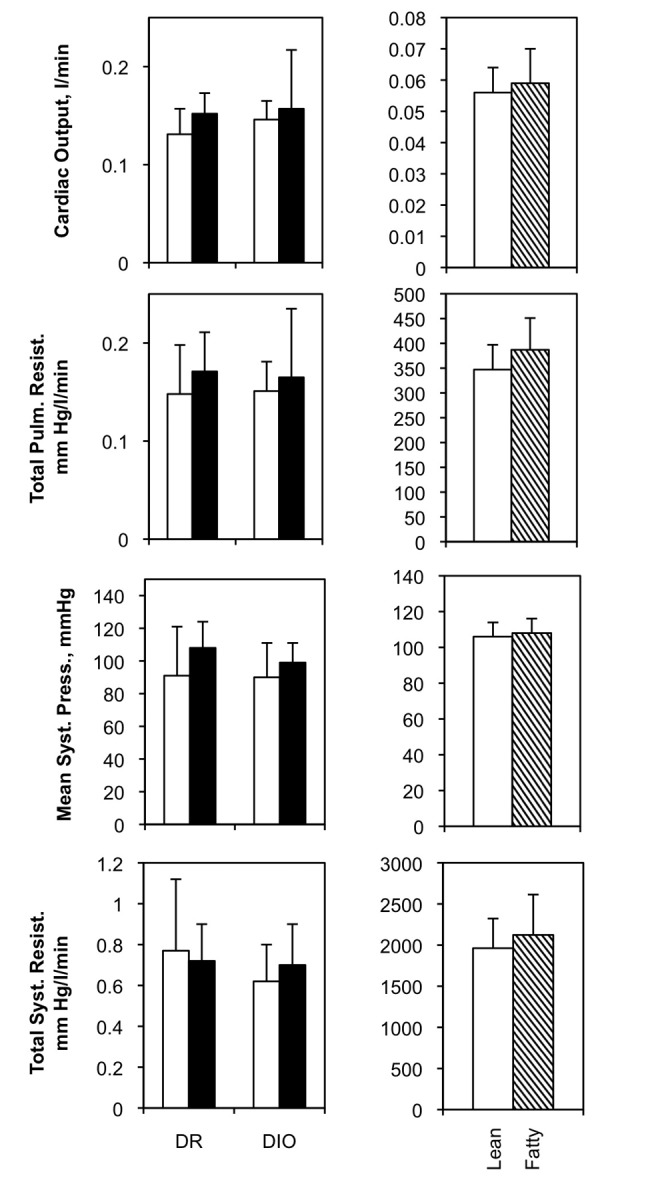
Hemodynamic evaluation of diet-resistant/diet-induced obesity (DR/DIO) and Zucker lean/fatty rat models. Left, eight-week-old male DR and DIO rats were fed either a low-fat (white bars) or a high-fat (black bars) diet ad lib. for 5 months. Right, eight-week-old male Zucker lean and fatty rats were fed standard chow ad lib. for 5 months. The indicated hemodynamic parameters were measured as described in “Methods.” Three experiments, N = 6 per group for each experiment. Pulm. resist.: pulmonary resistance; syst. press.: systemic pressure; syst. resist.: systemic resistance.
Figure S3.

Cardiac fibrosis in diet-resistant/diet-induced obesity (DR/DIO) rats. Eight-week-old male DR and DIO rats were fed either a low- or a high-fat diet ad lib. for 5 months. Representative bright-field micrographs of picrosirius red–stained heart regions are shown: right ventricle (RV), septum, and left ventricle (LV). No differences were noted in collagen deposition due to diet or genetic background.
Figure S4.
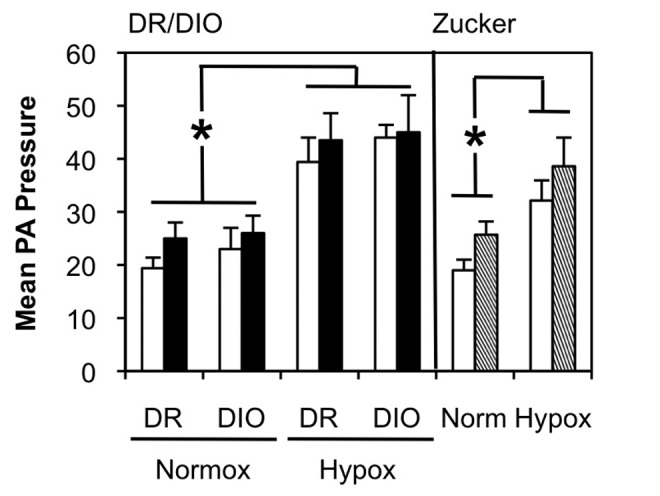
Hypoxia exacerbates pulmonary arterial hypertension in diet-resistant/diet-induced obesity (DR/DIO) and Zucker lean/fatty rat models. Left, eight-week-old male DR and DIO rats were fed either a low-fat (white bars) or a high-fat (black bars) diet ad lib. for 5 months. Right, eight-week-old male Zucker lean (white bars) or fatty (hatched bars) were fed standard chow for 5 months. Separate cohorts of rats were exposed to hypoxia (Hypox) for the last weeks of the treatment regimen. Mean pulmonary artery (PA) pressure (mmHg) was measured as described in “Methods.” There were 2 experiments, with 6 animals per group for each experiment. *P < .05 compared to the DR normoxic (Normox) cohort (left) or to the normoxic cohort (right).
Source of Support: This research was funded by National Institutes of Health grant P01 HL014985 (to DJK) and by Veterans Affairs MERIT and Colorado Clinical Translational Research Institute grant UL1RR025780 (to JEBR).
Conflict of Interest: None declared.
Supplements
AppendixPulmCirc-004-638.s001.pdf (6.1MB, pdf)
References
- 1.Centers for Disease Control and Prevention. Overweight and obesity. 2010. http://www.cdc.gov/nccdphp/dnpa/obesity/index.htm.
- 2.National Institute of Diabetes and Digestive and Kidney Diseases, Weight-Control Information Network. Overweight and obesity statistics. http://www.win.niddk.nih.gov/statistics/index.htm.
- 3.Bays H, Ballantyne C. Adiposopathy: why do adiposity and obesity cause metabolic disease? Future Lipidol 2006;1(4):389–420.
- 4.Brown WV, Fujioka K, Wilson PW, Woodworth KA. Obesity: why be concerned? Am J Med 2009;122(4 suppl.):S4–S11. [DOI] [PubMed]
- 5.Guh DP, Zhang W, Bansback N, Amarsi Z, Birmingham CL, Anis AH. The incidence of co-morbidities related to obesity and overweight: a systematic review and meta-analysis. BMC Public Health 2009;9:88. [DOI] [PMC free article] [PubMed]
- 6.Murugan AT, Sharma G. Obesity and respiratory diseases. Chronic Respir Dis 2008;5(4):233–242. [DOI] [PubMed]
- 7.Dela Cruz CS, Matthay RA. Role of obesity in cardiomyopathy and pulmonary hypertension. Clin Chest Med 2009;30(3):509–523. [DOI] [PubMed]
- 8.Friedman SE, Andrus BW. Obesity and pulmonary hypertension: a review of pathophysiological mechanisms. J Obes 2012:505274. doi:10.1155/2012/505274. [DOI] [PMC free article] [PubMed]
- 9.Taraseviciute A, Voelkel NF. Severe pulmonary hypertension in postmenopausal obese women. Eur J Med Res 2006;11(5):198–202. [PubMed]
- 10.Blankfield RP, Hudgel DW, Tapolyai AA, Zyzanski SJ. Bilateral leg edema, obesity, pulmonary hypertension, and obstructive sleep apnea. Arch Intern Med 2000;160(15):2357–2362. [DOI] [PubMed]
- 11.Haque AK, Gadre S, Taylor J, Haque SA, Freeman D, Duarte A. Pulmonary and cardiovascular complications of obesity: an autopsy study of 76 obese subjects. Arch Pathol Lab Med 2008;132(9):1397–1404. [DOI] [PubMed]
- 12.Mokhlesi B, Tulaimat A. Recent advances in obesity hypoventilation syndrome. Chest 2007;132(4):1322–1336. [DOI] [PubMed]
- 13.Olson AL, Zwillich C. The obesity hypoventilation syndrome. Am J Med 2005;118(9):948–956. [DOI] [PubMed]
- 14.Voelkel NF, Mizuno S, Bogaard HJ. The role of hypoxia in pulmonary vascular diseases: a perspective. Am J Physiol Lung Cell Mol Physiol 2013;304(7):L457–L465. [DOI] [PubMed]
- 15.Soumaya K. Molecular mechanisms of insulin resistance in diabetes. In: Ahman SI, ed. Diabetes: an old disease, a new insight. Advances in experimental medicine and biology, vol. 771. New York: Landes Bioscience and Springer, 2012:240–251. [DOI] [PubMed]
- 16.Engfeldt P, Arner P. Lipolysis in human adipocytes: effects of cell size, age and regional differences. In: Pfeiffer EF, ed. Proinsulin. Hormone and metabolic research supplement series, vol. 19. Stuttgart: Thieme, 1988:26–29. [PubMed]
- 17.Klop B, Elte JW, Cabezas MC. Dyslipidemia in obesity: mechanisms and potential targets. Nutrients 2013;5(4):1218–1240. [DOI] [PMC free article] [PubMed]
- 18.Majka SM, Barak Y, Klemm DJ. Adipocyte origins: weighing the possibilities. Stem Cells 2011;29(7):1034–1040. [DOI] [PMC free article] [PubMed]
- 19.Singer GM, Setaro JF. Secondary hypertension: obesity and the metabolic syndrome. J Clin Hypertens 2008;10(7):567–574. [DOI] [PMC free article] [PubMed]
- 20.Zamanian RT, Hansmann G, Snook S, Lilienfeld D, Rappaport KM, Reaven GM, Rabinovitch M, Doyle RL. Insulin resistance in pulmonary arterial hypertension. Eur Respir J 2009;33(2):318–324. [DOI] [PMC free article] [PubMed]
- 21.Fouty B. Diabetes and the pulmonary circulation. Am J Physiol Lung Cell Mol Physiol 2008;295(5):L725–L726. [DOI] [PMC free article] [PubMed]
- 22.Levin BE, Dunn-Meynell AA, Balkan B, Keesey RE. Selective breeding for diet-induced obesity and resistance in Sprague-Dawley rats. Am J Physiol Regul Integr Comp Physiol 1997;273(2):R725–R730. [DOI] [PubMed]
- 23.Zucker LM, Zucker TF. Fatty, a new mutation in the rat. J Hered 1961;52(6):275–278.
- 24.Brennan LA, Steinhorn RH, Wedgewood S, Mata-Greenwood E, Roark EA, Russell JA, Black SM. Increased superoxide generation is associated with pulmonary hypertension in fetal lambs: a role for NADPH oxidase. Circ Res 2003;92(6):683–691. [DOI] [PubMed]
- 25.Hoshikawa Y, Ono S, Suzuki S, Tanita T, Chida M, Song C, Noda M, Tabata T, Voelkel NF, Fujimura S. Generation of oxidative stress contributes to the development of pulmonary hypertension by hypoxia. J Appl Physiol 2001;90(4):1299–1306. [DOI] [PubMed]
- 26.Elmedal B, de Dam MY, Mulvany MJ, Simonsen U. The superoxide dismutase mimetic, Tempol, blunts right ventricular hypertrophy in chronic hypoxic rats. Br J Pharmacol 2004;141(1):105–113. [DOI] [PMC free article] [PubMed]
- 27.Klemm DJ, Majka SM, Crossno JT Jr., Psilas JC, Reusch JEB, Garat CV. Reduction of reactive oxygen species prevents hypoxia-induced CREB depletion in pulmonary artery smooth muscle cells. J Cardiovasc Pharmacol 2011;58(2):181–191. [DOI] [PMC free article] [PubMed]
- 28.Lachmanová V, Hniličková O, Povýšilová V, Hampl V, Herget J. N-acetylcysteine inhibits hypoxic pulmonary hypertension most effectively in the initial phase of chronic hypoxia. Life Sci 2005;77(2):175–182. [DOI] [PubMed]
- 29.Gutierrez J, Ballinger SW, Darley-Usmar VM, Landar A. Free radicals, mitochondria, and oxidized lipids: the emerging role in signal transduction in vascular cells. Circ Res 2006;99(9):924–932. [DOI] [PubMed]
- 30.Shen GX. Oxidative stress and diabetic cardiovascular disorders: roles of mitochondria and NADPH oxidase. Can J Physiol Pharmacol 2010;88(3):241–248. [DOI] [PubMed]
- 31.Meyrick BO, Perkett EA. The sequence of cellular and hemodynamic changes of chronic pulmonary hypertension induced by hypoxia and other stimuli. Am Rev Respir Dis 1989;140(5):1486–1489. [DOI] [PubMed]
- 32.Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 2010;121(25):2747–2754. [DOI] [PubMed]
- 33.Gomez-Arroyo JG, Farkas L, Alhussaini AA, Farkas D, Kraskauskas D, Voelkel NF, Bogaard HJ. The monocrotaline model of pulmonary hypertension in perspective. Am J Physiol Lung Cell Mol Physiol 2012;302(4):L363–L369. [DOI] [PubMed]
- 34.Rondelet B, Dewachter C, Kerbaul F, Kang X, Fesler P, Brimioulle S, Naeije R, Dewachter L. Prolonged overcirculation-induced pulmonary arterial hypertension as a cause of right ventricular failure. Eur Heart J 2012;33(8):1017–1026. [DOI] [PubMed]
- 35.Huisamen B, Dietrich D, Bezuidenhout N, Lopes J, Flepisis B, Blackhurst D, Lochner A. Early cardiovascular changes occurring in diet-induced, obese insulin-resistant rats. Mol Cell Biochem 2012;368(1):37–45. [DOI] [PubMed]
- 36.Maarman G, Marais E, Lochner A, du Toit EF. Effect of chronic CPT-1 inhibition on myocardial ischemia-reperfusion injury (I/R) in a model of diet-induced obesity. Cardiovasc Drugs Ther 2012;26(3):205–216. [DOI] [PubMed]
- 37.Smith PM, Ferguson AV. Cardiovascular actions of leptin in the subfornical organ are abolished by diet-induced obesity. J Neuroendocrinol 2012;24(3):504–510. [DOI] [PubMed]
- 38.Johnson PR, Stern JS, Horwitz BA, Harris RE Jr., Greene SF. Longevity in obese and lean male and female rats of the Zucker strain: prevention of hyperphagia. Am J Clin Nutr 1997;66(4):890–903. [DOI] [PubMed]
- 39.Lam CS, Borlaug BA, Kane GC, Enders FT, Rodeheffer RJ, Redfield MM. Age-associated increases in pulmonary artery systolic pressure in the general population. Circulation 2009;119(20):2663–2670. [DOI] [PMC free article] [PubMed]
- 40.Bachschmid MM, Schildknecht S, Matsui R, Zee R, Haeussler H, Cohen RA, Pimental D, van der Loo B. Vascular aging: chronic oxidative stress and impairment of redox signaling—consequences for vascular homeostasis. Ann Med 2013;45(1):17–36. [DOI] [PMC free article] [PubMed]
- 41.Christon R, Drouin O, Marette A. Redox modulation of insulin signaling and endothelial function. Antioxid Redox Signal 2005;7(7–8):1062–1070. [DOI] [PubMed]
- 42.Hennig B, Toborek M, McClain CJ. High-energy diets, fatty acids and endothelial function: implications for atherosclerosis. J Am Coll Nutr 2001;20(2):97–105. [DOI] [PubMed]
- 43.Bell JA, Reed MA, Consitt LA, Martin OJ, Haynie KR, Hulver MW, Muoio DM, Dohm GL. Lipid partitioning, incomplete fatty acid oxidation, and insulin signal transduction in primary human muscle cells: effects of severe obesity, fatty acid incubation, and fatty acid translocase/CD36 overexpression. J Clin Endocrinol Metab 2010;95(7):3400–3410. [DOI] [PMC free article] [PubMed]
- 44.Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab 2008;7(1):45–56. [DOI] [PubMed]
- 45.Moore KH, Radloff JF, Hull FE, Sweeley CC. Incomplete fatty acid oxidation by ischemic heart: β-hydroxy fatty acid production. Am J Physiol Heart Circ Physiol 1980;239(2):H257–H265. [DOI] [PubMed]
- 46.Davi G, Falco A, Patrono C. Lipid peroxidation in diabetes mellitus. Antioxid Redox Signal 2005;7(1–2):256–268. [DOI] [PubMed]
- 47.Praticò D. Prostanoid and isoprostanoid pathways in atherogenesis. Atherosclerosis 2008;201(1):8–16. [DOI] [PubMed]
- 48.Bockhkov VN, Oskolkova OV, Birukov KG, Levonen AL, Binder CJ, Stöckl J. Generation and biological activities of oxidized phospholipids. Antioxid Redox Signal 2010;12(8):1009–1059. [DOI] [PMC free article] [PubMed]
- 49.Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res 2009;104(2):236–244. [DOI] [PMC free article] [PubMed]
- 50.Savale L, Tu L, Rideau D, Izziki M, Maitre B, Adnot S, Eddahibi S. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir Res 2009;10:6. [DOI] [PMC free article] [PubMed]
- 51.Chaouat A, Savale L, Chouaid C, Tu L, Sztrymf B, Canuet M, Maitre B, et al. Role for interleukin-6 in COPD-related pulmonary hypertension. Chest 2009;136(3):678–687. [DOI] [PubMed]
- 52.Ikeda Y, Yonemitsu Y, Kataoka C, Kitamoto S, Yamaoka T, Nishida KI, Takeshita A, Egashira K, Sueishi K. Anti-monocyte chemoattractant protein-1 gene therapy attenuates pulmonary hypertension in rats. Am J Physiol Heart Circ Physiol 2002;283(5):H2021–H2028. [DOI] [PubMed]
- 53.Li M, Riddle SR, Frid MG, El Kasmi KC, McKinsey TA, Sokol RJ, Strassheim D, et al. Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe pulmonary hypertension. J Immunol 2011;187(5):2711–2722. [DOI] [PMC free article] [PubMed]
- 54.Sanchez O, Marcos E, Perros F, Tu L, Humbert M, Dartevelle P, Simonneau G, Adnot S, Eddahibi S. Role of endothelium-derived CC chemokine ligand 2 in idiopathic pulmonary hypertension. Am J Respir Crit Care Med 2007;176(10):1041–1047. [DOI] [PubMed]
- 55.Cao H. Adipocytokines in obesity and metabolic disease. J Endocrinol 2014;220(2):T47–T59. [DOI] [PMC free article] [PubMed]
- 56.del Aguila LF, Claffey KP, Kirwan JP. TNF-α impairs insulin signaling and insulin stimulation of glucose uptake in C2C12 muscle cells. Am J Physiol Endocrinol Metab 1999;276(5):E849–E855. [DOI] [PubMed]
- 57.Senn JJ, Klover PJ, Nowak IA, Mooney RA. Interleukin-6 induces cellular insulin resistance in hepatocytes. Diabetes 2002;51(12):3391–3399. [DOI] [PubMed]
- 58.Summer R, Walsh K, Medoff BD. Obesity and pulmonary arterial hypertension: is adiponectin the molecular link between these condition? Pulm Circ 2011;1(4):440–447. [DOI] [PMC free article] [PubMed]
- 59.Hansmann G, Rabinovitch M. The protective role of adiponectin in pulmonary vascular disease. Am J Physiol Lung Cell Mol Physiol 2010;298(1):L1–L2. [DOI] [PubMed]
- 60.Summer R, Fiack CA, Ikeda Y, Sato K, Dwyer D, Ouchi N, Fine A, Farber HW, Walsh K. Adiponectin deficiency: a model of pulmonary hypertension associated with pulmonary vascular disease. Am J Physiol Lung Cell Mol Physiol 2009;297(3):L432–L438. [DOI] [PMC free article] [PubMed]
- 61.Nakagawa Y, Kishida K, Kihara S, Funahashi T, Shimomura I. Adiponectin ameliorates hypoxia-induced pulmonary arterial remodeling. Biochem Biophys Res Commun 2009;382(1):183–188. [DOI] [PubMed]
- 62.Weng M, Raher MJ, Leyton P, Combs TP, Scherer PE, Bloch KD, Medoff BD. Adiponectin decreases pulmonary arterial remodeling in murine models of pulmonary hypertension. Am J Respir Cell Mol Biol 2011;45(2):340–347. [DOI] [PMC free article] [PubMed]
- 63.Sutherland LN, Capozzi LC, Turchinsky NJ, Bell RC, Wright DC. Time course of high-fat diet-induced reductions in adipose tissue mitochondrial proteins: potential mechanisms and the relationship to glucose intolerance. Am J Physiol Endocrinol Metab 2008;295(5):E1076–E1083. [DOI] [PubMed]
- 64.Ribot J, Rodríguez AM, Rodríguez E, Palou A. Adiponectin and resistin response in the onset of obesity in male and female rats. Obesity 2008;16(4):723–730. [DOI] [PubMed]
- 65.Kwon EY, Shin SK, Cho YY, Jun UJ, Kim E, Park T, Park JHY, et al. Time-course microarrays reveal early activation of the immune transcriptome and adipokine dysregulation leads to fibrosis in visceral adipose tissue depots during diet-induced obesity. BMC Genomics 2012;13:450. [DOI] [PMC free article] [PubMed]
- 66.Duval C, Thissen U, Keshtkar S, Accart B, Stienstra R, Boekschoten MV, Roskams T, Kersten S, Müller M. Adipose tissue dysfunction signals progression of hepatic steatosis towards nonalcoholic steatohepatitis in C57BL/6 mice. Diabetes 2010;59(12):3181–3191. [DOI] [PMC free article] [PubMed]
- 67.Moral-Sanz J, Menendez C, Moreno L, Moreno E, Cogolludo A, Perez-Vizcaino F. Pulmonary arterial dysfunction in insulin resistant obese Zucker rats. Respir Res 2011;12:51. [DOI] [PMC free article] [PubMed]