

Abstract
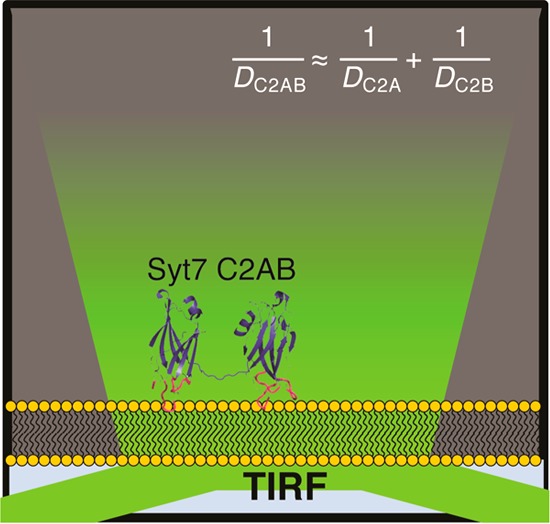
The synaptotagmin (Syt) family of proteins contains tandem C2 domains, C2A and C2B, which bind membranes in the presence of Ca2+ to trigger vesicle fusion during exocytosis. Despite recent progress, the role and extent of interdomain interactions between C2A and C2B in membrane binding remain unclear. To test whether the two domains interact on a planar lipid bilayer (i.e., experience thermodynamic interdomain contacts), diffusion of fluorescent-tagged C2A, C2B, and C2AB domains from human Syt7 was measured using total internal reflection fluorescence microscopy with single-particle tracking. The C2AB tandem exhibits a lateral diffusion constant approximately half the value of the isolated single domains and does not change when additional residues are engineered into the C2A–C2B linker. This is the expected result if C2A and C2B are separated when membrane-bound; theory predicts that C2AB diffusion would be faster if the two domains were close enough together to have interdomain contact. Stopped-flow measurements of membrane dissociation kinetics further support an absence of interdomain interactions, as dissociation kinetics of the C2AB tandem remain unchanged when rigid or flexible linker extensions are included. Together, the results suggest that the two C2 domains of Syt7 bind independently to planar membranes, in contrast to reported interdomain cooperativity in Syt1.
The mammalian synaptotagmin (Syt) family of proteins consists of 17 isoforms, 8 of which are known to bind membranes in the presence of Ca2+ and trigger membrane fusion events.1−4 These proteins function as Ca2+ sensors for SNARE-dependent membrane fusion, with differing Ca2+ sensitivities due to different membrane affinities and kinetics tuned to the Ca2+ concentrations of their respective fusion events.4 For example, Syt1 binds to and releases from the membrane rapidly and is involved in fast synchronous neurotransmitter release, whereas Syt7 has biophysical properties tuned to its roles in slow asynchronous neurotransmitter release and neuroendocrine secretion.5−8
Syt consists of a transmembrane helix near the N-terminus, a flexible region of variable length, and two C2 domains joined by a short linker (Figure 1).9 Each C2 domain contains 8 antiparallel β-strands arranged in a compact β-barrel; in Ca2+-binding Syt isoforms, the C2A domain binds three Ca2+ ions and the C2B domain binds two (Syt1) or three (Syt7).3,10,11 The transmembrane helix anchors the protein in the membrane; Syt1 is anchored in secretory vesicle membranes, whereas Syt7 is found on either the plasma membrane or secretory vesicles, depending on cell type.12 The two C2 domains trigger membrane fusion upon binding Ca2+ and associating peripherally with vesicular and/or plasma membranes.5 Although the length between the transmembrane helix and the C2A domain is highly variable among the isoforms, the linker length between the C2A and C2B domains is conserved as 7 or 8 residues.1,4 Recently, it was shown that the insertion of long polyproline extensions to the C2A–C2B linker alters the kinetics of Syt1-triggered neurotransmitter release, suggesting that the relative orientation and/or interactions between the two domains are important for physiological function.13
Figure 1.
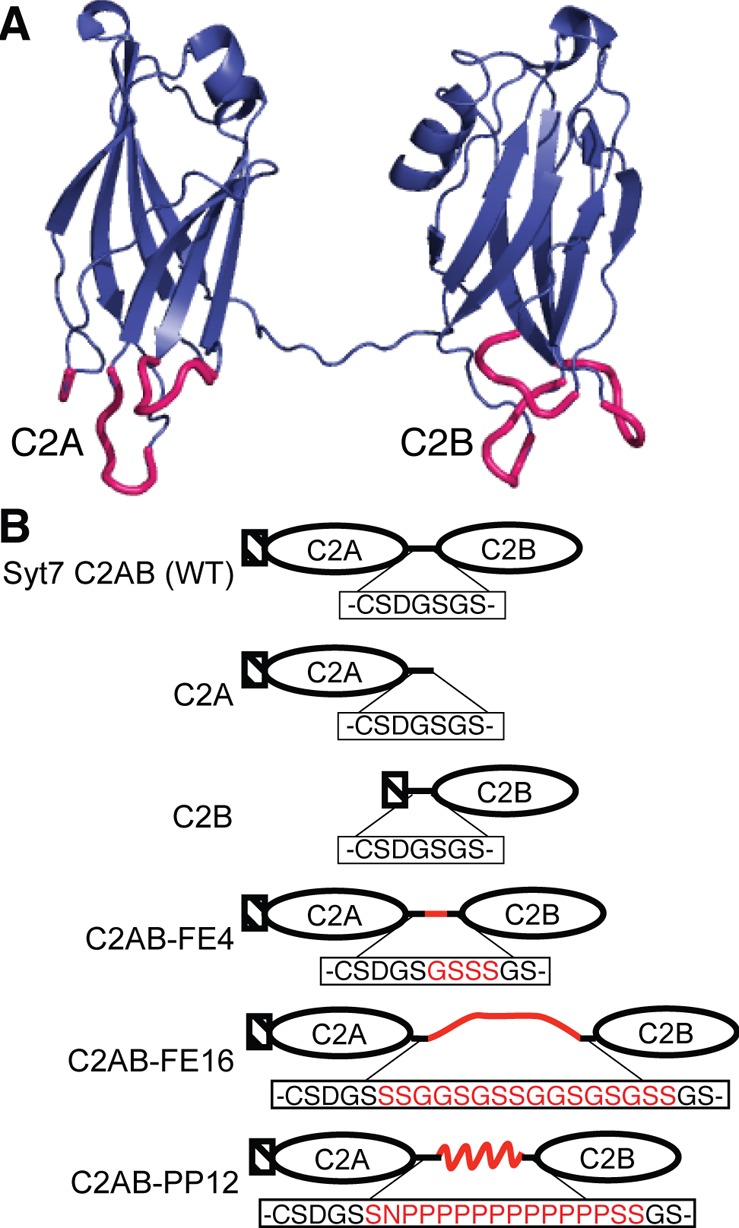
Protein constructs used in this study. (A) Composite of published structures from Syt7 C2A (PDB ID: 2D8K) and Syt7 C2B (PDB ID: 3N5A)11 joined by the native 7-residue linker sequence. Ca2+- and membrane-binding loops are shown in pink. The domain orientation shown here represents a relatively extended linker conformation and is intended for illustration purposes only. (B) Schematic of constructs used in this study, with C2A-C2B linker sequences indicated below each cartoon. The native linker sequence is shown in black, and engineered insertions are in red. Hashed boxes represent the 11-amino acid sequence DSLEFIASKLA, used for enzymatic labeling with fluorophore at the underlined serine.30
Both Syt C2 domains bind to membranes containing anionic lipids in the presence of sufficient Ca2+ and contribute to tight Syt membrane binding.14−16 Syt C2AB tandem domains have the ability to dock either to the same membrane or to opposing membranes, depending on the experimental system.17−19 Brunger and colleagues have shown that within the Syt1 C2AB tandem the C2 domains orient randomly relative to each other in solution.20 Upon interacting with SNARE complexes, C2AB adopts a more restricted ensemble of relative C2 domain orientations, which is hypothesized to facilitate simultaneous binding of the two domains to the plasma membrane.20
Two sets of observations suggest that the two C2 domains of Syt1 co-penetrate target membranes cooperatively, suggesting possible interactions between C2A and C2B during membrane binding.21,22 First, EPR depth measurements show that both Syt1 C2A and C2B penetrate membranes more deeply in the context of the C2AB tandem than they do as isolated individual domains.21 Second, fluorescence measurements similarly demonstrate a deeper membrane penetration of Syt1 C2B in the context of the C2AB tandem relative to the isolated domain, even when the C2AB tandem is mutated so that C2A cannot bind Ca2+.22 These observations suggest that C2AB membrane binding may involve some sort of favorable thermodynamic interaction between the two domains, which would allow the C2AB tandem to overcome barriers to deep membrane insertion experienced by individual domains. However, no specific interdomain interactions have been reported.
The purpose of this study is to gain insight into the membrane-bound structure of Syt C2AB using a recently developed analytical approach based on diffusion of peripheral membrane proteins and complexes. The diffusion of a peripheral protein is determined by the friction of the portion of protein embedded in the membrane and not on the portion that is moving through solution, as the viscosity of aqueous solutions is much lower than that of the membrane bilayer.23−25 The diffusion constant D is related to a frictional coefficient f by
| 1 |
where kB is Boltzmann’s constant and T is temperature.26
We and others have shown that the diffusion of multiple linked, membrane-bound protein domains can be explained by simple additive friction among the domains.25,27,28 For a C2AB tandem, the predicted diffusion constant (Dpred) from this model can be formulated as follows
| 2 |
where DA and DB are the diffusion constants of C2A and C2B, respectively. This approximation is valid when the two domains are separated by a minimum distance known as the free draining limit. Tandem domains are predicted to diffuse faster if the domains are separated by shorter distances.29 Here, we measure such diffusion constants using single-molecule fluorescence tracking in order to test for additive friction between Syt7 C2A and C2B in the membrane-bound state. In addition, we use stopped-flow fluorescence kinetic measurements in an alternative approach to test for cooperativity and interdomain interaction. Both approaches suggest independent membrane binding by the two domains without significant interdomain contact.
Experimental Methods
Materials
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phospho-l-serine (sodium salt) (DOPS), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) (LRB-DOPE) were from Avanti Polar Lipids (Alabaster, AL); Alexa Fluor 488 C2-maleimide and N-[5-(dimethylamino)naphthalene-1-sulfonyl]-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (dansyl-PE) were from Life Technologies. 2-Mercaptoethanol (BME) was from Fisher. All reagents were ACS grade or higher.
DNA Constructs
The C2A, C2B, and C2AB domains from human Syt7 were subcloned from cDNA (IMAGE: 11045721) into a previously described glutathione S-transferase (GST) expression plasmid with N-terminal enzymatic labeling recognition sequence.27,30 Site-directed mutagenesis (QuikChange II XL, Agilent) was used to insert a unique XhoI restriction site within the C2A–C2B linker, in a frame chosen to encode two serine residues, using the following primers: fwd, CAGCGATGGGAGCTCGAGTGGGAGCCGAG; rev, CTCGGCTCCCACTCGAGCTCCCATCGCTG. In order to generate the Syt7C2AB-FE4 plasmid, a second round of site-directed mutagenesis was performed with the following primers: fwd, CAGCGATGGGAGCGGGAGCTCGAGTGGGAG; rev, CTCCCACTCGAGCTCCCGCTCCCATCGCTG. In order to generate the Syt7C2AB-FE16 and Syt2C2AB-PP12 plasmids, synthetic duplex oligonucleotides were ligated into the XhoI-digested plasmid described above. (FE16: fwd, TCGAGTGGTGGCAGCGGTAGTTCCGGAGGCTCAGGTAGCGGC; rev, TCGAGCCGCTACCTGAGCCTCCGGAACTACCGCTGCCACCAC; PP12: fwd, TCGAACCCGCCTCCGCCGCCGCCGCCACCCCCACCTCCACCC; rev, TCGAGGGTGGAGGTGGGGGTGGCGGCGGCGGCGGAGGCGGGT; IDT, Coralville, IA) Correct insertion was verified via primer extension sequencing. Plasmid DNA encoding Sfp phosphopantetheinyl transferase was a gift from Christopher Walsh (Harvard), and this protein was expressed and purified as described.31
Protein Expression and Purification
Plasmids were transformed into Escherichia coli BL21 cells for protein expression. Bacterial suspensions were cultured at 37 °C until the absorbance at 600 nm reached approximately 0.6, and then expression was induced by addition of 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and allowed to proceed for 8 h at room temperature. Following lysis, the proteins were purified using glutathione sepharose affinity columns essentially as previously described27,32 and were eluted into labeling buffer (10 mM MgCl2, 50 mM l-glutamic acid, 50 mM l-arginine, 20 mM BME, 50 mM HEPES, pH 7.5) following cleavage of the N-terminal GST tag with thrombin, leaving GST on the column. Due to the large positive net charge on Syt proteins, nucleic acid contaminants have been observed in bacterial preparations;33 therefore, protein purification procedures incorporated nuclease digestion and high-salt washes to minimize such contamination.32 The absence of nucleic acid in purified proteins was quantified using absorbance measurements on a PerkinElmer λ-650 spectrophotometer, and all purified proteins had an A260/A280 ratio of 0.65 or lower (Supporting Information, Figure S1).
After initial purification, the proteins were enzymatically labeled with Alexa Fluor 488 (AF488) using Sfp phosphopantetheinyl transferase as described previously.27,31 The purity of the labeled protein was determined using SDS-PAGE with fluorescent imaging, and the C2AB tandem domain preparations were found to contain detectable amounts of single C2 domain, presumably from hydrolysis at the interdomain linker. To remove these contaminants and any residual thrombin, the C2AB tandem domains were further purified over a Superdex G75 10/300 GL gel filtration column (GE Healthcare, Uppsala, Sweden) in buffer A (150 mM KCl, 15 mM NaCl, 0.5 mM MgCl2, 20 mM HEPES, pH 7.4) with 500 mM NaCl added, and purity was determined using SDS-PAGE (Figure 2). The purified proteins were flash-frozen in buffer A, with 8% glycerol and 20 mM BME added to tandem domain preparations to aid in protein stability. Diffusion data for individual protein domains are from a single purified batch of protein. Each tandem domain was purified and labeled twice, with indistinguishable results between the two batches.
Figure 2.
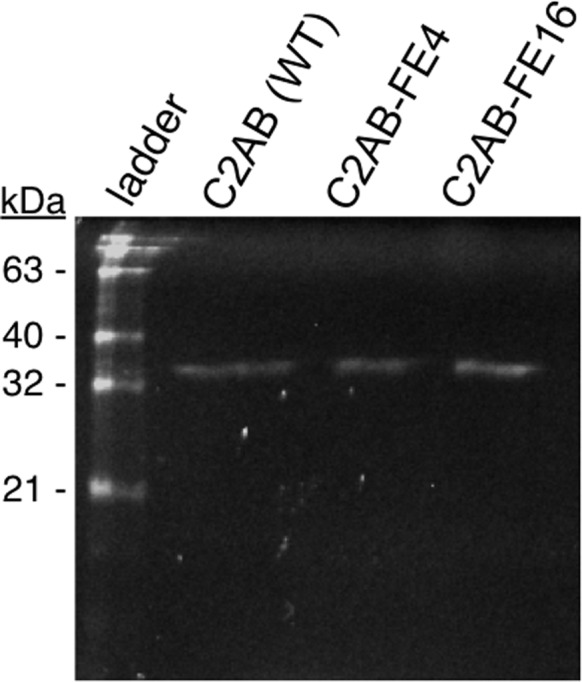
Purity of tandem domain species for TIRFM experiments. AF488-labeled Syt7 C2AB domain variants (expected mass: 34–35 kDa) were purified using gel filtration chromatography in order to remove cleavage products and analyzed using SDS-PAGE. The gel includes the BenchMark fluorescent protein ladder (Life Technologies) and was imaged under ultraviolet excitation.
Lipid Preparation
Synthetic phospholipids were combined in chloroform at the desired molar ratio. After evaporation of the chloroform, the lipid films were dried under vacuum for ≥2 h, reconstituted in buffer A containing 20 mM BME to a concentration of 3 mM total lipid, and sonicated on ice 4 min (intervals of 1 s separated by 2 s pauses) using a Sonics Vibra-Cell probe sonicator to yield small unilamellar vesicles (SUVs).24 Diffusion data are averages among measurements performed over 10 days of experiments, with a fresh batch of liposomes prepared for each.
Total Internal Reflection Fluorescence Microscopy
Glass coverslips (25 mm × 25 mm, no. 1.5, VWR) were rinsed in freshly drawn Milli-Q water, soaked in 3:1 H2SO4 to 30% aqueous H2O2 for 1 h to clean and etch the glass surface, rinsed extensively in Milli-Q water, and dried under a stream of N2 gas. Perfusion chambers (Life Technologies, 60 μL volume) were rinsed in Milli-Q water, bath-sonicated at room temperature for 1 h in a 20% ethanol/water solution, dried under a stream of N2, and adhered to the coverslips. SUVs containing 3:1 mixture of DOPC/DOPS with approximately 1 to 5 ppb LRB-DOPE were diluted 1:1 into buffer A containing 1 M NaCl and introduced to the perfusion chambers to form supported lipid bilayers (SLBs). After 30 min, SLBs were rinsed with ∼20 mL Milli-Q water and exchanged into buffer A containing 200 μM CaCl2.24 In a subset of experiments, decreasing the Ca2+ concentration to 10 μM was found not to influence diffusion constants, whereas addition of 2 mM EDTA removed nearly all protein from the membrane (Figure S2).
Samples were imaged using an Axio Observer total internal reflection fluorescence microscope (TIRFM) with a 100×, 1.46 NA objective (Zeiss, Germany), with illumination from a 50 mW 488 nm laser and 20 mW 552 nm laser (Intelligent Imaging Innovations, Denver, CO). First, movies of the LRB-DOPE were imaged at 552 nm to verify bilayer fluidity and at 488 nm to verify negligible levels of fluorescent contaminants. AF488-tagged Syt7C2A, Syt7C2B, Syt7C2AB WT, Syt7C2AB-FE4, or Syt7C2AB-FE16 was then added to the bilayers at 10–100 pM, allowed to equilibrate to room temperature (20–24 °C) for 5 min, and imaged with 488 nm excitation for single-particle tracking. All movies were acquired with an Evolve 512 × 512 EMCCD camera (Photometrics) for 500 frames at 19.8 frames/s (exposure time of 50.0 ms, with a frame transfer time of 0.4 ms). For each sample, three movies of LRB-DOPE and five movies of AF488-labeled protein were acquired on different regions of the bilayer.
Single-Particle Tracking and Diffusion Analysis
The LRB-DOPE movies were exported from Slidebook 5.5 as TIFF stacks and imported into ImageJ for tracking analysis using the Particle Tracker plugin as described.24 The AF488–protein images were tracked in Slidebook 5.5 using a Laplacian transformation to enhance contrast and a mask to filter background noise and keep only fluorescent objects that persisted for at least five frames. Centroid positions of particles were defined on the basis of the intensity distributions of the original untransformed images.
All trajectory data were exported as text files for subsequent analysis in Mathematica (Wolfram Research). Each text file was filtered to exclude trajectories fewer than 5 frames in length. Except where otherwise noted, steps from all trajectories in each movie were pooled, and statistical analysis was performed on the combined pool of steps. Spurious detections were removed with filters to identify unusually bright or immobile particles, likely from contaminants or rapidly dissociating particles.
Subsequently, the bin was analyzed using methods developed by Schütz et al. for one and two components.24,34 For each movie, the cumulative probability distribution 1 – P(r2, Δt) of the square displacement r2 or greater over a given time interval Δt was calculated for Δt values from three to eight frames, using r2 values of 0.2 μm2 or larger in order to exclude immobile contaminants. Single-component fits are given by eq 3
| 3 |
where m is the mobile fraction and ⟨r2⟩ is the mean-square displacement (MSD). For tandem domains, data were also fit to a two-component model
| 4 |
where α is the fraction of particles in the slow-diffusing state, ⟨r12⟩ is the MSD of the slow-diffusing state, and ⟨r2⟩ was constrained to the expected MSD for a fast-diffusing state with a D of 1.8 μm2/s, corresponding to D for a single C2 domain (Table 1).
Table 1. Diffusion Parameters of Syt7 C2 Species.
| species | D (μm2/s)a | major diffusing population (%)a,b | fast-diffusing population (%)a,b | n |
|---|---|---|---|---|
| C2A | 1.82 ± 0.12 | 10 | ||
| C2B | 1.82 ± 0.08 | 14 | ||
| C2AB-WT | 0.99 ± 0.06 | 83 ± 5 | 15 ± 4 | 8 |
| C2AB-FE4 | 1.03 ± 0.07 | 87 ± 6 | 11 ± 6 | 9 |
| C2AB-FE16 | 1.00 ± 0.07 | 93 ± 2 | 5 ± 2 | 8 |
| Dpred | 0.91 ± 0.08c |
Mean ± 95% CI among n independent samples (supported bilayers) are reported.
From two-component fits to C2AB tandem domain species. D from the major component is reported here. Populations total less than 100% due to the presence of a small population (∼2%) of immobile particles.
Dpred was calculated using eq 2 and propagating error from DC2A and DC2B.
The MSD is linearly related to Δt as described by eq 5
| 5 |
where D is the measured two-dimensional diffusion constant and till is the illumination time (50 ms).35 Finally, the trajectory-by-trajectory distribution of diffusion constants was analyzed for selected movies by fitting the probability density to a Lorentzian distribution or the sum of two Lorentzian distributions (eq 6 or 7)
| 6 |
| 7 |
where D1 and D2 are the characteristic diffusion constants, ϒi are scale parameters of the Lorentzian distributions, and αi are normalization and scaling factors for the components.
Statistical Analysis
Statistical analysis was performed in Mathematica. First, in order to determine whether the variability between bilayers was statistically significant, one-way ANOVA was performed comparing diffusion constants of Syt7C2AB-WT from eight different supported bilayers acquired on three different days, on which five movies were acquired per bilayer sample. The variability between bilayers was statistically significant compared to the variability between movies from the same bilayer (P < 0.001). Therefore, for all subsequent analysis, the diffusion constants calculated from the five movies on each bilayer were averaged and recorded as the diffusion constant from that sample.
Stopped-Flow Spectroscopy
Stopped-flow fluorescence kinetic measurements were performed on a BioLogic SFM-3000 (Knoxville, TN) using 284 nm excitation and a 455 nm long-pass emission filter. C2 domains (0.5 μM) bound to SUVs containing 3:1 DOPC/DOPS and 5% dansyl-PE (100 μM total accessible lipid) in the presence of 200 μM CaCl2 were rapidly mixed with a solution containing 1 mM EDTA (all concentrations listed are after mixing). The observed kinetics correspond to a loss of protein-to-membrane FRET upon membrane dissociation as described.36 Data were fit using Mathematica to either a single-exponential decay (eq 8) or a two-step model (eq 9)
| 8 |
| 9 |
where koff is a dissociation rate constant, k1 and k2 are rate constants of two sequential steps in which a fluorescence change is observed only after both steps occur, ΔFmax is the amplitude of the fluorescence change, and C is an offset.
Results
Protein Purification and Fluorescent Labeling
Individual and tandem domains from Syt7 were purified and tagged with fluorophore, and their lateral diffusion constants were measured on supported lipid bilayers composed of a 3:1 mixture of DOPC and DOPS, a simplified approximation of the anionic lipid content of the plasma and/or secretory vesicle membranes to which Syt C2 domains bind.37−39 In order to accurately predict tandem domain diffusion constants using eq 2, it is necessary to precisely measure diffusion constants for the individual species. Syt7 was chosen for this study, rather than the better-studied isoform Syt1, because the membrane-bound lifetime of the Syt1 C2A domain was too short for accurate diffusion measurements on these membranes (Supporting Information, Movie S1). However, both individual and tandem C2 domains from Syt7 remained visible for tracking for up to several seconds.
Each C2 domain or C2AB tandem was expressed as a GST fusion, purified using glutathione affinity chromatography, and eluted following cleavage of the N-terminal GST tag with thrombin. The purified proteins were verified to be free of contaminating nucleic acids, which have been found to alter membrane-binding properties of synaptotagmins.33 Each protein species contained an 11-residue N-terminal recognition sequence for Sfp phosphopantheinyltransferase, which was used for enzymatic fluorescent labeling with AF488.27,30 The purification and labeling procedure produced tandem domains free of detectable fluorescent protein contaminants using SDS-PAGE with fluorescence imaging (Figure 2). On the basis of the dilution factors necessary to achieve an adequate density of fluorescent spots in the single-molecule experiments, the concentrations of purified fluorescent tandem domains are estimated to be ∼100 nM. A significant advantage of single-molecule detection is the ability to work with picomolar concentrations of multidomain proteins.
Lipid Diffusion as an Internal Standard for Bilayer Fluidity
In order to ensure uniformity in bilayer fluidity among protein diffusion samples, lissamine rhodamine-DOPE (LRB-DOPE) was used as a qualitative internal standard in each supported bilayer prepared. Inclusion of ∼5 ppb LRB-DOPE in the bilayers provided enough fluorescent lipid for qualitative measurements of lipid diffusion without inducing significant spectral bleed-through to the AF488 channel used for protein diffusion measurements. This is a much lower particle density than the ∼150 ppb we have used for lipid diffusion measurements in previous reports and therefore the diffusion constants so measured are inherently less precise than those from our previous reports and our current protein diffusion measurements. Nevertheless, the lipid diffusion constants generally ranged from 2.2 to 3.0 μm2/s, consistent with fluid bilayers as reported previously.24,27 Rare samples with lipid diffusion constants outside this range were discarded. We also note a second population of LRB-DOPE present in all coverslips that visibly diffuses extremely slowly and comprises approximately half of the total fluorescent lipid (Movie S2). This population is likely LRB-DOPE in the lower leaflet of the bilayer, next to the glass support. Its slow diffusion likely does not reflect the diffusion rate of DOPC and DOPS in the lower leaflet, but rather stems from its bulky and ionic fluorescent headgroup bridging with or near the glass surface. This slow-diffusing population was not observed in our previous studies, likely due to an experimental protocol that included prebleaching of slow and immobile populations prior to data acquisition.24,29
As a test for diffusion of the lower leaflet, we performed simple FRAP measurements using tail-labeled NBD-PC as the fluorophore (Figure S3). These revealed complete recovery with a diffusion constant of approximately 0.6 ± 0.2 μm2/s, 4–5-fold slower than LRB-DOPE in the upper leaflet but >100-fold faster than the slow population of LRB-DOPE. Thus, the slow population of LRB-DOPE is not representative of bulk lipids in the lower leaflet.
Diffusion of Syt7 C2A, C2B, and C2AB Domains
Following imaging of lipid diffusion, fluorescent-tagged protein (10–100 pM) was added to the bilayer. Because Syt7 C2 domains bind with high affinity and slow off-rate to the 3:1 PC/PS bilayer,2,32,40 each protein appeared as a field of mobile fluorescent spots with very little background contamination (Figure 3 and Movies S3–S8). Imaging parameters were chosen to minimize background fluorescence and bleed-through of LRB-DOPE into the 488 channel (Figure 3, bottom right panel). Rarely, samples with a large population of immobile particles or poor binding of fluorescent protein to the bilayer were observed. These defects indicate incomplete bilayer formation; therefore, such samples were discarded without analysis. Protein–membrane binding was Ca2+-dependent, as addition of 2 mM EDTA removed nearly all fluorescent particles from the surface (Figure S2).
Figure 3.

Images of protein diffusion. All images are taken from movies acquired with 50 ms exposure and illustrate random, uniform distributions of particles with negligible background contamination. The bottom right panel shows the same bilayer as that in the bottom left panel but prior to the addition of protein. Scale bars: 10 μm. Representative movies are available in the Supporting Information.
In order to quantify accurately and consistently the diffusion constant of each protein species, at least eight separate replicates acquired on two or more days of experiments were analyzed for each protein species. Four or five movies of AF488–protein diffusion were acquired from each sample. Most movies produced 500 to 2000 particle trajectories comprising a total of 105 or more steps, which were pooled and analyzed. For C2A and C2B, diffusion was well-described by fitting the squared displacement data to a single diffusing population, whereas the C2AB tandem domains were better fit to a combination of two populations (Figure 4). The faster-diffusing population was lower in abundance, consisting of 3–20% of the total steps (Table 1). Its abundance varied between C2AB species, with the wild-type tandem typically containing the largest fast-diffusing population, although the abundance also varied between days of experiments. The population of the faster-diffusing state was too low to accurately measure its diffusion constant; attempted fitting of data led to wide variability in best-fit parameters when the faster diffusion constant was allowed to float. However, consistent quality fits were obtained when the diffusion constant of this state was fixed at 1.8 μm2/s, on the assumption that the population corresponds to molecules with only one C2 domain bound to the membrane (see Experimental Methods).
Figure 4.

Representative fits of squared-displacement data to cumulative density functions. Fits are shown to a single-component model (eq 3, dashed lines) and a two-component model in which the faster-diffusing component was constrained to that of a single C2 domain (eq 4, solid lines). For each movie, such fitting was performed for Δt values from 3 (0.15 s) to 8 frames (0.4 s). Individual fits were improved slightly at the longest step sizes for C2AB-WT and C2AB-FE4 when the diffusion constant of the faster-diffusing component was unconstrained; however, the value of diffusion constants obtained in this manner was inconsistent among different Δt values from the same movie, which resulted in large uncertainties in the other fit parameters due to covariation. Thus, to improve consistency, the value of 1.8 μm2/s was chosen as D for the fast-diffusing species.
Analysis of mean-squared displacement of the major population as a function of lag time showed linear relationships, indicating free diffusion without barriers for all species (Figure 5). From the slope of these fits, diffusion constants were calculated for each movie. ANOVA analysis indicated that the sample-to-sample variability exceeded the movie-to-movie variability within each sample, suggesting that small differences in mobility exist between different supported bilayers.29,41 Therefore, the diffusion constant of each sample was calculated as the average D value from all 4 to 5 movies acquired on the same supported bilayer. The D values for each protein species were then averaged from among the sample diffusion coefficients and are reported in Table 1. In a control experiment, versions of Syt7 C2A labeled with three different fluorophores with different sized aromatic ring structures (AF488, AF555, and AF647) showed identical diffusion constants, within measurement error, when imaged on the same bilayer (Table S1). This indicates that the fluorophore has negligible impact on the diffusion constant. AF488 was chosen for the continued work because of its photostability relative to Alexa Fluor 647 and because the 552 nm channel was used for the control measurements of LRB-DOPE.
Figure 5.

MSD plots from representative movies of Syt7 C2A (open circles), C2B (filled circles), and C2AB-WT (open squares). Plots of other C2AB species overlay closely with C2AB-WT and are omitted for clarity. Solid lines show linear fits to eq 5, indicating diffusion without barriers on this time scale. Error bars were calculated as described.54 Where not visible, error bars on individual points are smaller than the symbols.
The Syt7 C2A and C2B domains have identical (within error) diffusion constants of 1.8 μm2/s, comparable to those of other C2 domains, whereas the C2AB tandems exhibited significantly slower diffusion of 1.0 μm2/s (Figure 5 and Table 1). This is the expected result for a scenario in which both C2 domains experience significant membrane contact.25,27,28 Indeed, the measured diffusion constant of the wild-type C2AB tandem is 54% of the individual domains’ diffusion constants and is within error of the diffusion constant predicted using eq 2 (Dpred, Table 1). Increasing the length of the C2A–C2B linker had no effect on diffusion, within experimental error (Table 1). Together, these results are consistent with the two C2 domains docked independently and simultaneously to the supported lipid bilayer.
As a qualitative probe of the lifetime of the fast-diffusing state, the diffusion constant of each trajectory was estimated, and the probability distribution was fitted to the sum of two Lorentzian distributions. This fitting approach describes a mixture of two long-lived states whose average lifetime is significantly longer than the average trajectory length.28 Indeed, the data fit well to the sum of two distributions with maxima near 1.0 and 1.8 μm2/s (Figure 6). This clear separation indicates that the observed heterogeneity in diffusion states does not arise from reversible transitions within individual trajectories. Rather, the fast-diffusing state is long-lived and most likely arises from a subpopulation of molecules with only one functional C2 domain.
Figure 6.

Bimodal distribution of diffusion constants among trajectories. Diffusion constants were estimated from the mean-square displacement of each trajectory using a Δt value of one frame. Distributions were fit to either a single Lorentzian (eq 6, dashed line) or the sum of two Lorentzian distributions (eq 7, solid line), omitting the small immobile population. Distributions of diffusion constants have been empirically found to fit well to Lorentzian functions.28 The distribution shown encompasses 806 trajectories from one movie of C2AB-WT and is representative of distributions analyzed similarly from three movies of this species.
In an attempt to directly test for interactions between C2A and C2B, diffusion of the isolated fluorescent-tagged C2B domain was measured in the presence of approximately equimolar unlabeled Syt7 C2A (∼100 pM total concentrations of each protein in the sample chamber). The unlabeled C2A domain did not slow diffusion of the C2B domain (Table S2), as would have been expected upon heterodimer formation.28 Thus, C2A–C2B heterodimers are not observed at these concentrations.
EDTA-Induced Dissociation of Single and Tandem Domains
Additive friction could be consistent either with independent membrane associations of the two domains or with a combination of both interdomain interaction (which would speed diffusion) and insertion a few angstroms deeper into the upper membrane leaflet (which would slow diffusion).25 Therefore, as an alternative approach to test for interdomain cooperativity, we measured membrane dissociation kinetics of Syt7 C2A, C2B, and C2AB domains induced by the rapid addition of the Ca2+ chelator EDTA. Addition of EDTA to preformed C2 domain–membrane complexes induces dissociation and prevents rebinding as each C2 domain loses its bound Ca2+ ions irreversibly.42
Syt7 C2A and C2B dissociation profiles from 3:1 PC/PS membranes were single-exponential, as observed on the basis of FRET between intrinsic Trp residues and dansyl-PE included in the liposomes (Figure 7A). C2AB dissociation proceeded on approximately the same overall time scale as that of the individual domains, but it did so with a kinetic profile that was not well fit by a single-exponential decay. Rather, these data included a lag phase and were better modeled as a two-step process, in which only the second step produces a FRET change. This profile is reasonable, as whichever domain dissociates first would remain in close enough proximity to the membrane for Trp-to-dansyl FRET to occur (R0 ≈ 21–24 Å) until the other domain dissociates.43 Alternatively, each domain may first transition to a weakly membrane-associated state, from which dissociation proceeds rapidly in the absence of Ca2+ (Figure 8; see Discussion).
Figure 7.

EDTA-induced membrane dissociation of individual and tandem Syt7 C2 domains. (A) Syt7 C2A and C2B dissociation profiles from 3:1 PC/PS membranes. (B) Dissociation kinetics for the FE16 linker extension variant of Syt7 C2AB. (C) Dissociation kinetics for a variant of the C2AB tandem (PP12) with a rigid polyproline sequence inserted into the C2A–C2B linker region that prohibits interdomain interactions. Dashed lines indicate single-exponential fits, and solid lines mark fits of C2AB dissociation to a two-step model (eq 9), as described in the text. Insets show expansions of the lag phase (gray boxed regions), illustrating improved fitting from the two-step model. Data points are averages of 12 mixing events using the same solutions. The indicated rate constants are from measurements performed in parallel and are representative of 2 or more replicate experiments performed on different days with different preparations of vesicles. Rate constants varied by ∼40% between liposome preparations, but the reported trends between the proteins were consistent.
Figure 8.
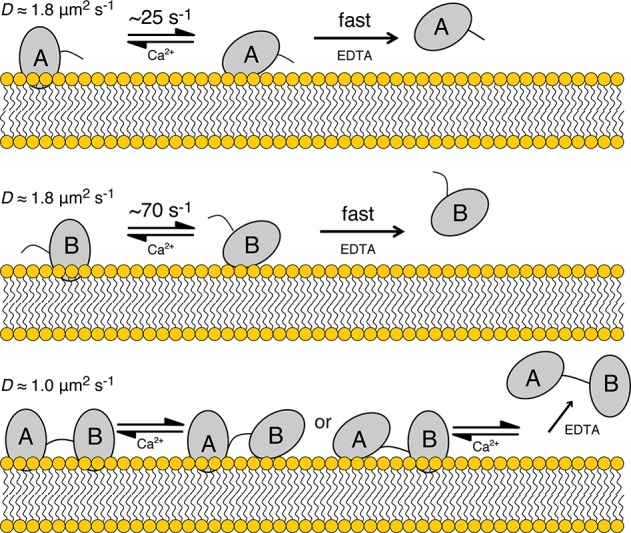
Model of Syt7 C2 membrane-bound states and dissociation. Membrane-bound states and dissociation mechanisms in the presence of excess Ca2+ are predicted on the basis of TIRFM measurements, whereas proposed mechanisms of EDTA-induced dissociation are based on koff measurements (Figure 7). (Top) C2A occupies a tightly bound, deeply inserted (left) and a weaker bound state in which Ca2+ ions are accessible to removal by EDTA (center). Thus, addition of EDTA results in a millisecond time scale depenetration followed by a rapid dissociation (right). (Middle) The isolated C2B domain may behave similarly to C2A but with faster depenetration kinetics. (Bottom) In the presence of Ca2+ (i.e., our TIRFM experiments), C2AB tandems are predominantly in a doubly inserted state (left) in which frictional coefficients of the two domains are additive. Upon addition of EDTA, depenetration and dissociation of C2A and C2B, in either sequential order, are observed with rate constants similar to those of the individual domains.
The best-fit rate constants of the two C2AB dissociation steps correspond well to those of the two individual domains. Importantly, the two-step model used for fitting does not specify the sequential order of the two events; eq 9 is identical if k2 and k1 are reversed. Either domain may dissociate first, or the sequence may be stochastic based on the individual domains’ dissociation kinetics. Thus, the data are fully consistent with a model in which C2A and C2B dissociate from the membrane independently (Figure 8).
In further support of independent dissociation, we observe that dissociation kinetics for the FE16 linker extension variant of Syt7 C2AB are virtually identical to the WT tandem domain (Figure 7B). Dissociation kinetics are also nearly identical for a variant of the C2AB tandem (PP12) with a rigid polyproline sequence inserted into the C2A–C2B linker region that prohibits interdomain interactions (Figure 7C).13 We note that the polyproline variant copurified with a lower-MW contaminant, likely a cleavage product (Figure S4). However, the dissociation kinetics clearly reveal a lag phase and overall kinetic profile closely similar to that of C2AB-WT. Thus, we conclude that binding of Syt7 C2AB to these target membranes does not include significant interdomain interactions but consists of independent membrane associations by the two domains.
Discussion
Analysis of Tandem Domain Diffusional States
Here, single-molecule diffusion measurements show that (a) the C2A and C2B domains of Syt7 diffuse similarly to that of other C2 domains on a 3:1 PC/PS supported lipid bilayer, (b) diffusion of the native C2AB tandem approaches the value predicted based on additive friction of the two individual domains, and (c) flexible extensions to the C2AB interdomain linker do not measurably impact diffusion constants. It is worth noting that different conclusions would have been reached if we had analyzed tandem domain diffusion using only one-component models (dashed lines in Figure 4), as these models produced significantly faster diffusion constants for C2AB-WT and C2AB-FE4 (1.12 ± 0.08 and 1.14 ± 0.11 μm2/s, respectively). Careful examination of the diffusion data revealed the population of fast-diffusing particles in these movies. Thus, the results underscore the importance of informed analysis of multiple states when analyzing single-molecule diffusion data.
Due to its relatively small population, the fast-diffusing state in the C2AB samples was most reproducibly modeled by fixing its diffusion constant at the value of a single domain and fitting to obtain the population in that state. The nature of this fast-diffusing state is unknown; however, SDS-PAGE results show that the fluorescent proteins were free of cleavage products (Figure 2). Protein absorbance spectra also demonstrate essentially complete removal of nucleic acid contaminants, which could compete with membranes for binding each C2 domain (Figure S1). Nevertheless, it is possible that small remaining contaminants could produce a population of molecules with only one membrane-active C2 domain. A reversible conformational equilibrium seems unlikely, as a breakdown of diffusion constant by trajectory (Figure 6) shows that the fast-diffusing population is stable on the time scale of entire trajectories.
Additive Friction
Previous studies have demonstrated additive friction for multidomain lipid-binding proteins using either (a) synthetic covalent multimers of a pleckstrin homology (PH) domain separated by 27-residue or longer flexible linkers,27 (b) noncovalent dimers induced by fusing the same PH domain to soluble interacting proteins,28 or (c) physiological combinations of C1 and C2 domains from protein kinase Cα (PKCα).25,44 PKCα contains a 15-residue flexible linker between its two C1 domains and a 20-residue linker between the C1b domain and the C2 domain.25 Thus, the present study of Syt C2AB tandems represents the shortest linker length examined for additive friction to date.
Camley and Brown have predicted that diffusion constants such as those measured here for multidomain proteins on supported lipid bilayers approach apparent frictional additivity when their bound lipids are separated by a few nanometers or more but diffuse significantly faster at shorter separation distances.29 It has not yet been possible to experimentally validate this prediction, as all tandem domains measured thus far have exhibited additive friction. Comparing single-domain to C2AB-WT diffusion yields a Ddimer/Dmonomer ratio of 0.54, with an uncertainty of ±0.05 obtained by propagating error from the 95% CI values of the individual measurements (Table 1). By contrast, Camley and Brown predict that protein domains ∼2 nm apart, each bound to a single lipid with a footprint radius of 0.45 nm, would have Ddimer/Dmonomer > 0.60.29 Deeply inserted C2 domains have a footprint radius of ∼1 nm, so a 2 nm center-to-center separation represents the distance of closest approach, at which the Ddimer/Dmonomer ratio is expected to be even higher.28,45 The relatively extended linker configuration illustrated in Figure 1A corresponds to a center-to-center separation distance of ∼4 nm. The observed near-additive friction suggests that the two domains remain well separated when bound to a PC/PS supported bilayer.
Dissociation Kinetics in Stopped-Flow and TIRFM Experiments
The time scale of dissociation induced by EDTA addition (Figure 7) is much shorter than the average dwell times of individual molecules on the membrane observed in the TIRFM experiments in the presence of Ca2+. Dissociation rate constants were not measured in TIRFM experiments due to photobleaching, but most trajectories of all domains had membrane-bound lifetimes of several hundred milliseconds or longer. Different off rates in the presence and absence of Ca2+ have been observed for other C2 domains as well and may arise from cooperative binding of Ca2+ ions and membranes.25,42,46,47 A simple model that accounts for this behavior is presented in Figure 8. Here, each individual domain exists in both a tightly membrane-bound (likely embedded) state and a loosely associated (likely surface-bound) state, the latter of which may or may not be bound to Ca2+ ions. In the presence of excess Ca2+, the two states are in equilibrium, with the tightly bound state predominating; thus, the TIRFM data show long-lived membrane-bound particles. In the presence of EDTA, loss of bound Ca2+ and membrane dissociation proceeds rapidly from the loosely associated state; thus, the transition from the tightly to loosely bound state is rate-limiting for EDTA-induced dissociation.
Implications for Structure of Membrane-Bound Syt7 C2AB and Relation to Syt1
Overall, our data are most consistent with independent membrane binding by the two C2 domains of Syt7. Previous studies have reported an absence of interdomain interactions for Syt1 C2AB in solution via NMR and single-molecule FRET.20,48 An alternative explanation of our diffusion results is that the two domains directly interact when bound to a supported bilayer but simultaneously insert more deeply into the bilayer than individual domains.17 However, such a direct interaction with deeper insertion would be expected to stabilize the membrane-bound state and significantly slow membrane dissociation, which we do not observe. Interdomain cooperativity in membrane insertion by Syt1 C2AB has been reported, although specific interdomain protein contacts have not been identified.14,22 However, it has also been observed that Syt1 C2AB can induce curvature, cluster lipids, and bridge between membranes, suggesting the forces that drive cooperativity may be more complex than interdomain interactions in a planar membrane.17,48−50 The supported bilayer system used in the TIRFM experiments precludes curvature and bridging, and the small sonicated vesicles used for stopped-flow likely prevent the induction of negative curvature, as has been implicated for Syt1.49,51,52 Further studies are needed to determine whether membrane curvature influences cooperativity in membrane binding.
Finally, analogous measurements could not be performed with Syt1, as Syt1 C2A dissociates too rapidly to measure lateral diffusion or dissociation kinetics accurately under the conditions used here. Thus, it is possible that the properties measured are unique to Syt7. Syt7 C2 domains dissociate from membranes much more slowly than the corresponding segments of other Syt isoforms, suggesting that they may have a larger membrane-binding interface.32,40 Furthermore, a hybrid form of Syt1 containing the C2B domain from Syt7 cannot rescue fast fusion in neurons from Syt1 knockout mice, suggesting that these two C2B domains function differently.11 Also, it was recently shown that chromaffin cell vesicles display dramatically different postfusion behavior depending on whether they contain Syt1 or Syt7, although it is not known whether the disparity stems from Syt–membrane or protein–protein interactions.53 Additional measurements, under conditions that favor longer-lived membrane association, may reveal whether additive friction and independent membrane association are unique to Syt7 or are common to other synaptotagmins.
Acknowledgments
We thank Dr. Joseph Falke for helpful discussions, Dr. Christopher Walsh for providing Sfp expression plasmid, and Drs. Michael Lerner and Brian Ziemba for critical comments on this manuscript.
Glossary
Abbreviations
- Syt
synaptotagmin
- SNARE
soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor
- GST
glutathione S-transferase
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- BME
β-mercaptoethanol
- AF488
Alexa Fluor 488
- DOPC or PC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- DOPS or PS
1,2-dioleoyl-sn-glycero-3-phospho-l-serine
- LRB-DOPE
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl)
- dansyl-PE
N-[5-(dimethylamino)naphthalene-1-sulfonyl]-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine
- SUV
small unilamellar vesicle
- SLB
supported lipid bilayer
- TIRFM
total internal reflection fluorescence microscopy
- PH
pleckstrin homology
- EDTA
ethylenediamine tetraacetic acid
- MSD
mean square displacement
- FRET
fluorescence resonance energy transfer
- PKC
protein kinase C
- CI
confidence interval
- FRAP
fluorescence recovery after photobleaching
Supporting Information Available
Representative movies of single-molecule membrane diffusion are available for LRB-DOPE and each of the protein constructs listed in Table 1. Table S1: Diffusion of Syt7 C2A is independent of fluorophore. Table S2: Diffusion of Syt7 C2B before and after addition of unlabeled C2A. Figure S1: Syt7 C2AB absorbance data. Figure S2: Ca2+ dependence of lipid binding and diffusion. Figure S3: FRAP analysis of supported lipid bilayers. Figure S4: Impurity in C2AB-PP12. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† (J.K.V.) Department of Chemistry, University of Wisconsin–Madison, Madison, Wisconsin 53706, United States.
Author Present Address
‡ (M.D.C.) Department of Medicine, Yale University School of Medicine, New Haven, Connecticut 06510, United States.
This work was supported by NIH R15GM102866 to J.D.K.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Rickman C.; Craxton M.; Osborne S.; Davletov B. (2004) Comparative analysis of tandem C2 domains from the mammalian synaptotagmin family. Biochem. J. 378, 681–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita S.; Shin O. H.; Han W.; Lao Y.; Sudhof T. C. (2002) Synaptotagmins form a hierarchy of exocytotic Ca2+ sensors with distinct Ca2+ affinities. EMBO J. 21, 270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J.; Chen X.; Arac D. (2006) Unraveling the mechanisms of synaptotagmin and SNARE function in neurotransmitter release. Trends Cell Biol. 16, 339–350. [DOI] [PubMed] [Google Scholar]
- Gustavsson N.; Han W. (2009) Calcium-sensing beyond neurotransmitters: functions of synaptotagmins in neuroendocrine and endocrine secretion. Biosci. Rep. 29, 245–259. [DOI] [PubMed] [Google Scholar]
- Chapman E. R. (2008) How does synaptotagmin trigger neurotransmitter release?. Annu. Rev. Biochem. 77, 615–641. [DOI] [PubMed] [Google Scholar]
- Gustavsson N.; Lao Y.; Maximov A.; Chuang J. C.; Kostromina E.; Repa J. J.; Li C.; Radda G. K.; Sudhof T. C.; Han W. (2008) Impaired insulin secretion and glucose intolerance in synaptotagmin-7 null mutant mice. Proc. Natl. Acad. Sci. U.S.A. 105, 3992–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A.; Sudhof T. C. (2005) Autonomous function of synaptotagmin 1 in triggering synchronous release independent of asynchronous release. Neuron 48, 547–554. [DOI] [PubMed] [Google Scholar]
- Liu H.; Bai H.; Hui E.; Yang L.; Evans C. S.; Wang Z.; Kwon S. E.; Chapman E. R. (2014) Synaptotagmin 7 functions as a Ca2+-sensor for synaptic vesicle replenishment. eLife 3, e01524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perin M. S.; Brose N.; Jahn R.; Südhof T. C. (1991) Domain structure of synaptotagmin (p65). J. Biol. Chem. 266, 623–629. [PubMed] [Google Scholar]
- Bhalla A.; Chicka M. C.; Chapman E. R. (2008) Analysis of the synaptotagmin family during reconstituted membrane fusion. Uncovering a class of inhibitory isoforms. J. Biol. Chem. 283, 21799–21807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M.; Craig T. K.; Shin O. H.; Li L.; Brautigam C. A.; Tomchick D. R.; Sudhof T. C.; Rosenmund C.; Rizo J. (2010) Structural and mutational analysis of functional differentiation between synaptotagmins-1 and -7. PLoS One 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof T. C. (2013) Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80, 675–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Bai H.; Xue R.; Takahashi H.; Edwardson J. M.; Chapman E. R. (2014) Linker mutations reveal the complexity of synaptotagmin 1 action during synaptic transmission. Nat. Neurosci. 17, 670–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J.; Wang P.; Chapman E. R. (2002) C2A activates a cryptic Ca2+-triggered membrane penetration activity within the C2B domain of synaptotagmin I. Proc. Natl. Acad. Sci. U.S.A. 99, 1665–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.; Wang C.-T.; Bai J.; Jackson M. B.; Chapman E. R. (2003) Mutations in the effector binding loops in the C2A and C2B domains of synaptotagmin I disrupt exocytosis in a nonadditive manner. J. Biol. Chem. 278, 47030–47037. [DOI] [PubMed] [Google Scholar]
- Schiavo G.; Gu Q. M.; Prestwich G. D.; Sollner T. H.; Rothman J. E. (1996) Calcium-dependent switching of the specificity of phosphoinositide binding to synaptotagmin. Proc. Natl. Acad. Sci. U.S.A. 93, 13327–13332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrick D. Z.; Kuo W.; Huang H.; Schwieters C. D.; Ellena J. F.; Cafiso D. S. (2009) Solution and membrane-bound conformations of the tandem C2A and C2B domains of synaptotagmin 1: evidence for bilayer bridging. J. Mol. Biol. 390, 913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennekate W.; Schroder S.; Lin C. C.; van den Bogaart G.; Grunwald M.; Jahn R.; Walla P. J. (2012) Cis- and trans-membrane interactions of synaptotagmin-1. Proc. Natl. Acad. Sci. U.S.A. 109, 11037–11042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Liu H.; Gu Y.; Chapman E. R. (2011) Reconstituted synaptotagmin I mediates vesicle docking, priming, and fusion. J. Cell Biol. 195, 1159–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi U. B.; Strop P.; Vrljic M.; Chu S.; Brunger A. T.; Weninger K. R. (2010) Single-molecule FRET-derived model of the synaptotagmin 1–SNARE fusion complex. Nat. Struct. Mol. Biol. 17, 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrick D. Z.; Sterbling S.; Rasch K. A.; Hinderliter A.; Cafiso D. S. (2006) Position of synaptotagmin I at the membrane interface: cooperative interactions of tandem C2 domains. Biochemistry 45, 9668–9674. [DOI] [PubMed] [Google Scholar]
- Hui E.; Bai J.; Chapman E. R. (2006) Ca2+-triggered simultaneous membrane penetration of the tandem C2-domains of synaptotagmin I. Biophys. J. 91, 1767–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffman P. G.; Delbruck M. (1975) Brownian motion in biological membranes. Proc. Natl. Acad. Sci. U.S.A. 72, 3111–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight J. D.; Falke J. J. (2009) Single-molecule fluorescence studies of a PH domain: new insights into the membrane docking reaction. Biophys. J. 96, 566–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemba B. P.; Falke J. J. (2013) Lateral diffusion of peripheral membrane proteins on supported lipid bilayers is controlled by the additive frictional drags of (1) bound lipids and (2) protein domains penetrating into the bilayer hydrocarbon core. Chem. Phys. Lipids 172–173, 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz W. L.; Clegg R. M.; Hallmann D. (1985) Translational diffusion of lipids in liquid crystalline phase phosphatidylcholine multibilayers. A comparison of experiment with theory. Biochemistry 24, 781–786. [DOI] [PubMed] [Google Scholar]
- Knight J. D.; Lerner M. G.; Marcano-Velazquez J. G.; Pastor R. W.; Falke J. J. (2010) Single molecule diffusion of membrane-bound proteins: window into lipid contacts and bilayer dynamics. Biophys. J. 99, 2879–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemba B. P.; Knight J. D.; Falke J. J. (2012) Assembly of membrane-bound protein complexes: detection and analysis by single molecule diffusion. Biochemistry 51, 1638–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camley B. A.; Brown F. L. H. (2013) Diffusion of complex objects embedded in free and supported lipid bilayer membranes: role of shape anisotropy and leaflet structure. Soft Matter 9, 4767–4779. [Google Scholar]
- Yin J.; Straight P. D.; McLoughlin S. M.; Zhou Z.; Lin A. J.; Golan D. E.; Kelleher N. L.; Kolter R.; Walsh C. T. (2005) Genetically encoded short peptide tag for versatile protein labeling by Sfp phosphopantetheinyl transferase. Proc. Natl. Acad. Sci. U.S.A. 102, 15815–15820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J.; Lin A. J.; Golan D. E.; Walsh C. T. (2006) Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nat. Protoc. 1, 280–285. [DOI] [PubMed] [Google Scholar]
- Brandt D. S.; Coffman M. D.; Falke J. J.; Knight J. D. (2012) Hydrophobic contributions to the membrane docking of synaptotagmin 7 C2A domain: mechanistic contrast between isoforms 1 and 7. Biochemistry 51, 7654–7664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubach J.; Lao Y.; Fernandez I.; Arac D.; Sudhof T. C.; Rizo J. (2001) The C2B domain of synaptotagmin I is a Ca2+-binding module. Biochemistry 40, 5854–5860. [DOI] [PubMed] [Google Scholar]
- Schütz G. J.; Schindler H.; Schmidt T. (1997) Single-molecule microscopy on model membranes reveals anomalous diffusion. Biophys. J. 73, 1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulian M.; Simon S. M. (2000) Tracking single proteins within cells. Biophys. J. 79, 2188–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalefski E. A.; Falke J. J. (2002) Use of fluorescence resonance energy transfer to monitor Ca2+-triggered membrane docking of C2 domains. Methods Mol. Biol. 172, 295–303. [DOI] [PubMed] [Google Scholar]
- Voelker D. R. (2008) Lipid assembly into cell membranes, In Biochemistry of Lipids, Lipoproteins and Membranes (Vance D. E., and Vance J. E., Eds.) 5th ed., pp 441–484, Elsevier, Amsterdam, The Netherlands. [Google Scholar]
- Takamori S.; Holt M.; Stenius K.; Lemke E. A.; Gronborg M.; Riedel D.; Urlaub H.; Schenck S.; Brugger B.; Ringler P.; Muller S. A.; Rammner B.; Grater F.; Hub J. S.; De Groot B. L.; Mieskes G.; Moriyama Y.; Klingauf J.; Grubmuller H.; Heuser J.; Wieland F.; Jahn R. (2006) Molecular anatomy of a trafficking organelle. Cell 127, 831–846. [DOI] [PubMed] [Google Scholar]
- Corbin J. A.; Dirkx R. A.; Falke J. J. (2004) GRP1 pleckstrin homology domain: activation parameters and novel search mechanism for rare target lipid. Biochemistry 43, 16161–16173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E.; Bai J.; Wang P.; Sugimori M.; Llinas R. R.; Chapman E. R. (2005) Three distinct kinetic groupings of the synaptotagmin family: candidate sensors for rapid and delayed exocytosis. Proc. Natl. Acad. Sci. U.S.A. 102, 5210–5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scomparin C.; Lecuyer S.; Ferreira M.; Charitat T.; Tinland B. (2009) Diffusion in supported lipid bilayers: influence of substrate and preparation technique on the internal dynamics. Eur. Phys. J. E: Soft Matter Biol. Phys. 28, 211–220. [DOI] [PubMed] [Google Scholar]
- Nalefski E. A.; Slazas M. M.; Falke J. J. (1997) Ca2+-signaling cycle of a membrane-docking C2 domain. Biochemistry 36, 12011–12018. [DOI] [PubMed] [Google Scholar]
- Wu P.; Brand L. (1994) Resonance energy transfer: methods and applications. Anal. Biochem. 218, 1–13. [DOI] [PubMed] [Google Scholar]
- Ziemba B. P.; Li J.; Landgraf K. E.; Knight J. D.; Voth G. A.; Falke J. J. (2014) Single-molecule studies reveal a hidden key step in the activation mechanism of membrane-bound protein kinase C-alpha. Biochemistry 53, 1697–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaud S.; Tobias D. J.; Falke J. J.; White S. H. (2007) Self-induced docking site of a deeply embedded peripheral membrane protein. Biophys. J. 92, 517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin J. A.; Evans J. H.; Landgraf K. E.; Falke J. J. (2007) Mechanism of specific membrane targeting by C2 domains: localized pools of target lipids enhance Ca2+ affinity. Biochemistry 46, 4322–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertz J. A.; Almeida P. F.; Frazier A. A.; Berg A. K.; Hinderliter A. (2007) The cooperative response of synaptotagmin I C2A. A hypothesis for a Ca2+-driven molecular hammer. Biophys. J. 92, 1409–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arac D.; Chen X.; Khant H. A.; Ubach J.; Ludtke S. J.; Kikkawa M.; Johnson A. E.; Chiu W.; Sudhof T. C.; Rizo J. (2006) Close membrane-membrane proximity induced by Ca2+-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat. Struct. Mol. Biol. 13, 209–217. [DOI] [PubMed] [Google Scholar]
- Hui E.; Johnson C. P.; Yao J.; Dunning F. M.; Chapman E. R. (2009) Synaptotagmin-mediated bending of the target membrane is a critical step in Ca2+-regulated fusion. Cell 138, 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A. L.; Tamm L. K.; Ellena J. F.; Cafiso D. S. (2011) Synaptotagmin 1 modulates lipid acyl chain order in lipid bilayers by demixing phosphatidylserine. J. Biol. Chem. 286, 25291–25300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens S.; McMahon H. T. (2008) Mechanisms of membrane fusion: disparate players and common principles. Nat. Rev. Mol. Cell Biol. 9, 543–556. [DOI] [PubMed] [Google Scholar]
- Zimmerberg J.; Akimov S. A.; Frolov V. (2006) Synaptotagmin: fusogenic role for calcium sensor?. Nat. Struct. Mol. Biol. 13, 301–303. [DOI] [PubMed] [Google Scholar]
- Rao T. C.; Passmore D. R.; Peleman A. R.; Das M.; Chapman E. R.; Anantharam A. (2014) Distinct fusion properties of synaptotagmin-1 and synaptotagmin-7 bearing dense core granules. Mol. Biol. Cell 25, 2416–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian H.; Sheetz M. P.; Elson E. L. (1991) Single particle tracking. Analysis of diffusion and flow in two-dimensional systems. Biophys. J. 60, 910–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.