

Abstract Abstract
The Third International Right Heart Failure Summit (Boston, MA) convened a group of international clinical and scientific experts in pulmonary vascular disease and right heart disease to explore cutting-edge developments in the mechanisms and clinical management of right-sided cardiovascular disease. The symposium was organized into three distinct sessions, the first of which was titled “Pulmonary Hypertension and the Right Ventricle—Thinking outside the Box” and will be the focus of this review. Three internationally renowned experts in pulmonary hypertension and right heart disease—Drs. Stuart Rich, Sean Gaine, and Harm Bogaard—each delivered provocative lectures. The first lecture, by Dr. Rich, was titled “Current Classification for Pulmonary Hypertension—Why Are We Ignoring the Structural Basis of the Disease?” Rich focused on the current classification system of pulmonary hypertension and provided a unique historical perspective. He also addressed the need to evolve the prevailing conceptual framework of our approach to pulmonary vascular diseases and right ventricular dysfunction, including the future design of pulmonary hypertension clinical trials. Dr. Gaine delivered the second lecture, titled “Treatment Algorithm for Pulmonary Hypertension: Tunnel Vision of our Current Approach.” Gaine emphasized the tripartite model of pulmonary hypertension management, namely, supportive measures, pharmacologic therapy, and rescue therapy. Specifically, he detailed how each of these entities is changing as our understanding of the unmet needs in the field of pulmonary hypertension is becoming increasingly apparent. Finally, Dr. Bogaard provided a lecture titled “Treating Right Heart Failure: Why Does the Art of Medicine Lead the Science?” Bogaard provided a stimulating review of cutting-edge translational research of right ventricular function and dysfunction. In particular, he described a variety of molecular and cellular changes that occur in the hypertrophied right ventricle and contrasted those changes that may be adaptive from those that are maladaptive and may be potential therapeutic targets.
Keywords: pulmonary hypertension, pulmonary vasculature, right ventricle, right heart failure
The Third International Right Heart Failure Summit in Boston, Massachusetts, convened a group of international clinical and scientific experts in pulmonary vascular disease and right heart disease to explore cutting-edge developments in the mechanisms and clinical management of right-sided cardiovascular disease. The summit built on the previous year’s summit, which among its agendas sought to define a standardized vocabulary by which to discuss right heart disease that has subsequently been published.1 The major aims of this year’s symposium were to present contemporary developments and data relevant to right ventricular (RV)–pulmonary vascular pathophysiology and to encourage a free exchange of novel and provocative ideas with regard to the various approaches to the care of patients with right heart failure syndromes. To accomplish these objectives, the summit was divided into three sections, titled (1) “Pulmonary Hypertension and the Right Ventricle—Thinking outside the Box,” (2) “Emerging Hemodynamic Signatures of the Right Heart,” and (3) “Transplantation in End-Stage Pulmonary Hypertension.” The salient scientific and clinical revelations of each section will be the feature of a review article series in Pulmonary Circulation, with section 1 being the focus of the current work.
Welcome from Drs. Mandeep Mehra and Myung Park
The Third International Right Heart Failure Summit opened with a welcoming message from Drs. Mandeep Mehra and Myung Park. Specifically, they reiterated the mission of the International Right Heart Failure Foundation, whose main objective is to increase awareness and promote interdisciplinary and innovative education, training, and research in right heart failure syndromes. They proceeded to outline three short-term strategic goals: (1) to develop strong partnerships with diverse societies and bring them together toward a common mission, (2) to support innovative educational and training curricula to develop the next generation of right heart failure specialists, and (3) to advance right heart failure by creating standardized and systematic approaches to its phenotyping and management. Following their introductory remarks, the summit began with a series of three lectures under the heading “Pulmonary Hypertension and the Right Ventricle—Thinking outside the Box.”
Current classification for pulmonary hypertension (PH)—why are we ignoring the structural basis of the disease? Dr. Stuart Rich
The keynote address of the symposium was delivered by Dr. Stuart Rich, who addressed the current classification of PH and provided a thought-provoking discourse on the need to reexamine the current state of the field and to begin to adapt our efforts to the changing landscape and our evolving understanding of PH. In 1973, the World Health Organization (WHO) convened a meeting on PH and established the original—albeit relatively simplistic—classification of PH as being either (a) of unknown etiology (primary PH) or (b) of known etiology (left heart disease, intrinsic lung disease, pulmonary emboli, etc.). A morphological classification was also provided to characterize the disease as (a) plexogenic pulmonary arteriopathy, (b) pulmonary venoocclusive disease, or (c) pulmonary thromboembolism.2 Because of the limited clinical utility of this original classification, the classification system was significantly revised in 1998 during the Second World Symposium on Pulmonary Hypertension held in Evian, France, into a new schema, the core of which remains largely unchanged to this very day.3 Specifically, this new classification system was not designed to guide treatment of the various forms of PH; rather, it was developed as a diagnostic schema to provide clinicians with a stepwise, diagnostic approach to the clinical evaluation of the patient with PH of uncertain etiology so that underlying disease(s) that might be causing or contributing to the PH could be identified. The end product was the classification of PH into five distinct groups, each containing multiple subgroups (Table 1).
Table 1.
The 1998 clinical classification of pulmonary hypertension from Evian, France
| 1. Pulmonary arterial hypertension |
|---|
| 1.1. Primary pulmonary hypertension |
| a) Sporadic |
| b) Familial |
| 1.2. Related to |
| a) Collagen vascular disease |
| b) Congenital systemic-to-pulmonary shunts |
| c) Portal hypertension |
| d) HIV infection |
| e) Drugs/toxins |
| 1) Anorexigens |
| 2) Other |
| f) Persistent pulmonary hypertension of the newborn |
| g) Other |
| 2. Pulmonary venous hypertension |
| 2.1. Left-sided atrial or ventricular heart disease |
| 2.2. Left-sided valvular heart disease |
| 2.3. Extrinsic compression of central pulmonary veins |
| a) Fibrosing mediastinitis |
| b) Adenopathy/tumors |
| 2.4. Pulmonary venoocclusive disease |
| 2.5 Other |
| 3. Pulmonary hypertension associated with disorders of the respiratory system or hypoxemia |
| 3.1. Chronic obstructive pulmonary disease |
| 3.2. Interstitial lung disease |
| 3.3. Sleep-disordered breathing |
| 3.4. Alveolar hypoventilation disorders |
| 3.5. Chronic exposure to high altitude |
| 3.6. Neonatal lung disease |
| 3.7. Alveolar-capillary dysplasia |
| 3.8. Other |
| 4. Pulmonary hypertension caused by chronic thrombotic or embolic disease |
| 4.1. Thromboembolic obstruction of proximal pulmonary arteries |
| 4.2. Obstruction of distal pulmonary arteries |
| a) Pulmonary embolism (thrombus, tumor, ova or parasites, foreign material) |
| b) In situ thrombosis |
| c) Sickle cell disease |
| 5. Pulmonary hypertension caused by disorders directly affecting the pulmonary vasculature |
| 5.1. Inflammatory |
| a) Schistosomiasis |
| b) Sarcoidosis |
| c) Other |
| 5.2. Pulmonary capillary hemangiomatosis |
Source: Adapted from Simmoneau et al.4
However, the approach to this PH diagnostic classification system began to morph into one used to dictate treatment options when the Food and Drug Administration (FDA) approved bosentan, the first available oral PH-specific therapy, for all PH entities captured by group 1 pulmonary arterial hypertension (PAH), a departure from how epoprostenol, the first-ever approved PH-specific drug, gained FDA approval for use restricted to particular PH syndromes.5-7 This pivotal decision by the FDA had the downstream consequence of creating a partition in the approach to PH treatment by in essence suggesting that group 1 PH and its subgroup entities constitute a disease distinct from the other four PH groups. This resulted in future drug development and subsequent approval of nearly all PH-specific drugs largely for group 1 PAH only, to the exclusion of all other PH syndromes. Among the unintended consequences of this paradigm shift was the implication that the mechanisms driving the development and progression of PH in group 1 PH were distinct from those in groups 2–5 PH. It also implied that the mechanistic basis of disease among the subgroups within group 1 itself (idiopathic, connective tissue disease, congenital right to left shunts, etc.) was largely the same and thus that these entities should be treated the same way. Finally, it suggested that the approved therapies are both safe and effective for the entirety of group 1 PAH, irrespective of the associated condition, and conversely it has been interpreted by clinicians that such therapies may not be safe and effective for all of the other PH syndromes.8
However, it is not clear that this conceptual framework of PH is accurate. Morphologically, studies from explanted lungs of patients with PH demonstrate that intimal proliferation with increased thickness in the pulmonary arterioles is uniform regardless of PH group. Medial hypertrophy, on the other hand, a long-standing focus of scientific research investigations and target of PH therapy, is actually only modestly increased and only adjunctively involved in the disease pathobiology (Figure 1).9 At the cellular and molecular levels, research efforts in PH have discovered that a multitude of mechanisms and complex, overlapping biologic pathways are at play in PH; that no single mechanism or pathway alone is likely sufficient to explain the disease; that certain mechanisms (i.e., inflammation) may play a more prominent role depending on the underlying disease state associated with the PH (i.e., connective tissue disease vs. idiopathic); and that little evidence currently exists to suggest that these mechanisms and pathways are unique to group 1 PAH only.10 Although survival has improved with the approval of PH-specific therapies, each of which targets a single pathway (nitric oxide, endothelin, prostaglandin, etc.), this may in part explain why survival in PH remains only modestly improved compared with that from the original National Institutes of Health (NIH) registry prior to the availability of treatments (Figure 2).11,12
Figure 1.

Histologic findings in severe pulmonary hypertension are nearly identical irrespective of World Health Organization pulmonary hypertension group. The pulmonary vasculature in lung tissue from four patients with severe pulmonary hypertension is shown. Patients were characterized clinically as category 1 (idiopathic pulmonary arterial hypertension), category 2 (left ventricular failure), category 3 (interstitial lung disease), and category 4 (chronic thromboembolic disease). However, each specimen reveals similar changes in the pulmonary arteriole, showing medial hypertrophy and intimal proliferation. Without knowing the clinical phenotype, it would not be possible to distinguish them on the basis of vascular pathology. Adapted with permission from Rich.8
Figure 2.
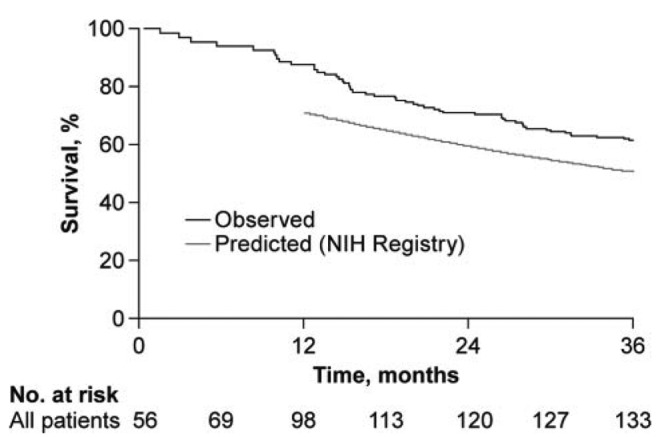
Survival in the modern pulmonary arterial hypertension (PAH) management era compared with that in the original National Institutes of Health (NIH) registry. Kaplan-Meier survival estimates in the combined population of patients with PAH (black line) are shown. When the predictive modeling approach of the NIH registry was used, the estimated survival (gray line) was ∼10% lower than what was actually observed. Adapted with permission from Humbert et al.12
From a hemodynamic perspective, with the exception of most cases of group 3 PH, the vast majority of patients with clinically significant PH, irrespective of etiology, have a mean pulmonary arterial pressure (mPAP) that approaches 50 mmHg.13 Future PH clinical trials should move away from the contemporary definition of a mPAP of >25 mmHg for trial eligibility (in the original NIH registry, not a single patient had a mPAP of <40 mmHg).11 Moreover, because mPAP lowering with drug therapy in most PH trials to date rarely exceeds 3 mmHg,13 future trial design should give consideration to focusing on more relevant and novel biomarkers, perhaps irrespective of WHO PH group, as a better way to discriminate patients with PH. In particular, secondary end points should include a variety of metrics of RV function (a reliable, strong predictor of outcomes in PH); changes in RV metabolism, energetics, and ischemia; and eventually an integration of validated discoveries using genomics and proteomics.14
Treatment algorithm for PH: tunnel vision of our current approach—Dr. Sean Gaine
The next presentation was provided by Dr. Sean Gaine, who delivered an exceptional talk about the current approach to PH treatment and the pressing need moving forward to escape from the current tunnel-vision approach. Specifically, the PH community needs to rethink the current clinical trial paradigm of short-duration trials with clinically limited end points, such as the 6-minute walk distance (6MWD); recognize the heterogeneity of PH by eliminating the dogma that “one size fits all”; and acknowledge that only modest, incremental improvements in outcomes have been realized in the current treatment era.
The current treatment algorithm for PAH can in many respects be reduced to three key components: general measures and supportive therapy, pharmacologic therapy, and ultimately rescue therapy when disease progression occurs.15 Progress has certainly been made in the field of PH by translating our understanding of three key pathways (nitric oxide, endothelin, and prostacyclin) into viable therapies that target each of these respective pathways. Yet while each of these classes of medications has been shown to modestly reduce PAPs and increase 6MWD, neither of these parameters is associated with survival, and in fact improvements in 6MWD can be misleading.16 Rather, the focus of future trials should be that they are of sufficient duration, that there is a shift in surrogate end points in the direction of indexes of RV function, and that the emphasis of funding of future drug trials is placed on drugs that target the RV, as it is RV function that ultimately determines prognosis in PH.17,18
In addition to currently available therapies and established supportive measures, such as prescriptions for oxygen, diuretics, and digoxin, there is a need to apply some of the lessons learned from left heart failure (Figure 3). Among these are evaluating how drugs such as neurohormonal antagonists (i.e., β-blockers, angiotensin-converting enzyme [ACE] inhibitors, and aldosterone antagonists) or device therapies (i.e., resynchronization therapy and intracardiac defibrillators) might be applied in PH. For instance, there is ample evidence that shows upregulation of the renin-angiotensin system in PH, and animal data and even small human studies suggest an improvement in RV-PA coupling with ACE inhibitors and/or angiotensin receptor blockade.19,20 Additionally, a recent provocative hypothesis-generating study found a synergistic benefit from combined use of spironolactone with ambrisentan in patients with PH from the ARIES (Ambrisentan for the Treatment of Pulmonary Arterial Hypertension) trials, where a post hoc analysis demonstrated that patients treated with both ambrisentan and spironolactone achieved a significantly greater 6MWD than those treated with ambrisentan alone. And there may indeed be biologic plausibility to support this clinical observation, as ambrisentan prevents excessive vasoconstriction by inhibiting endothelin type A receptors and spironolactone may promote improved pulmonary vasodilatation by inhibiting the downstream actions exerted by aldosterone to blunt endothelin receptor type B activation.21,22
Figure 3.

Potential pulmonary arterial hypertension (PAH) and right ventricular (RV) failure therapeutics currently in widespread use in the treatment of left heart failure syndromes. A schematic diagram indicating as yet unexplored pathophysiological mechanisms in PAH is shown. ETRA: endothelin receptor antagonist; PDE-5: phosphodiesterase 5; CRT: cardiac resynchronization therapy; SNS: sympathetic nervous system; RAAS: renin-angiotensin-aldosterone system; ACEI: angiotensin-converting enzyme inhibitor; ARB: angiotension receptor blockers; Aldo ant: aldosterone antagonist; MSNA: muscle sympathetic nervous activity; MIBG: metaiodobenzyl guanidine; HRV: heart rate variability; βAR: cardiomyocyte β1-adrenergic receptor; AT1R: cardiomyocyte angiotensin type 1 receptor; RHF: right heart failure; ICD: implantable cardioverter defibrillator. Adapted with permission from Handoko et al.17
Finally, when it comes to rescue therapies for PAH, the harsh reality is that when the disease progresses to late stage, lung transplantation is rarely a viable option. Of the 1,400 lung transplants performed annually worldwide, only 70 (5%) are performed in patients with PAH.23 On the other hand, atrial septostomy should not be a forgotten procedure—when performed by those experienced in this technique, it provides rather impressive improvements in functional capacity and survival. Moreover, recent improvements in imaging modalities and incorporating the use of stents across the septostomy may further enhance the safety and efficacy of this procedure.24,25 Last, the pursuit of developing innovative technologies that support the RV (and not only those that target the pulmonary vasculature) is critical if we are to take the next step in improving the prognosis of patients with PH.
Treating right heart failure: why does the art of medicine lead the science? Dr. Harm Jan Bogaard
Dr. Harm Jan Bogaard delivered the third and final presentation of the morning session with a major focus on evaluating RV function. He too echoed the common refrain of the morning session that changes in RV function but not PAPs or pulmonary vascular resistance consistently dictate outcomes in PH. Observations with both echocardiography and cardiac magnetic resonance imaging demonstrate that the RV contracts in both longitudinal and transverse planes, both of which are often impaired in PH. However, whereas tricuspid annular plane systolic excursion (TAPSE) is frequently impaired and its baseline value is indeed predictive of PH survival, the measured TAPSE, interestingly, does not change significantly over time (even in those who continue to develop progressive RV failure). On the other hand, transverse RV shortening not only correlates better with RV ejection fraction than TAPSE but changes in a more dynamic fashion, with decreases in transverse shortening often closely paralleling the progression to later stages of RV failure (Figure 4).26,27 Inefficiencies in RV contraction are also apparent in PH, as RV contraction has been shown to continue beyond the time of pulmonic valve closure. This results in septal bowing into the LV with impairment in LV filling and an expenditure of RV oxygen consumption without the benefit of further net ejection.28,29
Figure 4.

Dynamic changes in right ventricular (RV) transverse shortening and its association with pulmonary hypertension (PH) survival compared with RV longitudinal shortening. RV longitudinal and transverse shortening at baseline and follow-up are shown, indicating the average RV longitudinal (A) and transverse (B) shortening in survivors and nonsurvivors for baseline (black bars) and follow-up (white bars). A, Longitudinal shortening in nonsurvivors is significantly (P < .05, indicated by an asterisk) decreased at baseline compared with that in survivors, whereas no significant change during follow-up is seen in either group. B, Transverse shortening in nonsurvivors is significantly (P < .05, indicated by asterisks) decreased for levels 2–7 at baseline compared with that in survivors. During follow-up, RV transverse shortening for levels 2–6 further decreases significantly (P < .05, indicated by daggers) from baseline and even shows lengthening at the midventricular to apical level, whereas it did not decrease in survivors. Data are mean ± SEM. Base: baseline; fol: follow-up. Adapted with permission from Mauritz et al.27
Considerable research efforts are ongoing to better define and elucidate features associated with favorable, adaptive RV responses in PH to less favorable, maladaptive RV changes. An examination of the histologic changes of the RV in various animal models of PH provides insight into several differences between the adaptive RV compared with the maladaptive RV. In both cases, RV hypertrophy is apparent; however, whereas extensive fibrosis is a hallmark observation in the failing RV, only minimal degrees of fibrosis are typically seen in the adaptive RV.30 Emerging modalities that allow for the quantification of fibrosis may serve as a useful marker of RV status, and therapies that target adverse remodeling and collagen deposition may prove to be clinically useful. Another distinguishing feature of the healthy versus failing RV can be seen at the level of the RV microcirculation. In adaptive RV hypertrophy, there is a parallel increase in recruited vascular beds, which are arranged in an organized fashion. On the other hand, in models of a failing, maladaptive hypertrophied RV, there is evidence of both capillary rarefaction in a pattern of general disarray and striking heterogeneity of the appearance of the vasculature itself.30 Another apparent maladaptive RV response in PH is the finding that there may be a decrease in both number and function of mitochondria in RV cardiomyocytes. A downstream consequence of many of these aforementioned changes includes the observed metabolic shifts from oxidative to glycolytic metabolism.31 Taken together, alterations in the capillary network and a reduction in the number of functioning mitochondria may attenuate RV myocardial perfusion; in the setting of increased wall stress and myocardial oxygen demand in the RV, the net result is an ischemic state, a finding that has been demonstrated to occur in the failing human RV.32 In addition, similar to that seen in LV hypertrophy and failure, β-adrenergic receptor downregulation and loss of function has been described in animal models of PH, with lesser degrees of RV dilatation and fibrosis seen in those animals treated with β-blocking agents. Whether the careful use of β-blockers in the clinical setting will yield favorable long-term effects on clinical outcomes remains to be determined, although a recent study has demonstrated that the judicial use of carvedilol in PAH can be performed safely and may be associated with improvements in RV function.33
Finally, limited data on the direct actions of currently available PH-specific drugs on the RV itself exist. However, in a series of preclinical studies, Nagendran et al.34,35 demonstrated that phosphodiesterase 5 inhibitors, such as sildenafil, may act to increase cyclic guanosine monophosphate in RV cardiomyocytes, leading to an increase in RV contractility. On the other hand, endothelin receptor antagonists may have the converse result, exerting a negative inotropic effect on the hypertrophied RV, as optimal RV function may depend at least partially on endothelin stimulation of RV cardiomyocyte endothelin receptors. Preliminary animal data suggest that prostacyclins may attenuate collagen deposition and RV fibrosis.36
Conclusions
Considerable progress has been made in the field of PAH since the original WHO classification of the disease in 1973 and the establishment of the first PAH registry in 1989. Yet while overall survival has improved modestly with the availability of contemporary PAH-specific drugs, the overall prognosis remains generally poor due to progressive RV failure. Indeed, a common denominator of nearly all PAH-related epidemiologic studies is that RV function is what drives clinical outcomes. While the pulmonary vasculature itself must continue to be a target of future drug development, a major shift is (appropriately) occurring that focuses on maladaptive responses of the RV to a variety of pathologic stimuli and the development of novel therapeutic interventions aimed at either preserving RV function or reversing RV dysfunction.
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Mehra MR, Park MH, Landzberg MJ, Lala A, Waxman AB; International Right Heart Failure Foundation Scientific Working Group. Right heart failure: toward a common language. J Heart Lung Transplant 2014;33(2):123–126. [DOI] [PubMed]
- 2.Hatano S, Strasser T, eds. Primary pulmonary hypertension: report on a WHO meeting, Geneva, 15–17 October 1973. Geneva: World Health Organization, 1975.
- 3.Rich S, Rubin LJ, Abenhail L, eds. Primary pulmonary hypertension: executive summary from the World Symposium—Primary Pulmonary Hypertension 1998. Geneva: World Health Organization, 1998.
- 4.Simmoneau G, Galiè N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, Gibbs S, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004;43(12 suppl. S):5S–12S. [DOI] [PubMed]
- 5.Badesch DB, Tapson VF, McGoon MD, et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease: a randomized, controlled trial. Ann Internal Med 2000;132:425–434. [DOI] [PubMed]
- 6.Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 1996;334:296–301. [DOI] [PubMed]
- 7.Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002;346:896–903. [DOI] [PubMed]
- 8.Rich S. What is pulmonary arterial hypertension? Pulm Circ 2012;2:271–272. [DOI] [PMC free article] [PubMed]
- 9.Stacher E, Graham BB, Hunt JM, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;186:261–272. [DOI] [PMC free article] [PubMed]
- 10.Hassoun PM, Mouthon L, Barbera JA, et al. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol 2009;54:S10–S19. [DOI] [PubMed]
- 11.D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension: results from a national prospective registry. Ann Intern Med 1991;115:343–349. [DOI] [PubMed]
- 12.Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010;122:156–163. [DOI] [PubMed]
- 13.Rich S. Pulmonary hypertension. In: Bonow R, Mann D, Zipes D, Libby P, eds. Braunwald’s heart disease: a textbook of cardiovascular medicine. Philadelphia: Elsevier, 2012:1696–1718.
- 14.Dweik RA, Rounds S, Erzurum SC, et al. An official American Thoracic Society statement: pulmonary hypertension phenotypes. Am J Respir Crit Care Med 2014;189:345–355. [DOI] [PMC free article] [PubMed]
- 15.McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol 2009;53:1573–1619. [DOI] [PubMed]
- 16.Savarese G, Paolillo S, Costanzo P, et al. Do changes of 6-minute walk distance predict clinical events in patients with pulmonary arterial hypertension? a meta-analysis of 22 randomized trials. J Am Coll Cardiol 2012;60:1192–1201. [DOI] [PubMed]
- 17.Handoko ML, de Man FS, Allaart CP, Paulus WJ, Westerhof N, Vonk-Noordegraaf A. Perspectives on novel therapeutic strategies for right heart failure in pulmonary arterial hypertension: lessons from the left heart. Eur Respir Rev 2010;19:72–82. [DOI] [PMC free article] [PubMed]
- 18.van de Veerdonk MC, Dusoswa SA, Tim Marcus J, et al. The importance of trabecular hypertrophy in right ventricular adaptation to chronic pressure overload. Int J Cardiovasc Imaging 2014;30:357–365. [DOI] [PubMed]
- 19.Nootens M, Kaufmann E, Rector T, et al. Neurohormonal activation in patients with right ventricular failure from pulmonary hypertension: relation to hemodynamic variables and endothelin levels. J Am Coll Cardiol 1995;26:1581–1585. [DOI] [PubMed]
- 20.de Man FS, Handoko ML, Guignabert C, Bogaard HJ, Vonk-Noordegraaf A. Neurohormonal axis in patients with pulmonary arterial hypertension: friend or foe? Am J Respir Crit Care Med 2013;187:14–19. [DOI] [PubMed]
- 21.Maron BA, Zhang YY, White K, et al. Aldosterone inactivates the endothelin-B receptor via a cysteinyl thiol redox switch to decrease pulmonary endothelial nitric oxide levels and modulate pulmonary arterial hypertension. Circulation 2012;126:963–974. [DOI] [PMC free article] [PubMed]
- 22.Maron BA, Waxman AB, Opotowsky AR, et al. Effectiveness of spironolactone plus ambrisentan for treatment of pulmonary arterial hypertension (from the [ARIES] study 1 and 2 trials). Am J Cardiol 2013;112:720–725. [DOI] [PMC free article] [PubMed]
- 23.Levy RD, Estenne M, Weder W, Cosio MG. Lung transplantation: beyond palliation. Eur Respir J 2003;22:721–722. [DOI] [PubMed]
- 24.Roy AK, Gaine SP, Walsh KP. Percutaneous atrial septostomy with modified butterfly stent and intracardiac echocardiographic guidance in a patient with syncope and refractory pulmonary arterial hypertension. Heart Lung Circ 2013;22:668–671. [DOI] [PubMed]
- 25.Sandoval J, Gaspar J, Pena H, et al. Effect of atrial septostomy on the survival of patients with severe pulmonary arterial hypertension. Eur Respir J 2011;38:1343–1348. [DOI] [PubMed]
- 26.Kind T, Mauritz GJ, Marcus JT, van de Veerdonk M, Westerhof N, Vonk-Noordegraaf A. Right ventricular ejection fraction is better reflected by transverse rather than longitudinal wall motion in pulmonary hypertension. J Cardiovasc Magn Reson 2010;12:35. [DOI] [PMC free article] [PubMed]
- 27.Mauritz GJ, Kind T, Marcus JT, et al. Progressive changes in right ventricular geometric shortening and long-term survival in pulmonary arterial hypertension. Chest 2012;141:935–943. [DOI] [PubMed]
- 28.Marcus JT, Gan CT, Zwanenburg JJ, et al. Interventricular mechanical asynchrony in pulmonary arterial hypertension: left-to-right delay in peak shortening is related to right ventricular overload and left ventricular underfilling. J Am Coll Cardiol 2008;51:750–757. [DOI] [PubMed]
- 29.Wong YY, Ruiter G, Lubberink M, et al. Right ventricular failure in idiopathic pulmonary arterial hypertension is associated with inefficient myocardial oxygen utilization. Circ Heart Fail 2011;4:700–706. [DOI] [PubMed]
- 30.Bogaard HJ, Natarajan R, Henderson SC, et al. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 2009;120:1951–1960. [DOI] [PubMed]
- 31.Gomez-Arroyo J, Mizuno S, Szczepanek K, et al. Metabolic gene remodeling and mitochondrial dysfunction in failing right ventricular hypertrophy secondary to pulmonary arterial hypertension. Circ Heart Fail 2013;6:136–144. [DOI] [PMC free article] [PubMed]
- 32.Gomez A, Bialostozky D, Zajarias A, et al. Right ventricular ischemia in patients with primary pulmonary hypertension. J Am Coll Cardiol 2001;38:1137–1142. [DOI] [PubMed]
- 33.Grinnan D, Bogaard HJ, Grizzard J, et al. Treatment of group I pulmonary arterial hypertension with carvedilol is safe. Am J Respir Crit Care Med 2014;189:1562–1564. [DOI] [PubMed]
- 34.Nagendran J, Archer SL, Soliman D, et al. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation 2007;116:238–248. [DOI] [PubMed]
- 35.Nagendran J, Sutendra G, Paterson I, et al. Endothelin axis is upregulated in human and rat right ventricular hypertrophy. Circ Res 2013;112:347–354. [DOI] [PubMed]
- 36.Gomez-Arroyo J, Sakagami M, Syed AA, et al. Iloprost reverses established fibrosis in experimental right ventricular failure. Eur Respir J. doi:10.1183/09031936.00188013. Electronically published September 26, 2014. [DOI] [PubMed]